

Abstract
Varicella-zoster virus (VZV) open reading frame 61 (ORF61) encodes a protein that transactivates viral and cellular promoters in transient-transfection assays and is the ortholog of herpes simplex virus ICP0. In this report, we mapped the ORF61 promoter and investigated its regulation by viral and cellular proteins in transient-expression experiments and by mutagenesis of the VZV genome (parent Oka strain). The 5′ boundary of the minimal ORF61 promoter required for IE62 transactivation was mapped to position −95 relative to the mRNA start site, and three noncanonical GT-rich Sp1-binding sites were documented to occur within the region comprising positions −95 to −45. Contributions of the three Sp1-binding-site motifs, designated Sp1a, Sp1b, and Sp1c, to ORF61 expression and viral replication were varied despite their similar sequences. Two sites, Sp1a and Sp1c, functioned synergistically. When both sites were mutated in the pOka genome to produce pOka-61proΔSp1ac, the mutant virus expressed significantly less ORF61 protein. Using this mutant to investigate ORF61 functions resulted in reductions in the expression levels of IE proteins, viral kinases ORF47 and ORF66, and the major glycoprotein gE, with the most impact on gE. Virion morphogenesis appeared to be intact despite minimal ORF61 expression. Pretreating melanoma cells with sodium butyrate enhanced titers of pOka-61proΔSp1ac but not pOka, suggesting that ORF61 has a role in histone deacetylase inhibition. Growth of pOka-61proΔSp1ac was impaired in SCIDhu skin xenografts, indicating that the regulation of the ORF61 promoter by Sp1 family proteins is important for ORF61 expression in vivo and that ORF61 contributes to VZV virulence at skin sites of replication.
Varicella-zoster virus (VZV) is a human alphaherpesvirus that causes varicella (chicken pox) during primary infection and establishes lifelong latency in sensory ganglia; reactivation results in herpes zoster (shingles) (4). During lytic infection, VZV genes are presumed to be transcribed as a cascade of three kinetic classes: immediate early (IE), early (E), and late (L). VZV depends on host RNA polymerase II and cellular transcription factors for viral gene transcription, along with at least five VZV-encoded gene products, including ORF10, IE4, ORF61, IE62, and IE63 (22, 45). Among the viral regulatory proteins, IE62 is the major transactivator and induces all three kinetic classes of viral genes (38, 46, 70). The direct binding of IE62 to DNA, in association with cellular transcription factors, including the specificity protein 1 (Sp1) family, upstream stimulatory factor (USF), the TATA binding protein (TBP), TFIIB, and upstream cis-activating elements in VZV promoters, is involved in IE62 activation of VZV genes (22, 68, 69, 88). IE4, IE63, and ORF61 modulate IE62-mediated transactivation in transient-transfection assays, and physical interactions between IE62 and IE63 or IE4 occur in VZV-infected cells (7, 38, 40, 52, 59, 61, 70, 81, 82). IE62 alone, or with IE4 or ORF61, induced ORF61 promoter activity in transfections of a T-cell line (70).
ORF61 is the ortholog of herpes simplex virus (HSV) ICP0, but comparatively little is known about ORF61 regulation and functions. Transient expression of ORF61 protein enhanced VZV DNA infectivity, and deletion of a large portion of ORF61 impaired VZV replication in cell culture (21, 59). Our recent single-cell analysis of the kinetics of VZV protein expression demonstrated that ORF61 is expressed at very early times after virus entry into human fibroblasts; of interest, ORF61 localized to nuclear compartments that expressed promyelocytic leukemia protein (PML) and were distinct from those where IE62 protein accumulated at the same time after virus entry (73). ORF61 promiscuously transactivated a number of viral and cellular promoters, including its own, in transient-expression assays, and depending on the cell line used, ORF61 has been reported to either repress or enhance the activation of VZV gene promoters by IE4 and IE62 (38, 59, 61, 70). Since ORF61 is not a DNA-binding protein (27), its regulatory activity is presumed to be indirect.
ORF61 provided in trans functionally complemented an HSV ICP0 deletion mutant, suggesting that it has functions related to those of ICP0 (58). Among these, ICP0 stimulates the expression of IE, E, and L viral genes and many heterologous genes in transfection assays (26, 32, 64); upregulates E and L but not necessarily IE gene expression during productive infection (12, 17); and causes degradation of PML, Sp100, DNA-dependent protein kinase, cdc34, and centromere proteins (16, 28, 29, 34, 49, 50, 60, 66, 67). These ICP0 functions require the zinc RING finger domain, which is conserved among alphaherpesvirus orthologs, including ORF61. ICP0 binds to class II but not class I histone deacetylases (HDACs), can overcome HDAC-induced repression, and interacts with CoREST, displacing HDAC1 from the CoREST-REST-HDAC1/2 repression complex (33, 51). In addition, ICP0 interacts with cellular proteins in multiple pathways, including transcription (41), translation (42), cell cycle control (9, 43), and protein degradation (30, 35, 85) pathways. In vivo, ICP0 enhanced HSV-1 reactivation efficiency (11, 19, 36, 48).
Analyses of specific VZV gene promoters have identified functional motifs for cis- as well as trans-regulating factors. Sp1 regulates the gE and gI promoters in transient-transfection assays, and GC-rich elements have been identified in IE4 and IE63 promoters (37, 47, 72). Sp1 binds directly to the gI promoter and recruits IE62 (68). The importance of cellular and viral transactivator interactions for VZV virulence in vivo was first demonstrated by showing that disrupting the Sp1 and USF sites in the gI promoter attenuated viral growth in human skin and T-cell xenografts in the SCIDhu mouse model (39). VZV gE, which is an essential protein, also has Sp1 motifs in its promoter that are necessary for viral replication (7). Of interest, these gI and gE promoter studies indicated that noncanonical Sp1-binding sites were more important for VZV gene regulation than the canonical sites characteristic of cell gene promoters (7, 76). Another cellular factor, USF, recognizes 5′-CACGTG-3′ consensus sequences and regulates IE62-dependent activation of the ORF28/29 promoter (53, 78). USF also contributes to the activation of the VZV promoters of IE4, gI, and ORF10 (15, 37, 54). Binding of USF to the ORF10 promoter was dispensable in vitro but contributed to VZV virulence in skin xenografts in vivo (15).
In this study, we conducted a detailed analysis of ORF61 promoter regulation by VZV proteins and Sp1 family proteins. The contribution of Sp1 family proteins to viral replication and VZV gene expression in vitro was evaluated by introducing Sp1 site mutations into the ORF61 promoter in the VZV genome. The very limited ORF61 expression of one of these mutants allowed an assessment of the effects of ORF61 protein on viral gene expression and virion morphogenesis in vitro and the contribution of ORF61 to VZV pathogenesis in SCIDhu skin xenografts in vivo.
MATERIALS AND METHODS
Cell lines.
Human melanoma cells and human embryonic lung fibroblasts (HELF) were cultured in minimal essential medium (Mediatech, Washington, DC) supplemented with 10% fetal bovine serum, nonessential amino acids, and antibiotics.
Antibodies and chemicals.
Rabbit polyclonal antibodies against IE4, ORF61, IE62, and IE63 were generous gifts from P. R. Kinchington, University of Pittsburgh. Polyclonal antibodies against ORF47 and ORF66 were generated from rabbit immunization with glutathione S-transferase fusion proteins (8, 79). Mouse monoclonal anti-gE antibody 3B3 was kindly provided by C. Grose, University of Iowa (77). Sodium butyrate (NaBu) and trichostatin A (TSA) (Sigma) were prepared as 500 mM NaBu stock solution in double-distilled water and 2 mM TSA stock solution in dimethyl sulfoxide.
Plasmids.
Plasmids pCMV4, pCMV10, pCMV61, pCMV62, and pCMV63, in which VZV transactivators IE4, ORF10, ORF61, IE62, and IE63, respectively, are expressed under the control of the CMV IE promoter, were kindly provided by P. R. Kinchington (University of Pittsburgh, Pittsburgh, PA). ORF61 promoter luciferase reporter plasmids were constructed from the pGL3-Basic vector (Promega). The full-length and stepwise deletions of the intergenic region between ORF61 and ORF62 were amplified by PCR using primers containing NcoI sites at the 5′ end and MluI sites at the 3′ end. The PCR product was cloned into the pGL3-Basic plasmid upstream of the firefly luciferase reporter gene. Sp1-binding-site mutant constructs were made in the context of pGL3-61pro(−299) by using a two-round PCR method. In the first-round PCR, two PCRs were performed (one with the 5′ primer covering the NcoI restriction site and the antisense primer containing mutated nucleotides and one with the 3′ primer covering the MluI restriction site and the sense primer containing mutated nucleotides). The second-round PCR was done using the two first-round PCR products as templates and the 5′ and 3′ primers described above. The mutated fragment was digested by NcoI and MluI and ligated to NcoI/MluI-digested pGL3-Basic vector. All constructs were confirmed by nucleotide sequencing (Elim Biopharm, Inc., Hayward, CA).
Transfection and luciferase assay.
Melanoma cells (2 × 105) were seeded in each well in 24-well plates on the day before transfection. Transfections were done with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) in accordance with the manufacturer's instructions. For each experiment, three independent transfections were performed. In experiments studying the effects of VZV transactivators on ORF61 promoter activity, 0.9 μg of pGL3-61proFL was transfected either alone or together with 0.05 μg of pCMV4, pCMV10, pCMV61, pCMV62, or pCMV63, individually or in combination. Empty pBluescript vector (Stratagene, La Jolla, CA) was used to adjust the total DNA to 1 μg. In experiments mapping the promoter boundaries and identifying cis-regulatory elements, 0.05 μg pCMV62 was transfected together with 0.95 μg of each ORF61 promoter construct. In transfection and superinfection experiments, infected cells were added to the transfected cells at a ratio of 1 infected cell per 8 uninfected cells at 6 h posttransfection; the luciferase assay was performed 20 h after infection. In all transfections, 0.07 ng of the plasmid pRL-TK(−), from which the TK promoter has been removed, was included to normalize transfection efficiency. Cells were lysed in 1× passive lysis buffer (Promega) at 24 h posttransfection. Luciferase assays were performed using a dual luciferase kit (Promega) in accordance with the manufacturer's recommendations.
Electrophoretic mobility shift assay (EMSA).
Two wild-type complementary oligonucleotides covering each putative Sp1 site were synthesized as follows: Sp1a-wtF, 5′-CCACGTTTTAGTGGGTGGGACTTAAAAGAA-3′; Sp1a-wtR, 5′-TTCTTTTAAGTCCCACCCACTAAAACGTGG-3′; Sp1b-wtF, 5′-CTTAAAAGAAATGGGTGGAGGGATATAGG-3′; Sp1b-wtR, 5′-CCTATATCCCTCCACCCATTTCTTTTAAG-3′; Sp1c-wtF, 5′-GGAGGGATATAGGGGTGTGTCTTCGTTGGT-3′; and Sp1c-wtR, 5′-ACCAACGAAGACACACCCCTATATCCCTCC-3′. Single-stranded oligonucleotide was labeled with biotin by using a biotin 3′-end DNA labeling kit (Pierce, Rockford, IL) in accordance with the manufacturer's instructions. Complementary oligonucleotides were denatured at 95°C for 5 min and annealed. One hundred fifty nanograms of purified human Sp1 protein was incubated with 40 fmol biotin-labeled double-stranded wild-type probe in 1× binding buffer [4% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM dithiothreitol, 50 mM NaCl, 10 mM Tris-HCl (pH 7.5), and 50 μg/ml poly(dI-dC)] at 23°C for 20 min. For competition experiments, a 5-fold or 50-fold molar excess of unlabeled wild-type probe was preincubated with Sp1 protein for 5 min before biotin-labeled wild-type probe was added and incubated for another 15 min. For mutant probe binding experiments, six mutant oligonucleotides were synthesized (Sp1a-muF, 5′-CCACGTTTTAGTGAACTAGACTTAAAAGAA-3′; Sp1a-muR, 5′-TTCTTTTAAGTCTAGTTCACTAAAACGTGG-3′; Sp1b-muF,5′-CTTAAAAGAAATGAACTGAGGGATATAGG-3′; Sp1b-muR, 5′-CCTATATCCCTCAGTTCATTTCTTTTAAG-3′; Sp1c-muF, 5′-GGAGGGATATAGCAAGATGTCTTCGTTGGT-3′; and Sp1c-muR, 5′-ACCAACGAAGACATCTTGCTATATCCCTCC-3′). They were biotin labeled and annealed as described above. One hundred fifty nanograms of purified human Sp1 protein was incubated with 40 fmol biotin-labeled double-stranded mutant probe in 1× binding buffer. The binding samples were resolved on a 1.5% agarose gel in 0.5× Tris-borate-EDTA. DNA probes were then transferred to positively charged nylon membrane (Ambion, Inc.) via a capillary transfer method and detected by a chemiluminescence nucleic acid detection module (Pierce, Rockford, IL).
Generation of pOka recombinant viruses with mutations in the ORF61 promoter.
Recombinant viruses were generated using cosmids derived from pOka (63). The entire pOka genome is covered in four overlapping cosmids, designated Fsp73 (pOka nucleotides [nt] 1 to 33128), Spe14 (pOka nt 21795 to 61868), Pme2 (pOka nt 53755 to 96035), and Spe23 (pOka nt 94055 to 125124). The ORF61 promoter is located in the unique long region in the cosmid Spe23, upstream of the ORF61 start codon (pOka nt 104445). In order to make mutations in the ORF61 promoter, a 12.7-kb EcoRI-AvrII fragment from Spe23 was subcloned into the pLitmus28 plasmid (Invitrogen, Inc.) to generate pLit(ORF59-65), in which PstI and PmlI are unique restriction sites. Mutations of ORF61 promoter were introduced to pLit(ORF59-65) by a two-round PCR method as described above. The outside primer P1 covers a pOka region (nt 101464 to 101481) in the coding strand and contains the unique PstI restriction site; the other outside primer, P2, covers a pOka region (nt 106187 to 106204) in the complementary strand and contains the unique PmlI restriction site. The primers used to mutate the Sp1-binding sites were the same as those used for EMSA. The PCR product containing the desired mutation was digested with PstI and PmlI before ligation to PstI/PmlI-digested pLit(ORF59-65). The mutations were subsequently introduced into Spe23 by ligating the NheI/AvrII fragment obtained from pLit(ORF59-65) to NheI/AvrII-digested Spe23.
Recombinant viruses, designated pOka-61proΔSp1a, pOka-61proΔSp1b, pOka-61proΔSp1c, and pOka-61proΔSp1ac, were isolated by transfection of melanoma cells with the mutated Spe23 cosmid and the other three intact cosmids, Fsp73, Spe14, and Pme2. To confirm the targeted mutations in the ORF61 promoter, genomic DNA was extracted from virus-infected melanoma cells or HELF with the DNAzol reagent (Invitrogen, Carlsbad, CA). A PCR fragment covering the mutated region was amplified from the genomic DNA by using Pfu polymerase (Stratagene, La Jolla, CA), gel purified with a QIAquick gel extraction kit (Qiagen, Inc., Valencia, CA), and sequenced (Elim Biopharm, Inc., Hayward, CA).
Growth kinetics and plaque size.
The replication kinetics of recombinant viruses was assessed by an infectious focus assay with immunostaining to detect plaques as previously described (14, 55). Images of immunostained plaques were photographed with a light microscope (Zeiss) at ×5 magnification. Plaque size was determined by measuring the surface area of 30 plaques per virus with ImageJ software (1). Statistical differences in growth kinetics and plaque sizes were determined by Student's t test.
Western blot analysis.
Mock- or virus-infected melanoma or HELF cells were harvested and lysed in 1× radioimmunoprecipitation assay lysis buffer. Proteins were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA) with a semidry transfer cell (Bio-Rad). The membranes were blocked overnight at 4°C in 5% nonfat milk in TBST (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween 20), incubated with specific antiserum at room temperature (RT) for 2 h, washed three times with TBST, incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse immunoglobulin (Amersham) at RT for 1 h, and washed three times with TBST. Proteins were detected using the enhanced chemiluminescence plus detection system (Amersham).
Immunofluorescence.
HELF were grown on chamber slides and infected with pOka and pOka-61proΔSp1ac. At 48 h postinfection, the cells were fixed with 4% paraformaldehyde in 1× phosphate-buffered saline (PBS) and permeabilized with 0.2% Triton X-100. Cells were then incubated with gE monoclonal antibody diluted in dilution buffer (PBS with 5% newborn calf serum) at RT for 2 h, washed with 1× PBS, and incubated with fluorescein isothiocyanate-conjugated anti-mouse immunoglobulin (Jackson ImmunoResearch, Inc.) in dilution buffer at 4°C for 1 h. For the cell tracker experiment, infected inoculum cells were labeled with the BODIPY-green cell tracker (Invitrogen) as described previously (73). For dual detection of gE and IE62, infected HELF were incubated with gE monoclonal antibody and IE62 polyclonal antibody, followed by incubation with fluorescein isothiocyanate-conjugated anti-mouse immunoglobulin and Texas Red-conjugated anti-rabbit immunoglobulin. Cell nuclei were stained with 2 μg/ml Hoechst33342 for 10 min after secondary antibody incubation. Images were obtained with a Leica confocal microscope.
Transmission electron microscopy (TEM).
HELF infected with pOka and pOka-61proΔSp1ac were fixed with 2% glutaraldehyde in 0.1 M phosphate buffer (PBS), pH 7.0, for 45 min. The specimens were washed twice in PBS and postfixed with 1% osmium tetroxide (Polysciences, Inc., Warrington, PA) in PBS for 1 h, and after two 10-min washes in double-distilled water, specimens were treated with 0.25% uranyl acetate (Polysciences, Inc., Warrington, PA) overnight. After 24 h, the specimens were washed with water and dehydrated through a graded series of alcohol and propylene oxide washes. Each sample was infiltrated sequentially with 2:1 and 1:1 propylene oxide-Epon (Resolution Performance Products, Houston, TX) for 4 h, incubated overnight with 100% Epon, transferred to fresh Epon, and embedded and polymerized at 60°C for 24 h. Thin sections were collected on copper grids, stained with uranyl acetate and lead citrate, and viewed using a Phillips CM-12 transmission electron microscope.
Infection of skin xenografts in SCIDhu mice.
Skin xenografts were made in homozygous CB-17scid/scid mice, using human fetal tissue supplied by Advanced Bioscience Resources (Alameda, CA) in accordance with federal and state regulations (55, 56). Animal use was in accordance with the Animal Welfare Act and approved by the Stanford University Administrative Panel on Laboratory Animal Care. pOka and ORF61 promoter mutant viruses were passed three times in primary HELF and their titers determined before inoculation. Skin xenografts were harvested on days 11 and 21 to determine titers by an infectious focus assay (14, 55). DNA was extracted from skin tissue with proteinase K and phenol chloroform (Invitrogen, Carlsbad, CA). PCR and sequencing were performed to confirm the expected mutations.
RESULTS
Transactivation of the ORF61 promoter by VZV regulatory proteins.
In the transactivation experiments, the 717-bp ORF61-ORF62 intergenic region was cloned into the pGL3-Basic firefly luciferase reporter plasmid (Promega) to generate pGL3-61proFL. The five plasmids encoding VZV transactivating proteins, pCMV4, pCMV10, pCMV61, pCMV62, and pCMV63, were individually cotransfected with pGL3-61proFL into melanoma cells for luciferase assays. The pRL-TK(−) plasmid, from which the HSV TK promoter has been removed, was included as an internal control. Renilla luciferase readings with the empty vector were similar to those with viral transactivators (data not shown), suggesting that these viral products do not have a trans effect on the Renilla luciferase gene. The transactivation results were presented as n-fold induction compared to the luciferase activity with the empty vector, which was normalized to 1 (Fig. 1a). As expected, IE62 potently transactivated the ORF61 promoter (about 134-fold). The effects of IE4, ORF61, and IE63 were modest (about 1.5-fold, 2-fold, and 1.5-fold, respectively). Transactivation by ORF10 was not detected. To evaluate the regulatory roles of the other transactivators on IE62-mediated transactivation of the ORF61 promoter, IE62 was coexpressed with IE4, ORF61, or IE63. IE4 and IE63 increased the induction of the ORF61 promoter by IE62, while ORF61 had a suppressing effect (Fig. 1a). The combined effect of ORF61 and IE63 on the ORF61 promoter was assessed, as ORF61 has been shown to facilitate IE63 nuclear import in primary neurons (86), but no significant effect was observed (data not shown). These results confirm that IE62 is the primary viral transactivator of the ORF61 promoter in melanoma cells and show that this effect is modulated by ORF61, IE4, and IE63.
FIG. 1.
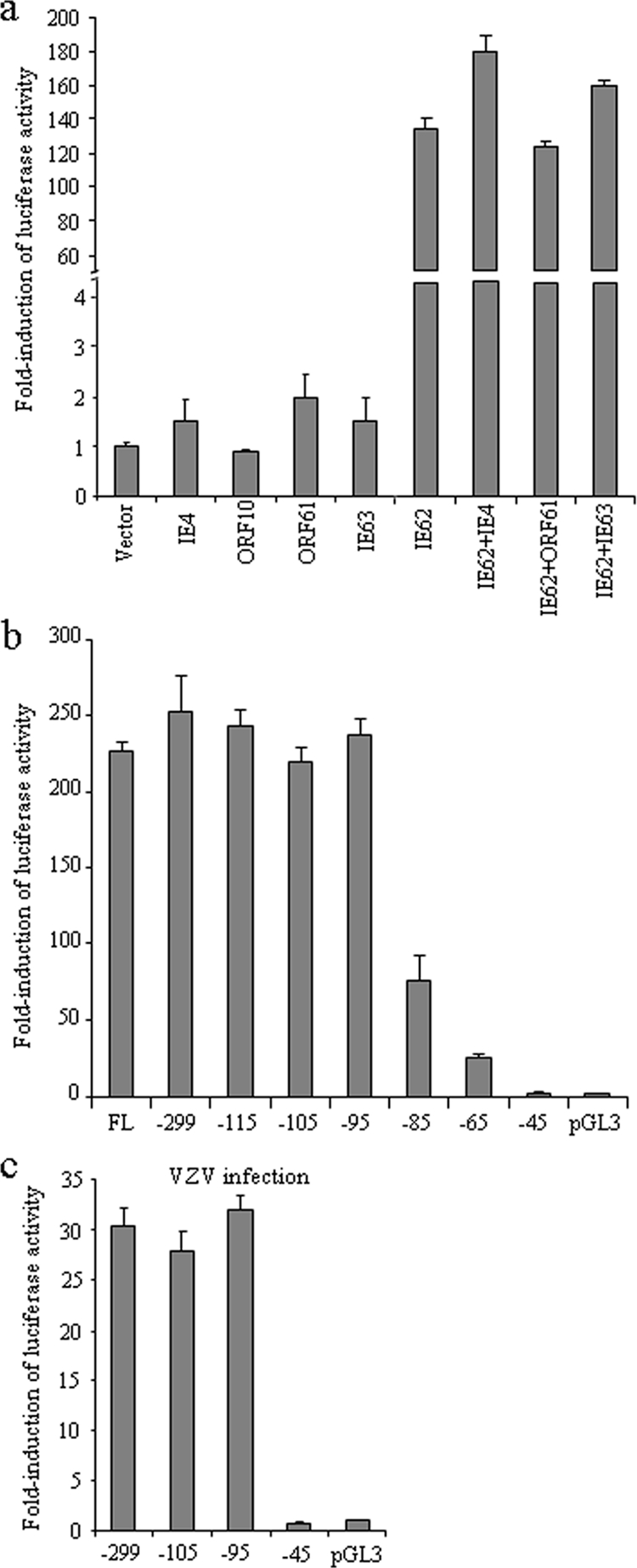
Transactivation of the ORF61 promoter by VZV proteins and mapping of the 5′ boundary of the ORF61 promoter. (a) Melanoma cells were transfected with pGL3-61proFL and plasmids expressing IE4, ORF10, ORF61, IE62, IE63, or IE62 together with IE4, ORF61, or IE63; empty pBluescript vector was used as the negative control. Renilla luciferase plasmid pRL-TK(−) was included in each transfection to normalize the transfection efficiency. Cells were harvested at 24 h posttransfection, and the dual luciferase assay was performed. The results are presented as n-fold induction compared to the luciferase activity with the empty vector, which was normalized to 1. (b) Stepwise deletions from the 5′ end of the ORF61 promoter were cloned into the pGL3-Basic luciferase reporter vector. Melanoma cells were cotransfected with full-length and deletion mutant constructs and pCMV62; pGL3-Basic was the promoter-null control. The luciferase activities of these promoter deletion constructs are presented as n-fold induction compared to that of the promoter-null control, which was set as 1. The bars indicate the means ± standard deviations (SD) of results from three independent transfections. (c) Melanoma cells were transfected with ORF61 promoter deletion constructs and superinfected with VZV at 6 h posttransfection. Luciferase assays were performed at 20 h after infection.
Mapping the 5′ boundary of the ORF61 promoter required for IE62 activation.
ORF61 mRNA initiates at 65 bp upstream from the translation start codon (83). To define the 5′ boundary of the promoter precisely, a series of promoter constructs with stepwise 5′ deletions were made and transfected into melanoma cells together with pCMV62. The luciferase activities of these promoter constructs were presented as n-fold induction compared to that of the pGL3-Basic promoter-null control, which was set as 1 (Fig. 1b). The induction remained approximately the same with the deletions at positions −299, −115, −105, and −95 relative to the mRNA initiation site. A reduction of ∼70% was observed with the position −85 deletion, and ∼90% reduction was observed with the position −65 deletion. With the position −45 deletion, luciferase activity dropped to the basal level observed with the pGL3-Basic control. A transfection and VZV superinfection experiment with position −299, −105, −95, and −45 deletion constructs was performed to map the promoter boundary in the context of virus infection and showed similar results (Fig. 1c). This deletion analysis suggested that an important cis-regulating element(s) was located in the region from position −95 to position −45 relative to the mRNA initiation site in the ORF61 promoter.
Identification of three putative Sp1-binding sites in the ORF61 promoter.
To identify potential cellular transcription factor binding motifs, the sequence from position −115 to the 3′ end of the ORF61 promoter was analyzed by the transcription factor motif prediction software programs TESS (80), MatInspector (13), and Motif (http://motif.genome.jp/). Putative binding sites for USF and Sp1 and TATA elements were identified (Fig. 2a). The position −95 deletion had no effect on either IE62-mediated or VZV infection induced-transactivation (Fig. 1b and c), indicating that USF is not essential for regulating the ORF61 promoter. Three GT-rich Sp1-binding sites were predicted to occur at positions −91 to −86, −72 to −67, and −56 to −51; these sites were designated Sp1a, Sp1b, and Sp1c, respectively (Fig. 2a). Since luciferase activity decreased gradually with stepwise deletions from position −95 to position −45 (Fig. 1b), it appeared that the three putative Sp1 sites might function together to mediate the activity of the ORF61 promoter. To verify this hypothesis, seven Sp1 mutation constructs were made in the context of pGL3-61pro(−299) (Fig. 2b), which retained the sequence required for the ORF61 promoter response to IE62 (Fig. 1b). These seven promoter constructs were transfected into melanoma cells with pCMV62, and dual luciferase assays were performed (Fig. 2c). Disruption of the Sp1b site alone had a minor effect on the promoter (∼25% reduction), whereas disruption of the Sp1a or Sp1c site caused a larger reduction in luciferase activity (∼60% and ∼40%, respectively). Luciferase activity was decreased by ∼85% when both Sp1a and Sp1b sites were mutated in the same construct and by ∼70% when a double mutation of the Sp1b and Sp1c sites was made. When the Sp1a and Sp1c sites were mutated together, more than 95% of the ORF61 promoter activity was lost. These observations suggest that Sp1a and Sp1c sites function to cis regulate the ORF61 promoter synergistically and account for most of the Sp1 contribution to ORF61 promoter activation. This assessment was supported by the observation that disrupting all three sites simultaneously had an effect similar to the dual mutation of Sp1a and Sp1c sites (Fig. 2c). A transfection and superinfection experiment was performed to evaluate the contribution of each Sp1 site to ORF61 promoter activity in the context of VZV infection. ORF61 promoter activity was decreased by 74.3% when Sp1a was disrupted, by 16.7% when Sp1b was disrupted, and by 50% when Sp1c was disrupted (data not shown).
FIG. 2.

Identification of three putative Sp1-binding sites in the ORF61 promoter. (a) Schematic representation of the ORF61 promoter from position −115 to position +65 relative to the mRNA initiation site. Potential transcription factor binding sites predicted by TESS, MatInspector, and Motif are distinguished with lowercase letters. Sp1-binding sites are underlined. The mRNA initiation site is shown by an arrow. (b) Individual or combinational mutations of the three putative Sp1-binding sites in the ORF61 promoter were made in plasmid pGL3-61pro(−299), in which the ORF61 promoter region from position −299 to position +65 was cloned upstream of the firefly luciferase reporter gene. Nucleotide changes in each mutant construct are indicated with bold. (c) Melanoma cells were cotransfected with Sp1 site mutant plasmids and pCMV62. The luciferase activities of these mutant promoter constructs are presented as n-fold induction compared to that of the pGL3-Basic promoter-null control, which was set as 1. The bars indicate means ± SD of results from three independent transfections.
Sp1 protein binds to the putative Sp1 sites in the ORF61 promoter in vitro.
The Sp1-binding elements that have been shown to occur in the VZV genome have the sequences 5′-GGGCGGG-3′ and 5′-ACGCCC-3′ (37, 72). Moreover, Sp1 sites in the ORF61 promoter were predicted only by TESS but not by the other two bioinformatics programs. Therefore, EMSA was done to determine whether Sp1 bound to any of the three GT boxes in the ORF61 promoter in vitro (Fig. 3). Three probes, Sp1a, Sp1b, and Sp1c, each containing only one Sp1-binding site, were labeled with biotin. In the presence of Sp1 protein, Sp1a, Sp1b, and Sp1c were shifted to a higher position (Fig. 3, lanes 1 and 2), demonstrating that purified human Sp1 protein binds to all three sites in the ORF61 promoter. This conclusion was reinforced by using nonlabeled probes in competition experiments and mutated probes. Binding of Sp1 protein to the biotin-labeled wild-type probes was inhibited by preincubation of Sp1 protein with 5-fold and 50-fold molar excesses of nonlabeled wild-type probes (Fig. 3, lanes 3 and 4). Mutated probes failed to bind to Sp1 protein (Fig. 3, lanes 5 and 6).
FIG. 3.
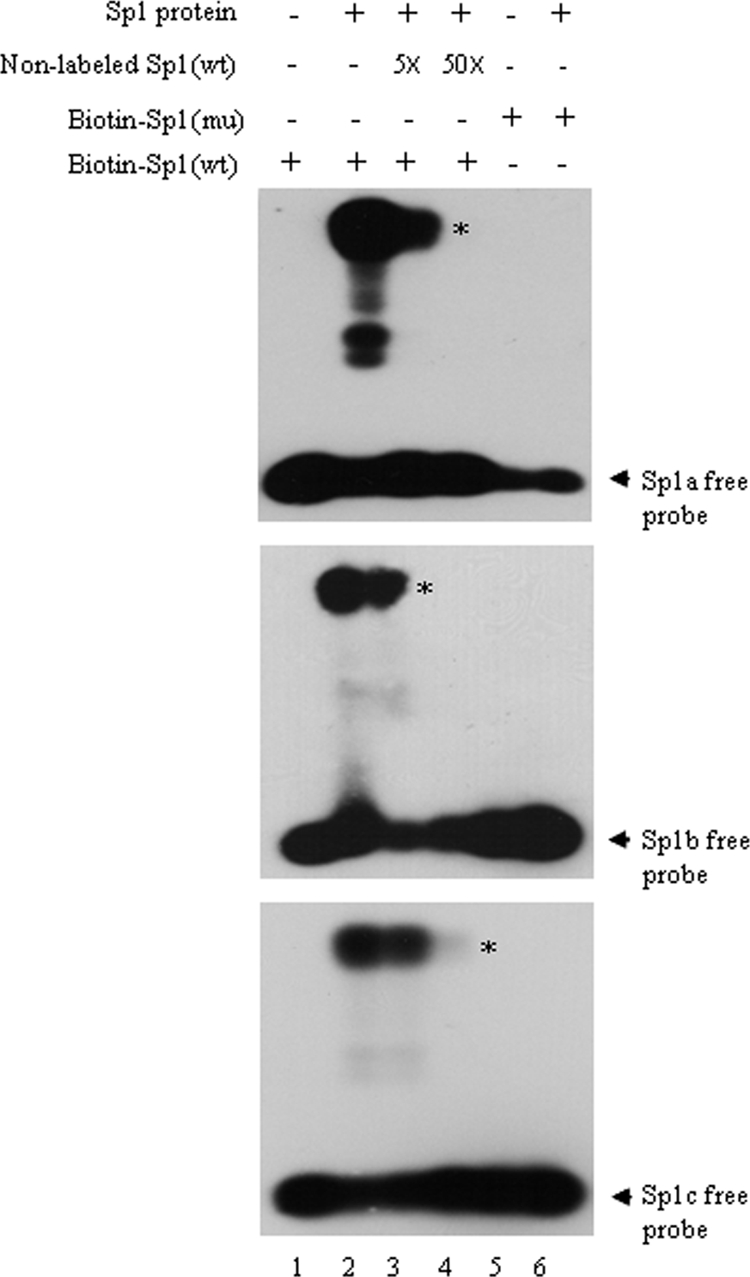
Binding of Sp1 protein to the ORF61 promoter in vitro. The binding of biotin-labeled probes Sp1a, Sp1b, and Sp1c with human recombinant Sp1 protein (Promega) was analyzed with a LightShift Chemiluminescent EMSA kit (Pierce). Lane 2 shows the binding of biotin-labeled wild-type (wt) probes with Sp1 protein. Lanes 3 and 4 show the results for the competition experiment: 5-fold and 50-fold molar excesses of nonlabeled wild-type probes were incubated with Sp1 protein before addition of biotin-labeled wild-type probes. Lane 6 shows the binding of biotin-labeled mutant (mu) probes with Sp1 protein. Lanes 1 and 5 show the binding reaction controls in the absence of Sp1 protein. Asterisks indicate the probes bound by Sp1 protein, and arrows indicate the free probes.
Sp1-binding sites in the ORF61 promoter cis regulate ORF61 expression in virus-infected cells.
To evaluate the contribution of the three Sp1-binding sites to regulation of ORF61 expression in the context of the VZV genome and to VZV replication, recombinant viruses that had mutations of these sites were made using the pOka cosmid system (63) (Fig. 4a and b). Four cosmids were constructed: Spe23-61proΔSp1a, Spe23-61proΔSp1b, and Spe23-61proΔSp1c, in which the Sp1a, Sp1b, or Sp1c site was disrupted individually, and Spe23-61ProΔSp1ac, in which both the Sp1a and the Sp1c sites were disrupted. Melanoma cells were transfected with the mutated cosmids, along with the other three pOka cosmids, Spe14, Fsp73, and Pme2. Four recombinant viruses were recovered and designated pOka-61proΔSp1a, pOka-61proΔSp1b, pOka-61proΔSp1c, and pOka-61proΔSp1ac. Expected mutations were confirmed by sequencing.
FIG. 4.

Construction of pOka recombinants with mutations in the ORF61 promoter. (a) Schematic representation of the VZV genome and the pOka cosmids Fsp73, Spe14, Pme2, and Spe23. The unique long (UL) and short (US) regions and the inverted sequences (internal repeat [IR] and terminal repeat [TR]) are indicated. (b) An EcoRI-AvrII fragment including the sequence from ORF59 to ORF65 was subcloned to pLitmus28 vector, resulting in pLit(ORF59-65). The genomic locations and directions of ORF59 to ORF65 are shown with block arrows. Mutagenesis of the ORF61 promoter was done in pLit(ORF59-65). Primers P1 and P2, containing unique restriction enzyme sites (PstI and PmlI), were used as the two outside primers for mutating Sp1-binding sites. (c) ORF61 protein expression in melanoma cells infected with pOka and Sp1 mutant viruses was analyzed by Western blotting with ORF61 antibody and quantified by densitometry.
Melanoma cells were inoculated with pOka and Sp1-binding-site mutant viruses at 1,000 PFU per 106 cells and harvested at 72 h postinfection. ORF61 protein expression was evaluated by Western blot analysis with ORF61 rabbit polyclonal antibody and densitometry with UN-SCAN-IT software (Silk Scientific, Inc.) (Fig. 4c). Compared to that of pOka-infected cells, ORF61 expression was reduced by 23.8% in cells infected with pOka-61proΔSp1a, by 29.3% in cells infected with pOka-61proΔSp1b, and by 81% in cells infected with pOka-61proΔSp1c. This pattern was confirmed in a second experiment comparing the mutants to pOka, which showed that ORF61 expression was decreased by 27.4% in pOka-61proΔSp1a-infected cells, by 14.8% in pOka-61proΔSp1b-infected cells, and by 78% in pOka-61proΔSp1c-infected cells. More strikingly, cells infected with pOka-61proΔSp1ac had a reduction of 95% in ORF61 expression. These observations were also confirmed with a second, separately derived pOka-61proΔSp1ac mutant (data not shown).
Growth kinetics and plaque sizes of the Sp1-binding-site mutant viruses.
The growth kinetics of pOka and the four Sp1-binding-site mutant viruses were evaluated for 6 days with melanoma cells (Fig. 5a). The pOka and pOka-61proΔSp1b titers were indistinguishable. pOka-61proΔSp1a was also similar to pOka, except at day 5 (titer lower than pOka; P < 0.05). The titers of both pOka-61proΔSp1c and pOka-61proΔSp1ac were significantly lower (∼0.2- to 0.4-log less) than those of pOka from day 1 to day 5 (P < 0.05). When 30 plaques in each infected monolayer were measured, the pOka-61proΔSp1a and pOka-61proΔSp1b plaques were found to be only marginally smaller than those of pOka (P > 0.05), while those of pOka-61proΔSp1c and especially pOka-61proΔSp1ac were significantly smaller than those of pOka (pOka-61proΔSp1c, P < 0.05; pOka-61proΔSp1ac, P < 0.0001) (Fig. 5b). These experiments indicate that the Sp1c and Sp1ac mutations, which caused the most-significant reductions in ORF61 expression, impaired VZV replication in vitro.
FIG. 5.
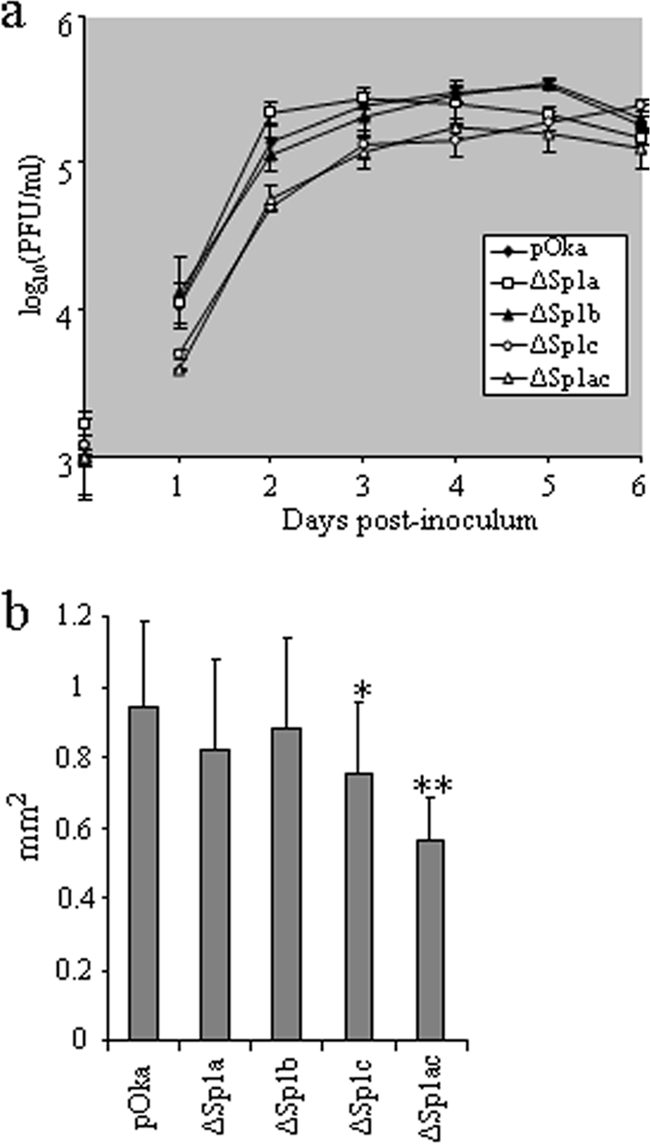
Growth kinetics and plaque sizes of Sp1-binding-site mutant viruses. (a) Growth curves of pOka and Sp1-binding-site mutant viruses. Melanoma cells (106) were inoculated with approximately 103 PFU of pOka and ORF61 promoter mutants on day 0. Titers at days 1 to 6 were determined by an infectious focus assay; each point represents the mean ± SD of titers from three wells. “PFU/ml” indicates the number of infectious centers per ml. (b) Plaque sizes of pOka and Sp1-binding-site mutants. Images of 30 individual plaques from each virus were obtained with a light microscope at ×5 magnification and measured with the aid of ImageJ software. The bars indicate the mean sizes ± SD for 30 plaques. One asterisk indicates a significant difference between pOka and pOka-61proΔSp1c (P < 0.05); two asterisks indicate a significant difference between pOka and pOka-61proΔSp1ac (P < 0.0001).
ORF61 is required for efficient expression of VZV proteins in all kinetic classes.
The expression of six VZV proteins (IE62, IE63, IE4, ORF47, ORF66, and gE) in melanoma cells infected with pOka and Sp1-binding-site mutant viruses at 1,000 PFU per 106 cells and harvested at 72 h postinfection was analyzed. The expression levels of all of these VZV gene products were reduced in melanoma cells infected with pOka-61proΔSp1c or pOka-61proΔSp1ac but not in pOka-61proΔSp1a- and pOka-61proΔSp1b-infected cells, compared with the level for pOka-infected cells (Fig. 6a, left panel). Among these proteins, the expression levels of ORF47, ORF66, and especially gE were reduced substantially, whereas those of IE62, IE63, and IE4 were less affected. The reductions in viral protein expression in pOka-61proΔSp1ac-infected cells, relative to the level for pOka, were assessed by densitometry analysis after normalization to the level for tubulin (Fig. 6a, right panel). Compared with those in pOka-infected cells, the IE62, IE63, and IE4 expression levels in pOka-61proΔSp1ac-infected cells were reduced by 42%, 56%, and 71%, respectively. ORF47 and ORF66 expression levels were reduced by 84% and 80%, respectively, and gE expression decreased by 91%. In HELF infected with pOka-61proΔSp1ac, very low ORF61 expression levels were associated with relatively little effect on IE62 (9% reduction); the expression levels of ORF66 and, again, especially gE were reduced (Fig. 6b).
FIG. 6.

Effect of deficient ORF61 expression on VZV protein expression and gE trafficking. (a) Immediate-early (IE), early (E), and late (L) gene expression in melanoma cells infected with pOka and Sp1-binding-site mutant viruses. Melanoma cells were inoculated with pOka and Sp1-binding-site mutant viruses at 103 PFU/106 cells and harvested at 72 h postinfection. Expression of IE62, IE63, IE4, ORF47, ORF66, gE, and tubulin was analyzed by Western blotting and densitometry. Viral protein expression levels were normalized to the tubulin amount, and the value of each viral protein in pOka-infected cells was set as 1. (b) Expression of ORF61, IE62, ORF66, and gE in HELF cells infected with pOka and Sp1-binding-site mutant viruses for 72 h was analyzed by Western blotting and densitometry as described for panel a. (c) An infected cell inoculum was labeled with BODIPY-green cell tracker and used to infect HELF; gE expression levels in pOka- and pOka-61proΔSp1ac-infected cells were compared at 4 h after infection. gE expression in 150 newly infected cells from each infected monolayer was assessed. (d) gE localization in HELF infected with pOka and pOka-61proΔSp1ac. HELF seeded on chamber slides were infected with pOka and pOka-61proΔSp1ac, fixed at 48 h postinfection, stained with monoclonal gE antibody, and visualized with Leica confocal microscopy.
The cell tracker labeling method for studying the spatiotemporal expression of VZV proteins within one VZV infection cycle, as described by Reichelt et al. (73), was used to assess the effect of intact or limited ORF61 on gE expression at the single-cell level (Fig. 6c). At 4 h after inoculation, newly infected cells were classified based on the number of IE62-positive nuclear punctae, which indicate formation of VZV replication compartments; cells were categorized as containing 3 to 5, 6 to 10, or >10 punctae. The pattern of gE expression in 50 cells in each category was recorded as either nondetectable, faint Golgi signal, or intense Golgi signal. Within newly infected cells that had three to five IE62 punctae, gE was not detectable in 48% of pOka-infected cells, compared to 68% of pOka-61proΔSp1ac-infected cells (P = 0.04; Fisher's exact test). Intense gE staining was noted to occur in 20% of this group of pOka-infected cells, compared to 8% of pOka-61proΔSp1ac-infected cells, with the same number of IE62 punctae. Among cells that had 6 to 10 IE62 punctae, gE was not detectable in 28% of pOka-infected cells, compared to 36% of pOka-61proΔSp1ac-infected cells; intense gE expression in the Golgi region was evident in 38% of this category of pOka-infected cells, compared to 26% of pOka-61proΔSp1ac-infected cells. Among cells that had more than 10 IE62 punctae, 72% of pOka-infected cells showed an intense gE Golgi signal, compared to 56% of the comparable subset of pOka-61proΔSp1ac-infected cells.
At 48 h after infection, gE had the typical Golgi region-like distribution and plasma membrane localization in pOka-61proΔSp1ac-infected cells, suggesting that although gE expression was reduced, its intracellular trafficking was intact (Fig. 6d).
Effect of deficient ORF61 expression on virion morphogenesis.
ORF61 was not associated with virus particles purified from infected melanoma cells (44). To analyze whether reduction of ORF61 expression had any overall consequences for virion morphogenesis, HELF infected with pOka and pOka-61proΔSp1ac were examined by TEM. More than 30 infected cells from each infected monolayer were assessed. No disruption in nucleocapsid formation or the structure of intracellular and extracellular virions was observed in HELF infected with pOka-61proΔSp1ac (Fig. 7). Abundant nucleocapsids containing DNA were observed in the nuclei of HELF infected with pOka or pOka-61proΔSp1ac (Fig. 7a and d). Cytoplasmic vacuoles containing viral particles in pOka-61proΔSp1ac-infected cells were indistinguishable from those in pOka-infected cells (Fig. 7b and e). Enveloped virions with DNA core and electron-dense tegument were present on the surfaces of cells infected with both viruses (Fig. 7c and f).
FIG. 7.

Virion morphogenesis of pOka-61proΔSp1ac. HELF were infected with pOka (a, b, and c) and pOka-61proΔSp1ac (d, e, and f) for 72 h and examined by TEM. (a and d) Nucleocapsid formation in nuclei. (b and e) Virions in cytoplasmic vacuoles. (c and f) Extracellular virions. Scale bars, 0.2 μm.
Effect of HDAC inhibitors on VZV replication.
HSV ICP0 and related proteins in other herpesviruses inhibit HDACs (51, 89); treatment with HDAC inhibitors compensated for the growth deficiencies of HSV-1 ICP0-null and HCMV IE1-deficient mutants (62, 71). To investigate whether HDACs limit VZV replication and the potential contribution of ORF61 to blocking of HDAC activity, melanoma cells were infected with pOka and pOka-61proΔSp1ac in the presence of the HDAC inhibitors (NaBu or TSA). Maintaining NaBu and TSA in the cell culture medium altered the normal morphology and viability of melanoma cells (data not shown), but these effects were not observed with pretreatment. Therefore, the titers of pOka and pOka-61proΔSp1ac in melanoma cells pretreated with NaBu or TSA were determined. Pretreating melanoma cells with 5 mM NaBu for 24 h increased the titers of pOka-61proΔSp1ac (P < 0.05) but not pOka (P > 0.05) (Fig. 8a). Pretreating melanoma cells with TSA for 12 h increased the titers of both pOka and pOka-61proΔSp1ac only slightly, but the differences were significant (P < 0.05) (Fig. 8b). No significant effect was observed with NaBu or TSA pretreatment at other time points. Pretreatment with both drugs was associated with increases in the plaque sizes of both pOka and pOka-61proΔSp1ac (data not shown). Pretreatment of HELF with NaBu or TSA induced changes in cell morphology that were visible at 2 h, and the growth rates of both pOka and pOka-61proΔSp1ac were inhibited (data not shown).
FIG. 8.
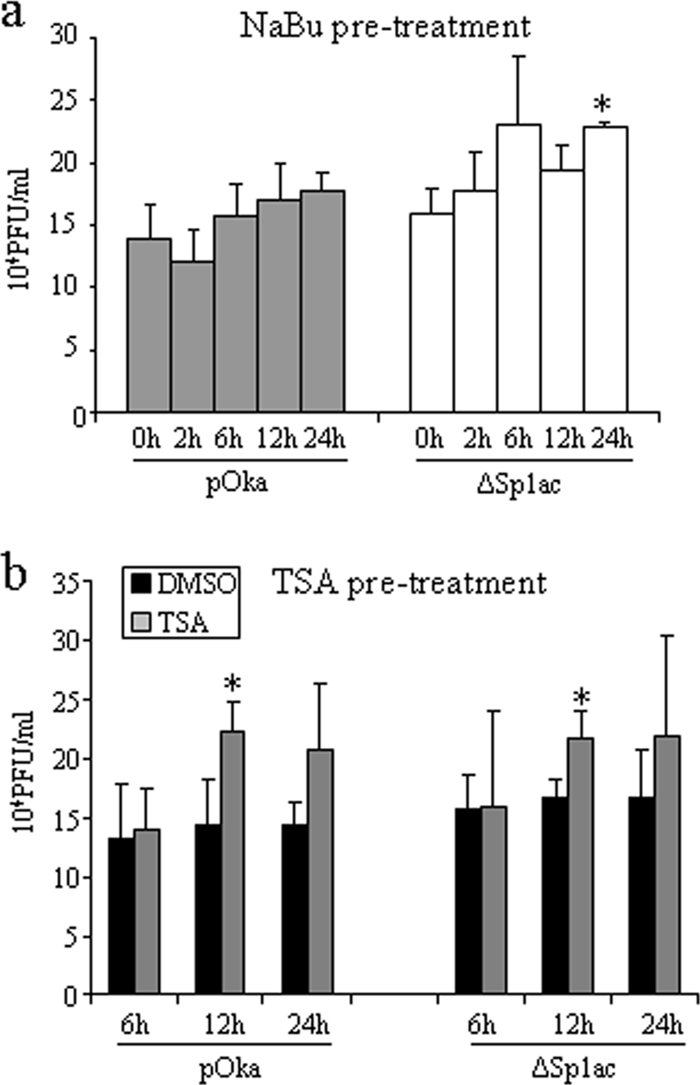
Effect of HDAC inhibitors on VZV replication. (a) Melanoma cells seeded on 24-well plates were incubated with 5 mM NaBu for 2, 6, 12, and 24 h before NaBu was removed and serial dilutions of pOka and pOka-61proΔSp1ac were added. (b) Melanoma cells seeded on 24-well plates were incubated with 500 nM TSA for 6, 12, and 24 h before TSA was removed and serial dilutions of pOka and pOka-61proΔSp1ac were added. Viral titers were determined by an infectious focus assay. Each bar represents the mean ± SD of titers from three wells. The asterisk indicates significant difference between mock-treated and drug-treated cells (P < 0.05). DMSO, dimethyl sulfoxide.
Replication of ORF61 promoter mutants in skin xenografts in vivo.
To examine whether reduced ORF61 expression affected VZV pathogenesis, the growth rates of pOka-61proΔSp1a and pOka-61proΔSp1ac were compared to that of pOka in skin xenografts (Fig. 9). The titers of all virus inocula were similar. The pOka titers were 2.5 × 103 PFU/implant at day 11 and 2.4 × 103 PFU/implant at day 21. The pOka-61proΔSp1a titer was 6 × 102 PFU/implant at day 11, which was slightly lower than that of pOka, but the difference was not statistically significant (P = 0.11), and the titer was equivalent to that of pOka at day 21 (3.0 × 103 PFU/implant). Infectious virus was recovered from all five implants inoculated with pOka-61proΔSp1a at both time points. The mean titers of pOka-61proΔSp1ac were 8.3 × 101 PFU/implant at day 11 and 4.7 × 101 PFU/implant at day 21; each of these titers is significantly lower than the corresponding pOka titer (P < 0.05). Four of five implants yielded pOka-61proΔSp1ac at day 11, but infectious virus was recovered from only two of five implants at day 21. These observations suggest that activation of the ORF61 promoter by Sp1 family proteins is important in vivo and that diminished ORF61 expression restricts VZV virulence in skin.
FIG. 9.
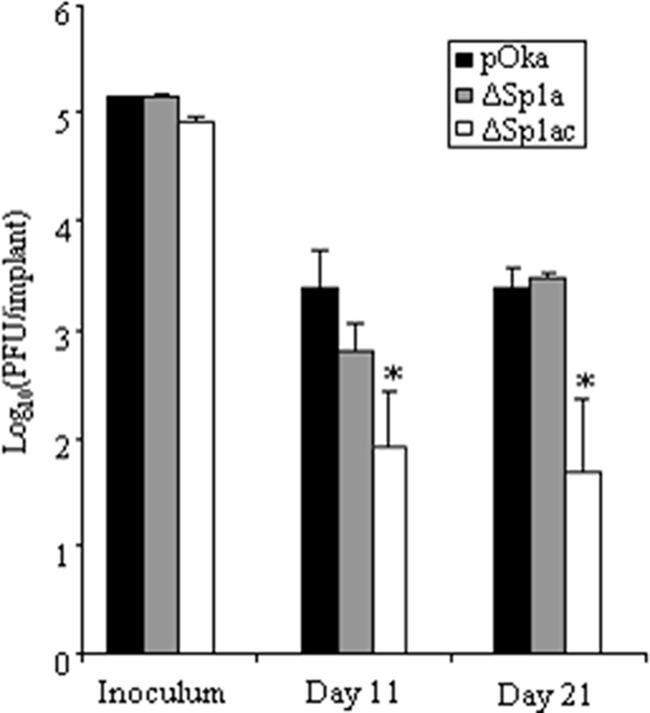
Growth of ORF61 promoter mutants in human skin xenografts in SCIDhu mice in vivo. Skin xenografts were infected with pOka, pOka-ΔSp1a, and pOka-ΔSp1ac and harvested at 11 and 21 days postinfection. Infectious virus yields were determined by an infectious focus assay with melanoma cells. Each bar represents the mean titer ± standard error. The asterisk indicates a statistically significant difference between pOka and the mutant virus (P < 0.05).
DISCUSSION
Our characterization of the VZV ORF61 promoter demonstrated that the minimal sequence for IE62 activation consists of the 95 residues proximal to the ORF61 transcription start site and includes three noncanonical GT-rich Sp1-binding sites. ORF61 promoter activity was induced by IE62 and to a much lesser extent by ORF61. IE4 and IE63 alone had minor effects on the ORF61 promoter, in addition to enhancing IE62-mediated transactivation. Mutagenesis of two of the Sp1 sites in combination in the context of the VZV genome caused a marked reduction in ORF61 expression and allowed an assessment of ORF61 functions during VZV replication in vitro and in skin xenografts in vivo. ORF61 contributed to synthesis of VZV regulatory proteins and the viral kinases and was particularly important for expression of gE, which is an essential protein in VZV. This phenotype of the ORF61-deficient virus was associated with a reduction in VZV virulence in human skin cells in vivo.
These experiments support the importance of the Sp1 family of cellular transactivating proteins for VZV gene transcription, showing that these proteins function in coregulation of ORF61 expression. Of interest, ORF61 is not present in the VZV virion (44), and we have shown that ORF61 protein is newly synthesized as early as 1 h after infection, with kinetics that parallel those of IE62 (73). Therefore, Sp1 regulates not only the putative late promoters (gE and gI) but also a critically important very early expressed promoter during VZV replication (7, 37, 39, 72). The prototype sequence targeted for binding by the Sp1 family proteins in cellular gene promoters is 5′-GGGCGGG-3′ (25). This motif is functional in the VZV gE promoter (7, 72). However, an atypical Sp1-binding motif, 5′-ACGCCC-3′, also regulates the gE and gI promoters (7, 37, 39, 72). Putative regulatory regions of 17 other ORFs, including ORF61, were predicted to have either or both of these motifs (76). We found that removing the two canonical 5′-GGGCGGG-3′ sites in the ORF61 promoter did not affect IE62-mediated transactivation. However, three other sites were identified when the ORF61 promoter was analyzed for atypical GT-rich Sp1-binding motifs. These motifs were mapped proximal to the ORF61 transcription start site, and human Sp1 protein bound to all three. The promoters of HSV ICP0 and pseudorabies virus EP0, which are ORF61 orthologs, contain putative Sp1-binding sites (23, 24, 87), and our comparative analysis of the simian varicella virus ORF61 and the bovine and equine herpesvirus ICP0 promoters also suggested the presence of canonical or noncanonical Sp1 sites (data not shown). Thus, Sp1 coregulation may be a common feature of the regulation of these conserved genes in the alphaherpesviruses. USF, another cellular transcription factor, is also important for VZV replication and pathogenesis, as demonstrated by mutagenesis of the gI and ORF10 promoters (15, 37, 39). A USF site was predicted to occur at positions −104 to −99 in the ORF61 promoter, but deleting sequences upstream of position −95 had no effect, suggesting that USF is not involved in ORF61 promoter regulation. Other minor cis-activating sequences may also be present in the ORF61 promoter, as basal activity was observed even when all three Sp1-binding sites were disrupted.
Although the sequences of three Sp1-binding sites in the ORF61 promoter are similar, the contribution of each individual site varied and the relative importance of each site for ORF61 expression during VZV replication was not predictable from luciferase assays. In transient-transfection assays, mutation of Sp1a, Sp1b and Sp1c led to reductions of ∼60%, 25%, and 40%, respectively, in IE62-mediated promoter activity, whereas disrupting Sp1c in the VZV genome resulted in ∼80% less ORF61 protein, compared to a ∼20% to 30% reduction with Sp1a or Sp1b mutations. The Sp1a and Sp1c sites appear to function synergistically; disrupting both resulted in minimal luciferase activity, and the pOka-61proΔSp1ac mutant produced the least ORF61 protein. The differences in function among the sites may reflect differences in their binding affinities for Sp1, which can be affected by minor variations in the nucleotide sequence, or the transcription complex architecture formed during VZV replication may make Sp1c more accessible to transcription factors. Other Sp family members, including Sp2, Sp3, and Sp4, and unrelated proteins, including Krüppel-like factors BTEB1, TIEG1, and TIEG2, can also bind to GC/GT boxes (84). Therefore, although recombinant human Sp1 protein binds to Sp1a, Sp1b, and Sp1c sequences, these sites may also interact with other transcription factors in infected cells. The inconsistency between the effects of disrupting Sp1a and Sp1c in the luciferase assay and in the VZV genome may reflect the presence of multiple viral transactivators and the more complex cellular environment in infected cells.
The substantial reductions of ORF61 protein in cells infected with pOka-61proΔSp1c and especially pOka-61proΔSp1ac were associated with delayed VZV replication and smaller plaques. Since the essential VZV glycoprotein, gE, which is important for cell-to-cell spread, was greatly reduced in cells infected with pOka-61proΔSp1ac, this phenotype may derive from impaired spread; VZV gE may also play a role in viral entry (2). These results, in which ORF61 expression in pOka was knocked down without altering the ORF61 coding sequence, are consistent with the deficient growth of a vaccine Oka ORF61 mutant from which the ORF61 sequence, except for that encoding amino acids 453 to 467, was deleted (21). However, alphaherpesviruses, including HSV, equine herpesvirus 1, pseudorabies virus, bovine herpesvirus 1, and simian varicella virus, have latency-associated transcripts that are antisense to the HSV ICP0 gene or its orthologs (5, 18, 65, 74, 75). Whether ORF61 has important antisense transcripts and whether such transcripts might function in lytic infection are not known, but manipulating ORF61 expression by mutation of promoter motifs reduces concern about unidentified effects on other transcripts. Disruption of Sp1a or Sp1b did not alter VZV growth, although ORF61 expression was diminished, suggesting that there is a threshold of ORF61 expression that is sufficient to support typical VZV replication.
Limiting ORF61 protein synthesis in VZV-infected melanoma cells and fibroblasts had differential consequences for the expression of other VZV proteins, exhibiting the least impact on IE62 and the most on gE. The effects on IE4, IE63, and the viral kinases ORF47 and ORF66 were intermediate. Reductions of IE62 and gE were also observed in melanoma cells infected with the vaccine Oka ORF61 partial deletion mutant (21). In our experiments with the pOka-61proΔSp1ac mutant in melanoma cells, the effect on gE can be attributed to a 42% decrease in IE62. However, gE was also markedly reduced in fibroblasts infected with pOka-61proΔSp1ac, despite a decrease of only 9% in IE62 compared the level for pOka-infected fibroblasts. Diminished and delayed gE expression was confirmed by single-cell analysis of fibroblasts infected with the ORF61-deficient mutant, even though the patterns of IE62 expression and nuclear localization were indistinguishable from those for pOka-infected fibroblasts. Restricting the availability of ORF61 as a viral transactivator seems unlikely to fully explain these dramatic effects on gE expression in the context of VZV replication since ORF61 activation of the gE promoter in reporter construct assays was only two- to threefold lower than its induction of the ORF4 and ORF62 promoters (7, 59). We have shown that within 1 hour after VZV entry, IE62, which is the major VZV transactivator, localizes to nuclear compartments that are distinct from those where ORF61 accumulates (73), suggesting that ORF61 transactivating function is not involved in initiating VZV gene transcription. However, since ORF61 is distributed diffusely in infected cell nuclei at 6 hours (73), it could have some role in regulating IE62-induced gene transcription at a later stage of infection. ORF61, like HSV-1 ICP0, associates with PML and may have a role in PML degradation (73). PML depletion increased HSV-1 ICP4 and UL42 expression in cells infected with an ICP0-null mutant (31). However, this ORF61 function might be expected to have a general effect on VZV gene transcription without affecting gE expression in particular. The difference in the impact of limiting ORF61 on IE62 production in melanoma cells and fibroblasts also points to cell-type-specific requirements for ORF61, as has been reported with ICP0 mutants (10).
Recent studies suggest that the effect of ICP0 and its orthologs in other alphaherpesviruses on transactivation is related to their interactions with HDACs (51, 89) and that ICP0 promotes both histone removal and acetylation on viral DNA (20). Gu and Roizman have proposed a model in which ICP0 blocks silencing of viral DNA by dissociating HDACs from the CoREST/REST repressor complex (33). Our data suggest that ORF61 might also function as a viral HDAC inhibitor to partially relieve repression of VZV gene transcription in melanoma cells. However, the effects observable in experiments with the ORF61-deficient mutant appeared to be relatively limited compared to those with HSV-1 ICP0 mutants. The interaction of IE63 with the human antisilencing function 1 (ASF1) protein indicates that VZV has other strategies for modulating histones and viral gene transcription (3). In HSV, the viral kinase Us3 also blocks histone deacetylation by a mechanism distinct from that of ICP0 (71). VZV ORF66, the Us3 ortholog, may have similar functions that were preserved despite the lower levels of ORF66 expression associated with ORF61 deficiency.
The pOka-61proΔSp1ac mutant exhibited reduced virulence in human skin xenografts in the SCIDhu mice, indicating that the regulation of the ORF61 promoter by Sp1 family proteins is important for ORF61 expression in vivo and that ORF61 contributes to the pathogenesis of VZV infection in skin. The impact of restricting ORF61 protein synthesis on VZV protein synthesis in vitro was consistent with these observations about altered growth in vivo. On the basis of our studies of other VZV mutants in the SCIDhu mouse model, the reductions of ORF47 and gE are likely to contribute most significantly to the impaired virulence of the ORF61-deficient mutant in skin (6, 8, 57).
In summary, Sp1 family proteins regulate the ORF61 promoter in the context of the VZV genome. During VZV replication, ORF61 is important for the expression of VZV regulatory proteins, viral kinases, and particularly gE in vitro. Despite these effects, VZV replication can occur in skin when ORF61 protein synthesis is limited but infectivity is attenuated.
Acknowledgments
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases, AI20459.
We thank Xibing Che, Mike Reichelt, and Stefan Oliver for their technical assistance and helpful discussions. We thank Leigh Zerboni for advice on skin xenograft experiments and Nafisa Ghori for assistance with TEM.
Footnotes
Published ahead of print on 20 May 2009.
REFERENCES
- 1.Abramoff, M. D., P. J. Magelhaes, and S. J. Ram. 2004. Image processing with ImageJ. Biophotonics Int. 1136-42. [Google Scholar]
- 2.Ali, M. A., Q. Li, E. R. Fischer, and J. I. Cohen. 2009. The insulin degrading enzyme binding domain of varicella-zoster virus (VZV) glycoprotein E is important for cell-to-cell spread and VZV infectivity, while a glycoprotein I binding domain is essential for infection. Virology 386270-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ambagala, A. P., T. Bosma, M. A. Ali, M. Poustovoitov, J. J. Chen, M. D. Gershon, P. D. Adams, and J. I. Cohen. 2009. Varicella-zoster virus immediate-early 63 protein interacts with human antisilencing function 1 protein and alters its ability to bind histones h3.1 and h3.3. J. Virol. 83200-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arvin, A. M. 2001. Varicella-zoster virus, p. 2731-2768. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 5.Baxi, M. K., S. Efstathiou, G. Lawrence, J. M. Whalley, J. D. Slater, and H. J. Field. 1995. The detection of latency-associated transcripts of equine herpesvirus 1 in ganglionic neurons. J. Gen. Virol. 763113-3118. [DOI] [PubMed] [Google Scholar]
- 6.Berarducci, B., M. Ikoma, S. Stamatis, M. Sommer, C. Grose, and A. M. Arvin. 2006. Essential functions of the unique N-terminal region of the varicella-zoster virus glycoprotein E ectodomain in viral replication and in the pathogenesis of skin infection. J. Virol. 809481-9496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berarducci, B., M. Sommer, L. Zerboni, J. Rajamani, and A. M. Arvin. 2007. Cellular and viral factors regulate the varicella-zoster virus gE promoter during viral replication. J. Virol. 8110258-10267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Besser, J., M. H. Sommer, L. Zerboni, C. P. Bagowski, H. Ito, J. Moffat, C. C. Ku, and A. M. Arvin. 2003. Differentiation of varicella-zoster virus ORF47 protein kinase and IE62 protein binding domains and their contributions to replication in human skin xenografts in the SCID-hu mouse. J. Virol. 775964-5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boutell, C., and R. Everett. 2003. The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and ubiquitinates p53. J. Biol. Chem. 27836596-36602. [DOI] [PubMed] [Google Scholar]
- 10.Boutell, C., R. Everett, J. Hilliard, P. Schaffer, A. Orr, and D. Davido. 2008. Herpes simplex virus type 1 ICP0 phosphorylation mutants impair the E3 ubiquitin ligase activity of ICP0 in a cell type-dependent manner. J. Virol. 8210647-10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai, W., T. L. Astor, L. M. Liptak, C. Cho, D. M. Coen, and P. A. Schaffer. 1993. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 677501-7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai, W., and P. A. Schaffer. 1992. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J. Virol. 662904-2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cartharius, K., K. Frech, K. Grote, B. Klocke, M. Haltmeier, A. Klingenhoff, M. Frisch, M. Bayerlein, and T. Werner. 2005. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 212933-2942. [DOI] [PubMed] [Google Scholar]
- 14.Chaudhuri, V., M. Sommer, J. Rajamani, L. Zerboni, and A. M. Arvin. 2008. Functions of varicella-zoster virus ORF23 capsid protein in viral replication and the pathogenesis of skin infection. J. Virol. 8210231-10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Che, X., B. Berarducci, M. Sommer, W. T. Ruyechan, and A. M. Arvin. 2007. The ubiquitous cellular transcriptional factor USF targets the varicella-zoster virus open reading frame 10 promoter and determines virulence in human skin xenografts in SCIDhu mice in vivo. J. Virol. 813229-3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chelbi-Alix, M. K., and H. de The. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18935-941. [DOI] [PubMed] [Google Scholar]
- 17.Chen, J., and S. Silverstein. 1992. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. J. Virol. 662916-2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung, A. K. 1989. Detection of pseudorabies virus transcripts in trigeminal ganglia of latently infected swine. J. Virol. 632908-2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clements, G. B., and N. D. Stow. 1989. A herpes simplex virus type 1 mutant containing a deletion within immediate early gene 1 is latency-competent in mice. J. Gen. Virol. 702501-2506. [DOI] [PubMed] [Google Scholar]
- 20.Cliffe, A. R., and D. M. Knipe. 2008. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 8212030-12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohen, J. I., and H. Nguyen. 1998. Varicella-zoster virus ORF61 deletion mutants replicate in cell culture, but a mutant with stop codons in ORF61 reverts to wild-type virus. Virology 246306-316. [DOI] [PubMed] [Google Scholar]
- 22.Cohen, J. I., S. E. Straus, and A. M. Arvin. 2007. Varicella-zoster virus proteins that regulate transcription, p. 2782-2783. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 23.Davido, D. J., and D. A. Leib. 1996. Role of cis-acting sequences of the ICP0 promoter of herpes simplex virus type 1 in viral pathogenesis, latency and reactivation. J. Gen. Virol. 771853-1863. [DOI] [PubMed] [Google Scholar]
- 24.Davido, D. J., and D. A. Leib. 1998. Analysis of the basal and inducible activities of the ICP0 promoter of herpes simplex virus type 1. J. Gen. Virol. 792093-2098. [DOI] [PubMed] [Google Scholar]
- 25.Dynan, W. S., and R. Tjian. 1983. The promoter-specific transcription factor Sp1 binds to upstream sequences in the SV40 early promoter. Cell 35179-87. [DOI] [PubMed] [Google Scholar]
- 26.Everett, R. D. 1984. Trans activation of transcription by herpes virus products: requirement for two HSV-1 immediate-early polypeptides for maximum activity. EMBO J. 33135-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Everett, R. D., P. Barlow, A. Milner, B. Luisi, A. Orr, G. Hope, and D. Lyon. 1993. A novel arrangement of zinc-binding residues and secondary structure in the C3HC4 motif of an alpha herpes virus protein family. J. Mol. Biol. 2341038-1047. [DOI] [PubMed] [Google Scholar]
- 28.Everett, R. D., W. C. Earnshaw, J. Findlay, and P. Lomonte. 1999. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 181526-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Everett, R. D., P. Freemont, H. Saitoh, M. Dasso, A. Orr, M. Kathoria, and J. Parkinson. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 726581-6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Everett, R. D., M. Meredith, A. Orr, A. Cross, M. Kathoria, and J. Parkinson. 1997. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 161519-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Everett, R. D., S. Rechter, P. Papior, N. Tavalai, T. Stamminger, and A. Orr. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 807995-8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gelman, I. H., and S., Silverstein. 1985. Identification of immediate early genes from herpes simplex virus that transactivate the virus thymidine kinase gene. Proc. Natl. Acad. Sci. USA 825265-5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gu, H., and B. Roizman. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. USA 10417134-17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hagglund, R., and B. Roizman. 2003. Herpes simplex virus 1 mutant in which the ICP0 HUL-1 E3 ubiquitin ligase site is disrupted stabilizes cdc34 but degrades D-type cyclins and exhibits diminished neurotoxicity. J. Virol. 7713194-13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hagglund, R., and B. Roizman. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J. Virol. 782169-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halford, W. P., and P. A. Schaffer. 2001. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 753240-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He, H., D. Boucaud, J. Hay, and W. T. Ruyechan. 2001. Cis and trans elements regulating expression of the varicella zoster virus gI gene. Arch. Virol. Suppl. 1757-70. [DOI] [PubMed] [Google Scholar]
- 38.Inchauspe, G., S. Nagpal, and J. M. Ostrove. 1989. Mapping of two varicella-zoster virus-encoded genes that activate the expression of viral early and late genes. Virology 173700-709. [DOI] [PubMed] [Google Scholar]
- 39.Ito, H., M. Sommer, L. Zerboni, H. He, D. Boucaud, J. Hay, W. T. Ruyechan, and A. M. Arvin. 2003. Promoter sequences of varicella-zoster virus glycoprotein I targeted by cellular transactivating factors Sp1 and USF determine virulence in skin and T cells in SCIDhu mice in vivo. J. Virol. 77489-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackers, P., P. Defechereux, L. Baudoux, C. Lambert, M. Massaer, M. P. Merville-Louis, B. Rentier, and J. Piette. 1992. Characterizations of regulatory functions of the varicella-zoster virus gene 63-encoded protein. J. Virol. 663899-3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawaguchi, Y., M. Tanaka, A. Yokoymama, G. Matsuda, K. Kato, H. Kagawa, K. Hirai, and B. Roizman. 2001. Herpes simplex virus 1 alpha regulatory protein ICP0 functionally interacts with cellular transcription factor BMAL1. Proc. Natl. Acad. Sci. USA 981877-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawaguchi, Y., R. Bruni, and B. Roizman. 1997. Interaction of herpes simplex virus 1 α regulatory protein ICP0 with elongation factor 1δ: ICP0 affects translational machinery. J. Virol. 711019-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawaguchi, Y., C. Van Sant, and B. Roizman. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 717328-7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kinchington, P. R., D. Bookey, and S. E. Turse. 1995. The transcriptional regulatory proteins encoded by varicella-zoster virus open reading frames (ORFs) 4 and 63, but not ORF 61, are associated with purified virus particles. J. Virol. 694274-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kinchington, P. R., and J. I. Cohen. 2000. Viral proteins, p. 74-104. In A. M. Arvin and A. A. Gershon (ed.), Varicella zoster virus virology and clinical management. Cambridge University Press, Cambridge, United Kingdom.
- 46.Kinchington, P. R., J. K. Hougland, A. M. Arvin, W. T. Ruyechan, and J. Hay. 1992. The varicella-zoster virus immediate-early protein IE62 is a major component of virus particles. J. Virol. 66359-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kinchington, P. R., J. P. Vergnes, P. Defechereux, J. Piette, and S. E. Turse. 1994. Transcriptional mapping of the varicella-zoster virus regulatory genes encoding open reading frames 4 and 63. J. Virol. 683570-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leib, D. A., D. M. Coen, C. L. Bogard, K. A. Hicks, D. R. Yager, D. M. Knipe, K. L. Tyler, and P. A. Schaffer. 1989. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol. 63759-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lomonte, P., and E. Morency. 2007. Centromeric protein CENP-B proteasomal degradation induced by the viral protein ICP0. FEBS Lett. 581658-662. [DOI] [PubMed] [Google Scholar]
- 50.Lomonte, P., K. F. Sullivan, and R. D. Everett. 2001. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem. 2765829-5835. [DOI] [PubMed] [Google Scholar]
- 51.Lomonte, P., J. Thomas, P. Texier, C. Caron, S. Khochbin, and A. L. Epstein. 2004. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J. Virol. 786744-6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lynch, J. M., T. K. Kenyon, C. Grose, J. Hay, and W. T. Ruyechan. 2002. Physical and functional interaction between the varicella zoster virus IE63 and IE62 proteins. Virology 30271-82. [DOI] [PubMed] [Google Scholar]
- 53.Meier, J. L., X. Luo, M. Sawadogo, and S. E. Straus. 1994. The cellular transcription factor USF cooperates with varicella-zoster virus immediate-early protein 62 to symmetrically activate a bidirectional viral promoter. Mol. Cell. Biol. 146896-6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michael, E. J., K. M. Kuck, and P. R. Kinchington. 1998. Anatomy of the varicella-zoster virus open-reading frame 4 promoter. J. Infect. Dis. 178(Suppl. 1)S27-S33. [DOI] [PubMed] [Google Scholar]
- 55.Moffat, J. F., M. D. Stein, H. Kaneshima, and A. M. Arvin. 1995. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J. Virol. 695236-5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moffat, J. F., L. Zerboni, P. R. Kinchington, C. Grose, H. Kaneshima, and A. M. Arvin. 1998. Attenuation of the vaccine Oka strain of varicella-zoster virus and role of glycoprotein C in alphaherpesvirus virulence demonstrated in the SCID-hu mouse. J. Virol. 72965-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moffat, J. F., L. Zerboni, M. H. Sommer, T. C. Heineman, J. I. Cohen, H. Kaneshima, and A. M. Arvin. 1998. The ORF47 and ORF66 putative protein kinases of varicella-zoster virus determine tropism for human T cells and skin in the SCID-hu mouse. Proc. Natl. Acad. Sci. USA 9511969-11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moriuchi, H., M. Moriuchi, H. A. Smith, S. E. Straus, and J. I. Cohen. 1992. Varicella-zoster virus open reading frame 61 protein is functionally homologous to herpes simplex virus type 1 ICP0. J. Virol. 667303-7308. (Erratum, 69:2723.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moriuchi, H., M. Moriuchi, S. E. Straus, and J. I. Cohen. 1993. Varicella-zoster virus (VZV) open reading frame 61 protein transactivates VZV gene promoters and enhances the infectivity of VZV DNA. J. Virol. 674290-4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Müller, S., and A. Dejean. 1999. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J. Virol. 735137-5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagpal, S., and J. M. Ostrove. 1991. Characterization of a potent varicella-zoster virus-encoded trans-repressor. J. Virol. 65:5289-5296. (Erratum, 69:2723.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nevels, M., C. Paulus, and T. Shenk. 2004. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. USA 10117234-17239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Niizuma, T., L. Zerboni, M. H. Sommer, H. Ito, S. Hinchliffe, and A. M. Arvin. 2003. Construction of varicella-zoster virus recombinants from parent Oka cosmids and demonstration that ORF65 protein is dispensable for infection of human skin and T cells in the SCID-hu mouse model. J. Virol. 776062-6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O'Hare, P., and G. S. Hayward. 1985. Evidence for a direct role for both the 175,000- and 110,000-molecular-weight immediate-early proteins of herpes simplex virus in the transactivation of delayed-early promoters. J. Virol. 53751-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ou, Y., K. A. Davis, V. Traina-Dorge, and W. L. Gray. 2007. Simian varicella virus expresses a latency-associated transcript that is antisense to open reading frame 61 (ICP0) mRNA in neural ganglia of latently infected monkeys. J. Virol. 818149-8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parkinson, J., and R. D. Everett. 2000. Alphaherpesvirus proteins related to herpes simplex virus type 1 ICP0 affect cellular structures and proteins. J. Virol. 7410006-10017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parkinson, J., S. P. Lees-Miller, and R. D. Everett. 1999. Herpes simplex virus type 1 immediate-early protein Vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 73650-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peng, H., H. He, J. Hay, and W. T. Ruyechan. 2003. Interaction between the varicella zoster virus IE62 major transactivator and cellular transcription factor Sp1. J. Biol. Chem. 27838068-38075. [DOI] [PubMed] [Google Scholar]
- 69.Perera, L. P. 2000. The TATA motif specifies the differential activation of minimal promoters by varicella zoster virus immediate-early regulatory protein IE62. J. Biol. Chem. 275487-496. [DOI] [PubMed] [Google Scholar]
- 70.Perera, L. P., J. D. Mosca, W. T. Ruyechan, and J. Hay. 1992. Regulation of varicella-zoster virus gene expression in human T lymphocytes. J. Virol. 665298-5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Poon, A. P., H. Gu, and B. Roizman. 2006. ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc. Natl. Acad. Sci. USA 1039993-9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rahaus, M., and M. H. Wolff. 2000. Transcription factor Sp1 is involved in the regulation of varicella-zoster virus glycoprotein E. Virus Res. 6969-81. [DOI] [PubMed] [Google Scholar]
- 73.Reichelt, M., J. Brady, and A. M. Arvin. 2009. The replication cycle of varicella-zoster virus: analysis of the kinetics of viral protein expression, genome synthesis, and virion assembly at the single-cell level. J. Virol. 833904-3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rock, D. L., S. L. Beam, and J. E. Mayfield. 1987. Mapping bovine herpesvirus type 1 latency-related RNA in trigeminal ganglia of latently infected rabbits. J. Virol. 613827-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roizman, B., D. M. Knipe, and R. J. Whitley. 2007. Herpes simplex virus latency in experimental systems, p. 2544-2546. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 76.Ruyechan, W. T., H. Peng, M. Yang, and J. Hay. 2003. Cellular factors and IE62 activation of VZV promoters. J. Med. Virol. 70(Suppl. 1)S90-S94. [DOI] [PubMed] [Google Scholar]
- 77.Santos, R. A., C. C. Hatfield, N. L. Cole, J. A. Padilla, J. F. Moffat, A. M. Arvin, W. T. Ruyechan, J. Hay, and C. Grose. 2000. Varicella-zoster virus gE escape mutant VZV-MSP exhibits an accelerated cell-to-cell spread phenotype in both infected cell cultures and SCID-hu mice. Virology 275306-317. [DOI] [PubMed] [Google Scholar]
- 78.Sawadogo, M. 1988. Multiple forms of the human gene-specific transcription factor USF. II. DNA binding properties and transcriptional activity of the purified HeLa USF. J. Biol. Chem. 26311994-12001. [PubMed] [Google Scholar]
- 79.Schaap, A., J. F. Fortin, M. Sommer, L. Zerboni, S. Stamatis, C. C. Ku, G. P. Nolan, and A. M. Arvin. 2005. T-cell tropism and the role of ORF66 protein in pathogenesis of varicella-zoster virus infection. J. Virol. 7912921-12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schug, J. 2008. Using TESS to predict transcription factor binding sites in DNA sequence. Curr. Protoc. Bioinformatics 2008(March)2.6. [DOI] [PubMed] [Google Scholar]
- 81.Sommer, M. H., E. Zagha, O. K. Serrano, C. C. Ku, L. Zerboni, A. Baiker, R. Santos, M. Spengler, J. Lynch, C. Grose, W. Ruyechan, J. Hay, and A. M. Arvin. 2001. Mutational analysis of the repeated open reading frames, ORFs 63 and 70 and ORFs 64 and 69, of varicella-zoster virus. J. Virol. 758224-8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Spengler, M. L., W. T. Ruyechan, and J. Hay. 2000. Physical interaction between two varicella zoster virus gene regulatory proteins, IE4 and IE62. Virology 272375-381. [DOI] [PubMed] [Google Scholar]
- 83.Stevenson, D., K. L. Colman, and A. J. Davison. 1992. Characterization of the varicella-zoster virus gene 61 protein. J. Gen. Virol. 73521-530. [DOI] [PubMed] [Google Scholar]
- 84.Suske, G. 1999. The Sp-family of transcription factors. Gene 238291-300. [DOI] [PubMed] [Google Scholar]
- 85.Van Sant, C., R. Hagglund, P. Lopez, and B. Roizman. 2001. The infected cell protein 0 of herpes simplex virus 1 dynamically interacts with proteasomes, binds and activates the cdc34 E2 ubiquitin-conjugating enzyme, and possesses in vitro E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 988815-8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walters, M. S., C. A. Kyratsous, S. Wan, and S. Silverstein. 2008. Nuclear import of the varicella-zoster virus latency-associated protein ORF63 in primary neurons requires expression of the lytic protein ORF61 and occurs in a proteasome-dependent manner. J. Virol. 828673-8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Watanabe, S., E. Ono, H. Nikami, and H. Kida. 1998. Promoter activity of sequence located upstream of the pseudorabies virus early protein 0 gene. Vet. Microbiol. 617-19. [DOI] [PubMed] [Google Scholar]
- 88.Yang, M., H. Peng, J. Hay, and W. T. Ruyechan. 2006. Promoter activation by the varicella-zoster virus major transactivator IE62 and the cellular transcription factor USF. J. Virol. 807339-7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang, Y., Y. Jiang, V. Geiser, J. Zhou, and C. Jones. 2006. Bovine herpesvirus 1 immediate-early protein (bICP0) interacts with the histone acetyltransferase p300, which stimulates productive infection and gC promoter activity. J. Gen. Virol. 871843-1851. [DOI] [PubMed] [Google Scholar]