

Abstract
The Asian H5N1 highly pathogenic avian influenza (HPAI) viruses have been increasing in pathogenicity in diverse avian species since 1996 and are now widespread in Asian, European, and African countries. To better understand the basis of the increased pathogenicity of recent Asian H5N1 HPAI viruses in chickens, we compared the fevers and mean death times (MDTs) of chickens infected with the Asian H5N1 A/chicken/Yamaguchi/7/04 (CkYM7) strain with those infected with the H5N1 Duck/Yokohama/aq10/03 (DkYK10) strain, using a wireless thermosensor. Asian H5N1 CkYM7 caused peracute death in chickens before fever could be induced, whereas DkYK10 virus induced high fevers and had a long MDT. Real-time PCR analyses of cytokine mRNA expressions showed that CkYM7 quickly induced antiviral and proinflammatory cytokine mRNA expressions at 24 h postinfection (hpi) that suddenly decreased at 32 hpi. In contrast, these cytokine mRNA expressions increased at 24 hpi in the DkYK10 group, but decreased from 48 hpi onward to levels similar to those resulting from infection with the low-pathogenicity H5N2 A/chicken/Ibaraki/1/2004 strain. Sequential titrations of viruses in lungs, spleens, and kidneys demonstrated that CkYM7 replicated rapidly and efficiently in infected chickens and that the viral titers were more than twofold higher than those of DkYK10. CkYM7 preferentially and efficiently replicated in macrophages and vascular endothelial cells, while DkYK10 grew moderately in macrophages. These results indicate that the increased pathogenicity in chickens of the recent Asian H5N1 HPAI viruses may be associated with extremely rapid and high replication of the virus in macrophages and vascular endothelial cells, which resulted in disruption of the thermoregulation system and innate immune responses.
Since the first detection of the Asian lineage of highly pathogenic avian influenza (HPAI) virus (H5N1) in southern China in 1996, H5N1 virus infection in birds has continued for 13 years in Asia, acquiring pathogenicity not only in birds but also in mammals. In 1997, the H5N1 Hong Kong isolates caused illness and death in a variety of terrestrial birds and even in humans (9, 37, 48, 49). In 2001, emerging H5N1 Hong Kong isolates were more pathogenic to chickens and the mean death time (MDT) was 2 days without any prior clinical signs (12). In 2003 to 2004, the H5N1 epizootic occurred simultaneously in East Asian countries (22, 30). The 2003/2004 H5N1 isolates caused death in taxonomically diverse avian species, including domestic ducks (46, 47, 51), and humans (7, 55). Furthermore, the first indication of wild aquatic bird involvement occurred at recreational parks in Hong Kong in late 2002 to 2003 (46), and then migratory aquatic bird die-off occurred in 2005 at Qinghai Lake in China (6, 24). The broad host spectrum and increased pathogenicity of H5N1 viruses to diverse bird species raise serious concerns about the worldwide spread of the virus by migratory birds.
According to the international criteria, HPAI viruses are defined by over 75% mortality in 4- to 8-week-old chickens following an intravenous pathogenicity test or an intravenous pathogenicity index (IVPI) of more than 1.2 in 6-week-old chickens (34); however, there are some variations in pathogenicity intensity among the HPAI viruses in chickens (1, 3, 5, 12, 15, 28, 31, 48, 50-52, 57). Most of the HPAI viruses that were isolated before 1996 cause severe clinical signs (e.g., ruffled feathers, depression, labored breathing, and neurological signs) and severe gross lesions (e.g., head and face edema, cyanosis, subcutaneous hemorrhages in combs and leg shanks, and necrosis of combs and wattles) in chickens (1, 3, 15, 31, 50, 52, 57). These viruses usually kill chickens 3 to 6 days after intranasal inoculation. On the other hand, the recently emerged Asian H5N1 HPAI viruses are more virulent and kill chickens within 1 to 2 days without causing typical clinical signs and gross lesions (5, 12, 27, 33, 48, 51), although some Asian H5N1 viruses, such as A/Goose/Guangdong/2/96 (23), A/goose/Hong Kong/437-10/99 (17), and A/chicken/Indonesia/7/03 (58), were less virulent. To successfully control HPAI in poultry, it is important to better understand the mechanisms of increased pathogenicity of recent H5N1 HPAI viruses in chickens.
The Asian H5N1 HPAI virus has another important characteristic, which is its capability of crossing host-species barriers. It was reported that the H5N1 virus can infect and cause death in mammals such as mice (5, 9, 12, 14, 29), cats (21), tigers (2), ferrets (11, 26), monkeys (40), and humans (7, 49, 55). High-level inductions of proinflammatory cytokines in mammals infected with the H5N1 viruses, referred to as “cytokine storms,” have been hypothesized to contribute to the severity of pathological changes and ultimate death (4, 7, 13, 45, 55). Cytokine and chemokine dysregulation was detected in clinical cases of H5N1-infected humans (8, 13, 36) and also in monkeys experimentally infected with the H1N1 Spanish flu strain (20). In a mouse model, lymphocyte apoptosis and cytokine dysregulation have been proposed to contribute to the severity of the disease caused by H5N1 (56). Investigations with transgenic mice deficient in each cytokine gene suggest that tumor necrosis factor alpha (TNF-α) may contribute to morbidity and interleukin-1 (IL-1) may be important for virus clearance (53). However, mice deficient in TNF-α or IL-6 succumb to infection with H5N1, and cytokine inhibition treatment does not prevent death (42), suggesting that therapies targeting the virus rather than cytokines may be preferable. Thus, the significance of elevated proinflammatory cytokine responses in the pathogenesis of H5N1-infected mammals requires further studies.
In contrast, little is known about proinflammatory cytokine responses and their roles in pathogenicity in chickens infected with HPAI viruses, including the recent Asian H5N1 viruses. It was reported that infection with an HPAI virus results in upregulation of gene expression of gamma interferon (IFN-γ) and inducible nitric oxide synthase (58). However, the roles of proinflammatory cytokines in disease severity and outcomes in chickens infected systemically with HPAI viruses are largely unknown. The less-virulent Asian H5N1 virus, which causes severe clinical signs and gross lesions in chickens (17, 23, 27, 58), would be a valuable tool for investigating the role of proinflammatory cytokines in chickens infected with HPAI viruses, as well as for exploring the pathogenesis of the more-virulent Asian H5N1 HPAI virus, because of the antigenic and molecular similarities between them.
In this study, we compared the pathogenicities in chickens of the less-virulent and more-virulent Asian H5N1 HPAI viruses based on MDT, fever, cytokine responses, and viral replication. Our results suggest that the shift in the Asian H5N1 virus to increased virulence may be associated with efficient and rapid replication of the virus in chickens, accompanied by early destruction of host immune responses and followed by peracute death before fever can be induced. Finally, we discuss candidate genes that may account for the high pathogenicity of Asian H5N1 HPAI viruses in chickens.
MATERIALS AND METHODS
Viruses and titration.
Two HPAI virus isolates and one low-pathogenicity avian influenza (LPAI) virus isolate were used: A/chicken/Yamaguchi/7/2004 (H5N1) (CkYM7), isolated from a chicken during an outbreak in Japan (30), was used as the recent Asian H5N1 HPAI virus. A/duck/Yokohama/aq10/2003 (H5N1) (DkYK10), isolated in Japan from duck meat imported from China (27), was used as a less-virulent Asian H5N1 HPAI virus. A/chicken/Ibaraki/1/2005 (H5N2) (CkIB1) was an LPAI virus isolated from a chicken farm during the LPAI outbreaks in 2005 and 2006 in Japan (35). These viruses were propagated in the allantoic membrane of 10-day-old embryonated chicken eggs, and the virus fluids were harvested and stored at −80°C until use. A 50% egg infection dose (EID50) was determined using the method of Reed and Muench (39). The HPAI viruses were handled in a biosafety level 3 (BSL 3) facility following the protocols outlined in the biosafety manual for HPAI of our institute.
Chickens.
Four-week-old specific-pathogen-free (SPF) White Leghorn (Gallus gallus domesticus) chickens were purchased from the Nippon Institute of Biological Science (Kobuchizawa, Yamanashi, Japan). All of the chickens were housed in negative-pressure isolators with HEPA filters in the BSL 3 animal experimental facility in our institute in accordance with the biosafety manual.
Body temperature and MDT.
A wireless thermosensor (AirSense; Hitachi, Tokyo, Japan), which was designed for this experiment, was attached to the abdominal skin surface of each chicken by no-stretch sports tape. We also found that pathogenicity levels of two HPAI viruses were more clearly differentiated by the fever and MDT than by the INPI test (see Fig. S1 and S2 and Table 1 in the supplemental material). The wireless thermosensor signals were sent to a receiver placed outside of the isolator and then transferred to a computer outside the BSL 3 facility through an intranetworking system. The wireless thermosensor recorded the chickens' body temperature for 1 s every 20 s, and these data were processed to obtain the mean body temperature for each hour. The basal body temperature was estimated as the mean of the body temperature for 24 h before inoculation, and the basal body temperature varied from 39.9°C to 41.4°C among chickens. The fever in each chicken was estimated as above the basal body temperature for every 3 h. Individual death time was defined as the time when the body temperature dropped below 30°C after the virus inoculation.
A total of 34 chickens were divided into four groups of 7 to 10 chickens each. There were three inoculated groups for CkYM7 (n = 10), DkYK10 (n = 7), and CkIB1 (n = 10) and one uninoculated control group (n = 7). Each chicken was inoculated intranasally at a dose of 106.0 EID50/0.1 ml of AI virus with a micropipette, and chickens in each group were reared in a negative-pressure isolator as described above.
Chicken infectious dose.
To determine how viral dosage in inoculum affects fever and MDT in chickens, groups of 4-week-old SPF chickens were intranasally inoculated with different amounts of CkYM7 (n = 24) or DkYK10 (n = 24) (101 to 106.0 EID50 for each dilution). The chickens wore a wireless thermosensor to determine their exact body temperature and MDT and were observed for clinical signs twice a day until all of the chickens had died or for 7 days. The degree of fever and MDT for each group were estimated as described above.
Cytokine response experiment.
A total of 36 chickens were divided into four groups: groups inoculated with CkYM7 (n = 9), DkYK10 (n = 12), and CkIB1 (n = 12) and an uninoculated control group (n = 3). Each chicken was intranasally inoculated with 106.0 EID50/0.1 ml of CkYM7, DkYK10, or CkIB1, and chickens in each group were reared in a negative-pressure isolator as described above. Three chickens per group were euthanized with ether and sacrificed at each harvest time: 12, 24, and 32 h postinoculation (hpi) for the CkYM7 group; 12, 24, 48, and 84 hpi for the DkYK10 group; and 24, 48, 72, and 120 hpi for the CkIB1 group. The lungs were removed, soaked in RNA Later solution (Ambion, Austin, TX), and stored at −80°C until used.
Total tissue RNA was extracted from lung homogenate supernatants with the RNeasy minikit (Qiagen, MD) according to the manufacturer's instructions. The quality of the extracted total tissue RNA was assessed visually by agarose gel electrophoresis with 1.5% formamide to obtain pure RNA samples with a 28S/18S ratio of 2.0. Three micrograms of total tissue RNA per sample was reverse transcribed into cDNA at 42°C for 60 min using 200 U of avian myeloblastosis virus reverse transcriptase (Takara, Shiga, Japan) and 2.5 ng/μl random primers (Takara) in the presence of RNase inhibitor (Takara).
The cDNA performed on lung tissue was used to quantify cytokine mRNA expression levels by real-time PCR to compare the host immune responses among chickens infected with CkYM7, DkYK10, or CkIB1. A total of nine cytokine probe/primer sets used in this study were IFN-α, IFN-β, IFN-γ, IL-4, IL-6, IL-8, IL-10, IL-15, and IL-18 mRNAs (19, 38, 41, 59) (Table 1), with modification of the primer/probe concentrations. Briefly, the reaction mixture consisted of 5 μl of Taq polymerase master mixture (Perfect real time; Takara), 1 μl of cDNA (0.3 μg), 1 μl each of forward and reverse primers (100 μM), 1 μl of probes (50 μM), and 1 μl of ROX reference dye II (×50) (Takara). All of the reactions were run in triplicate with the ABI PRISM 7500 sequence detection system (Applied Biosystems, CA) using the following cycling parameters: 1 cycle at 50°C for 2 min and 95°C for 10 min followed by 40 cycles of 94°C for 15 s and 60°C for 1 min. Each cycle threshold (CT) value was an average of values obtained from the samples. The mRNA expression level was computed using the 2−ΔΔCT method with the formula 2−(ΔCT for sample − ΔCT for control) (25). The 28S rRNA was used as a control.
TABLE 1.
List of probes and primers for real-time quantitative reverse transcription-PCR for detection of chicken cytokine mRNAs
| Target RNA | Probe or primer sequence (5′→3′)a | Exon boundary | Accession no. | Reference |
|---|---|---|---|---|
| 28S | Probe: 5′-(FAM)-AGGACCGCTACGGACCTCCACCA-(BHQ)-3′ | X59733 | 38 | |
| Forward primer: 5′-GGCGAAGCCAGAGGAAACT-3′ | ||||
| Reverse primer: 5′-GACGACCGATTTGCACGTC-3′ | ||||
| IFN-α | Probe: 5′-(FAM)-CTCAACCGGATCCACCGCTACACC-(BHQ)-3′ | Intronless | U07868 | 38 |
| Forward primer: 5′-GACAGCCAACGCCAAAGC-3′ | ||||
| Reverse primer: 5′-GTCGCTGCTGTCCAAGCATT-3′ | ||||
| IFN-β | Probe: 5′-(FAM)-TTAGCAGCCCACACACTCCAAAACACTG-(BHQ)-3′ | Intronless | X92479 | 38 |
| Forward primer: 5′-CCTCCAACACCTCTTCAACATG-3′ | ||||
| Reverse primer: 5′-TGGCGTGCGGTCAAT-3′ | ||||
| IFN-γ | Probe: 5′-(FAM)-TGGCCAAGCTCCCGATGAACGA-(BHQ)-3′ | 3/4 | Y07922 | 19 |
| Forward primer: 5′-GTGAAGAAGGTGAAAGATATCATGGA-3′ | ||||
| Reverse primer: 5′-GCTTTGCGCTGGATTCTCA-3′ | ||||
| IL-4 | Probe: 5′-(FAM)-AGCAGCACCTCCCTCAAGGCACC-(BHQ)-3′ | 3/4 | AJ621735 | 41 |
| Forward primer: 5′-AACATGCGTCAGCTCCTGAAT-3′ | ||||
| Reverse primer: 5′-TCTGCTAGGAACTTCTCCATTGAA-3′ | ||||
| IL-6 | Probe: 5′-(FAM)-AGGAGAAATGCCTGACGAAGCTCTCCA-(BHQ)-3′ | 3/4 | AJ250838 | 19 |
| Forward primer: 5′-GCTCGCCGGCTTCGA-3′ | ||||
| Reverse primer: 5′-GGTAGGTCTGAAAGGCGAACAG-3′ | ||||
| IL-8 | Probe: 5′-(FAM)-TCTTTACCAGCGTCCTACCTTGCGACA-(BHQ)-3′ | 1/2 | AJ0099800 | 59 |
| Forward primer: 5′-GCCCTCCTCCTGGTTTCAG-3′ | ||||
| Reverse primer: 5′-TGGCACCGCAGCTCATT-3′ | ||||
| IL-15 | Probe: 5′-(FAM)-AAGTTGCAAATCTTGCATTTCCATTTTTCCA-(BHQ)-3′ | 4/5 | AJ416937 | 19 |
| Forward primer: 5′-TAGGAAGCATGATGTACGGAACAT-3′ | ||||
| Reverse primer: 5′-TTTTTGCTGTTGTGGAATTCAACT-3′ | ||||
| IL-18 | Probe: 5′-(FAM)-CCGCGCCTTCAGCAGGGATG-(BHQ)-3′ | 4/5 | AJ276026 | 19 |
| Forward primer: 5′-AGGTGAAATCTGGCAGTGGAAT-3′ | ||||
| Reverse primer: 5′-ACCTGGACGCTGAATGCAA-3′ |
FAM, 6-carboxyfluorescein; BHQ, Black Hole Quencher dye (Biosearch Technologies).
Virus replication in chickens.
To compare virus replication efficiencies among viruses in chickens, a total of 33 chickens were divided into three groups: CkYM7 (n = 12), DkYK10 (n = 12), and CkIB1 (n = 9). The chickens in each group were inoculated intranasally with 106.0 EID50/0.1 ml of each virus, and chickens in each group were reared in a negative-pressure isolator as described above. Three chickens per group were euthanized with ether and sacrificed at each of the harvest times: 12, 24, 30, and 32 hpi for the CkYM7 group; 24, 48, 72, and 81 hpi for the DkYK10 group; and 72, 120, and 168 hpi for the CkIB1 group. The lungs, spleens, and kidneys were removed and stored at −80°C until the titration. For the viral titration, 0.1 g of tissue was mixed with 900 μl of Dulbecco's modified Eagle's medium (Nissui, Tokyo, Japan) containing 10% fetal bovine serum (Invitrogen) and penicillin-streptomycin and then homogenized using a Multi-Beads shocker (Yasui Kikai, Osaka, Japan) to make the 10% tissue homogenate. After centrifugation at 10,000 rpm (8,000 × g) for 5 min at 4°C, the supernatant was used for virus titration with 10-day-old embryonated eggs. The virus titer was calculated as described previously (39).
Histopathology and immunohistochemistry.
The infected chickens used in the virus replication experiment were also processed for the histopathological study and immunostaining. These chickens were inoculated intranasally with CkYM7, DkYK10, or CkIB1, and their lungs, spleens, livers, kidneys, large intestines, pancreases, and tracheae were collected sequentially and fixed in 10% neutral buffered formalin. The tissues were dehydrated in a graded ethanol series (70, 80, 90, and 100% for 3 min in each) at room temperature and air dried. After the tissues were embedded in paraffin, 4-μm-thick sections were prepared and placed in hematoxylin-and-eosin solution (Sigma) for 20 min at room temperature, decolorized with 1% HCl in 70% ethanol, and stained with eosin solution (Sigma) for 10 min. Immunohistochemistry of the serial sections was performed to examine the distribution of the AI virus matrix protein (M) antigen with anti-M monoclonal antibody as described previously (54).
Detection of apoptotic cells.
To compare the apoptosis induced in chickens infected with the two HPAI viruses, each of three chickens was intranasally inoculated with CkYM7 or DkYK10 (106.0 EID50/0.1 ml), and each group of chickens was reared in a negative-pressure isolator. These chickens were euthanized with ether and sacrificed at 36 or 96 hpi for CkYM7 and DkYK10, respectively, and lungs and spleens were fixed in 10% neutral buffered formalin. These paraffin sections were stained with ApopTag peroxidase in situ apoptosis detection kit (S7100; Chemicon International, Inc.) according to their protocol within 7 days after being sacrificed.
Statistical analysis.
Student's t test was used to determine the statistical significance of fever and MDT in chickens infected with CkYM7, DkYK10, or CkIB1. Statistical significance was set at P < 0.05.
RESULTS
Clinical signs and gross lesions.
To compare the pathogenicities of the two HPAI viruses in 4-week-old chickens, each of 10 4-week-old SPF chickens was inoculated intranasally with each HPAI virus, and clinical signs, gross lesions, and mortality were observed every 12 h. Both of the two HPAI strains, CkYM7 and DkYK10, caused 100% mortality in the chickens, and the intranasal pathogenicity indices (INPI) were more than 2.3, whereas the LPAI strain, CkIB1 (H5N2), induced no mortality in chickens and the INPI was 0.0 (Fig. 1 and Table 2).
FIG. 1.
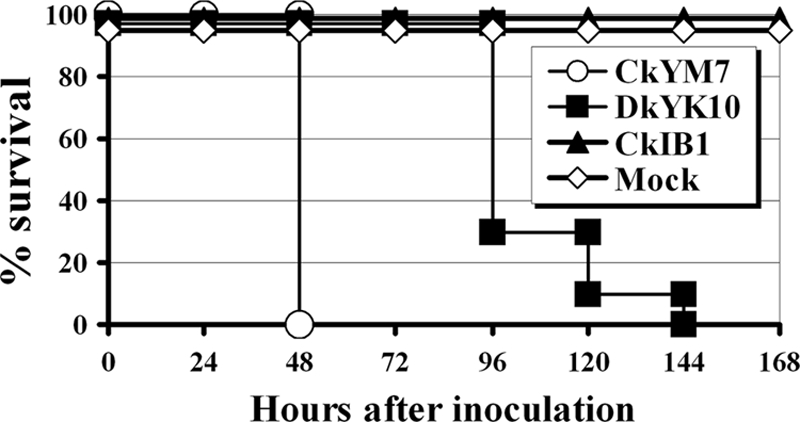
Survival of 4-week-old chickens after intranasal inoculation with one of two HPAI viruses or an LPAI virus. Shown is the percent survival of chickens inoculated with CkYM7 (H5N1), DkYK10 (H5N1), or CkIB1 (H5N2) or mock inoculated. The remaining chickens were observed for clinical signs and gross lesions twice a day through a 10-day observation period.
TABLE 2.
Pathogenicity of the two HPAI viruses and one LPAI virus in 4-week-old chickens after intranasal inoculationa
| Virus | No. of chickens sick/dead (total) | INPI | Reference |
|---|---|---|---|
| CkYM7 | 10/10 (10) | 2.7 | 30 |
| DkYK10 | 10/10 (10) | 2.3 | 27 |
| CkIB1 | 0/0 (10) | 0.0 | 35 |
Four-week-old White Leghorn chickens were inoculated intranasally with 106.0 EID50/0.1 ml of each virus.
There were some variations in the disease outcome among the two HPAI viruses. CkYM7, a recent Asian H5N1 virus, caused no typical clinical signs and no gross lesions, except for ruffled feathers and depression. All of the infected chickens (10/10) died within 2 days after inoculation with 106.0 EID50 (Fig. 1). In contrast, DkYK10 caused severe respiratory signs, along with hemorrhages in combs, wattles, and leg shanks and severe edema of the head and legs. The gross lesions were first observed after 48 hpi and then more frequently after 72 hpi. Chickens infected with DkYK10 died 4 to 6 days after inoculation (Fig. 1). Chickens infected with CkIB1 and uninfected control chickens showed no clinical signs, no gross lesions, and no mortality in the 10 days after inoculation.
Fever and MDT.
To further compare the pathogenicities of the two HPAI viruses in chickens, induction of fever and accurate MDT were determined with the wireless thermosensors. In chickens infected with the CkYM7 strain, slight fever (+0.6°C) was observed at 24 hpi, but it suddenly decreased from 30 hpi onward (Fig. 2A). No chickens had fevers of more than 1.0°C above their baseline temperature, and the MDT was 34 h ± 2.2 h (Fig. 3).
FIG. 2.

Body temperature kinetics of chickens after intranasal inoculation with one of two HPAI viruses or an LPAI virus. The average kinetics are shown. Each curve shows the mean ± standard deviation for 7 to 10 chickens. (A) CkYM7 (H5N1); (B) DkYK10 (H5N1); (C) CkIB1 (H5N2); and (D) mock.
FIG. 3.
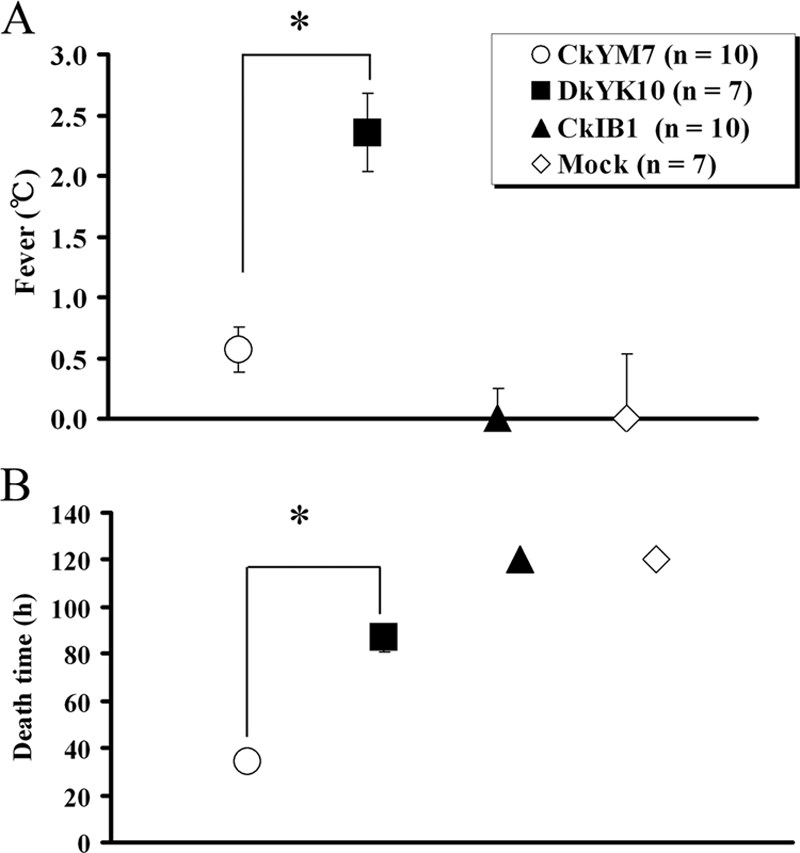
Fever and MDT of chickens after intranasal inoculation. Values represent the mean ± standard deviation for each group. (A) Fever is based on increases above the basal body temperature. Average fever per group is shown. (B) The time to death of each chicken was defined as the time when body temperature fell below 30°C after inoculation; the MDT for each group is shown. An asterisk denotes significant difference from the value for CkYM7 (H5N1) (P < 0.05).
In contrast, chickens infected with DkYK10 had high fevers (Fig. 2B). The fevers appeared at around 30 hpi and persisted for about 2 days with a peak of +2.4°C ± 0.3°C at around 60 hpi. All seven of the chickens had a fever of more than 2.0°C. Body temperatures started to drop at 72 hpi, and the MDT was 87 h ± 6.0 h (Fig. 3). The differences in fever and MDT between infection with CkYM7 and infection with DkYK10 were significant (P < 0.05).
CkIB1 did not induce fever (Fig. 2C), and the body temperature kinetics were similar to those of uninoculated control chickens (Fig. 2D). The slight increase in body temperature at 100 hpi in chickens infected with CkIB1 may be an artifact, because the change was not reproduced in the second trial (data not shown).
Averages of the fever and MDT in each virus group are shown in Fig. 3A and B, respectively.
Effect of virus amount on fever and MDT.
To determine the effect of virus dose on fever and MDT in chickens, four SPF chickens per group were intranasally inoculated with different amounts of CkYM7 or DkYK10, and fever and MDT were compared among groups infected with each virus strain (Fig. 4).
FIG. 4.

Effect of virus dose on fever and MDT of chickens infected with CkYM7, DkYK10, or CkIB1. Four-week-old SPF White Leghorn chickens (four chickens per dilution) were intranasally inoculated with 0.1 ml of diluted virus. MDT and average fevers of the chickens after inoculation with CkYM7 (A and B) and DkYK10 (C and D) are shown. Values represent the mean ± standard deviation for each dilution.
For CkYM7, all of the chickens inoculated intranasally with more than 103 EID50 died; the 50% chicken infectious dose was 102.5 EID50. The dead chickens did not show any respiratory signs or gross lesions. In parallel with the increase in the virus titer of the inoculum the MDT became shorter (∼7 h/log EID50): 103, 57 ± 4 h; 104, 49 ± 1.7 h; 105, 40 ± 1 h; and 106, 36 ± 2.4 h. Variation of the MDT within each dosage group was minimal for the CkYM7 strain (Fig. 4A), and the degrees of fever appeared to be similar among the CkYM7 groups: 103, +0.7 ± 0.1°C; 104, +1.0 ± 0.3°C; 105, +1.1 ± 0.2°C; and 106, +0.9 ± 0.2°C (Fig. 4B).
We also observed these trends in chickens inoculated with DkYK10. All of the chickens inoculated with more than 103 EID50 of DkYK10 died, and the 50% chicken infectious dose was 102.5 EID50. All of the dead chickens exhibited severe clinical signs and gross lesions before death. The MDT decreased in proportion with the increase in the virus titer of the inoculum (∼10 h/log EID50): 103, 110 ± 25 h; 104, 97 ± 16 h; 105, 97 ± 7 h; and 106, 88 ± 7 h (Fig. 4C). However, the MDTs within the DkYK10 groups varied markedly, particularly when the chickens were inoculated with low doses of the virus. The degree of fever did not differ substantially among the groups: 103, +2.7 ± 0.1°C; 104, +2.1 ± 0.3°C; 105, +2.4 ± 0.6°C; and 106, +2.4 ± 0.4°C (Fig. 4D).
These results indicate that MDT and fever, as well as clinical signs and gross lesions, were determined by intrinsic characteristics of the two virus strains, although MDT was influenced to some extent by the virus titer of the inoculum.
Comparison of cytokine responses in chickens.
To compare antiviral or proinflammatory cytokine responses in chickens infected with CkYM7, DkYK10, or CkIB1 of different pathogenicity, antiviral and proinflammatory cytokine expressions in lungs were sequentially quantified using a real-time PCR.
In chickens infected with CkYM7, quick increases in mRNA expression levels of antiviral (IFN-α and IFN-β) and proinflammatory (IL-6, IL-8, IL-15, and IL-18) cytokines were detected at 12 hpi compared with preinfection levels (Fig. 5). These cytokine expression levels then peaked at 24 hpi; however, suddenly the levels decreased at 32 hpi, just before death. On the contrary, the expression levels of IFN-γ and IL-4 mRNAs were decreased from 24 hpi and further at 32 hpi in the chickens.
FIG. 5.

Kinetic curves of mRNA expression of IFN-α, IFN-β, IFN-γ, IL-4, IL-6, IL-8, IL-10, IL-15, and IL-18 genes in the lungs of chickens infected with AI viruses. The data are the mean change (fold) ± standard deviation for cytokine mRNA expression levels in each group. In contrast with CkIB1, CkYM7 and DkYK10 induced higher levels of IFN-α, IFN-β, IL-6, IL-8, IL-15, and IL-18.
In chickens infected with DkYK10, expressions of antiviral (IFN-α and IFN-β) and proinflammatory (IL-6, IL-8, and IL-15) cytokine genes were rapidly upregulated at 12 hpi and then reached a maximum at 24 hpi, as was observed in chickens inoculated with CkYM7 (Fig. 5). These cytokine levels declined at 48 hpi to levels compatible with or lower than those induced by LPAI CkIB1; the lower levels persisted until the end of the experiment.
Together, IL-18 and IL-12 induce IFN-γ expression during cell-mediated immunity. IL-18 expression levels increased dramatically at 24 and 48 hpi in DkYK10-inoculated chickens (3 logs higher) and then decreased at 84 hpi to levels comparable with those of the CkIB1-infected chickens (Fig. 5). Nevertheless, the IFN-γ gene expression was not upregulated in the DkYK10 group (Fig. 5), and expression of IL-4 that stimulates proliferation of activated B cells was not increased.
Expression of IFN-α, IL-4, IL-6, IL-15, and IL-18 mRNAs moderately increased at 24 and/or 48 hpi in chickens infected with CkIB1, whereas IL-8 mRNA slowly increased with a peak at 72 hpi. IFN-β and IFN-γ expression levels were slightly upregulated or similar to those of uninfected chickens throughout the experiment (Fig. 5).
Comparison of virus replication efficiency.
To compare the viral growth curves following infection with CkYM7, DkYK10, or CkIB1, chicken lungs, kidneys, and spleens were collected sequentially and the tissue homogenates were subsequently assayed for virus titers.
As shown in Fig. 6, the viral titers of CkYM7 were very high compared with those of DkYK10. CkYM7 replicated very rapidly and efficiently in chickens (Fig. 6). There was almost no difference in viral titers among the three tissues. The virus was first detected at 12 hpi (102.0 and 103.0 EID50/g in two of three chickens); it reached 107.2 EID50/g at 24 hpi in the lungs, and then climbed to 108.8 EID50/g at 30 hpi, followed by 1010.5 EID50/g at 32 hpi, just before death. These virus titers were more than 100 times higher than those of DkYK10.
FIG. 6.

Kinetics of virus titers in chicken tissues. Titers of individual lungs, kidneys, and spleens were determined with embryonated chicken eggs. The mean virus titers ± standard deviations from three birds are shown (log10 EID50/g).
DkYK10 grew moderately well in chickens (Fig. 6). The virus was detected in lungs at 24 hpi (102.3 EID50/g in three of three chickens), and the virus titer increased to 106.0 EID50/g at 48 hpi, which was maintained until 84 hpi, just before death, except that virus titers at 72 hpi were slightly higher.
Replication of the LPAI CkIB1 in chickens was restrictive, and the virus titer was either low or not detected in tissues throughout the experiment. Viral titers in lung tissues were higher than those in spleens or kidneys. CkIB1 was recovered at a low titer from lungs at 72 (one of three chickens), 120 (102.0 EID50/g in 1/3 chickens), and 168 (102.0 EID50/g in one of three chickens) hpi, and hardly detected in spleens and kidneys from 120 hpi onward (Fig. 6).
Comparison of immunohistochemical staining.
To compare the yield and distribution of viral antigens in chickens inoculated with CkYM7, DkYK10, or CkIB1, the lungs, spleens, livers, kidneys, large intestines, pancreases, and tracheae were collected sequentially and stained immunohistochemically with anti-M antibody (Table 3). In the CkYM7 group, M antigen was first detected in lung and kidney tissues from one of the three chickens killed at 12 hpi and had spread to most tissues except for the intestines by 24 hpi. By 30 and 32 hpi, staining for M antigen was generalized and intense (Table 3). The M antigen was detected mainly in vascular endothelial cells and macrophages in lungs (Fig. 7A) and macrophages in spleen (Fig. 7C) in the CkYM7 group.
TABLE 3.
Distribution of viral antigen in 4-wk-old SPF chicken inoculated intranasally with CkYM7, DkYK10, or CkIB1
| Strain and tissuea | Time of AI virus matrix antigen detectionb
|
Location where AI virus membrane antigen was detected | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 12 hpi | 24 hpi | 30 hpi | 32 hpi | 48 hpi | 72 hpi | 81 hpi | 120 hpi | 168 hpi | ||
| CkYM7 | ||||||||||
| Lung | +, −, − | +, +, + | +++, +++, +++ | +++, +++, ++ | Vascular endothelial cells and macrophages | |||||
| Spleen | −, −, − | +, +, + | +++, +++, ++ | +++, +++, +++ | Vascular endothelial cells and macrophages | |||||
| Liver | −, −, − | +, ++, + | +++, +++, +++ | +++, +++, +++ | Vascular endothelial cells and Kupffer cells | |||||
| Kidney | +,−, − | +, +, + | +++, +++, ++ | +++, ++, +++ | Vascular endothelial cells and renal tubular epithelial cells | |||||
| Intestine | −, −, − | −, −, − | ++, +, + | +++, ++, − | Vascular endothelial cells and macrophages | |||||
| Pancreas | −, −, − | −, +, + | NT, +++, NT | +++, ++, +++ | Vascular endothelial cells and exocrine cells | |||||
| Trachea | −, −, − | +, +, + | +++, +++, ++ | +++, ++, +++ | Vascular endothelial cells | |||||
| DkYK10 | ||||||||||
| Lung | −, −, + | +, +, + | +, +, + | +, +, + | Macrophages | |||||
| Spleen | −, −, − | −, −, + | +, +, − | −, −, − | ||||||
| Liver | −, −, − | −, −, − | −, −, − | −, −, + | ||||||
| Kidney | −, −, − | −, −, + | +, −, + | −, +, + | ||||||
| Intestine | −, −, − | −, −, + | +, +, − | −, −, − | ||||||
| Pancreas | −, −, − | −, −, − | −, −, − | +, +, + | ||||||
| Trachea | −, −, − | −, +, + | +, −, + | −, −, − | ||||||
| CkIB1 | ||||||||||
| Lung | −, −, − | NT, −, − | −, −, − | |||||||
| Spleen | −, −, − | −, −, − | −, −, − | |||||||
| Liver | −, −, − | −, −, − | −, −, − | |||||||
| Kidney | −, −, − | −, −, − | −, −, − | |||||||
| Intestine | −, −, − | −, −, − | −, −, − | |||||||
| Pancreas | −, −, − | −, −, − | −, +, − | |||||||
| Trachea | −, −, − | −, −, − | −, −, − | |||||||
These tissues were obtained from the chicken used to obtain the virus growth curve in Fig. 6.
Each of three chickens was sacrificed at each point, and their tissues were examined for AI virus matrix antigen. Grading by level of AI virus matrix protein detected: −, none; +, mild, ++, moderate; +++, severe; NT, not tested.
FIG. 7.

Immunohistochemical staining of AI virus matrix protein in lungs and spleens of chickens intranasally inoculated with CkYM7 or DkYK10. The viral M antigen was detected mainly in vascular endothelial cells and macrophages of lung tissue 32 h after inoculation with CkYM7 (A), macrophages of lung tissue 81 h after inoculation with DkYK10 (B), and macrophages in splenic red pulp 32 h after inoculation with CkYM7 (C). (D) DkYK10 in spleen at 81 hpi. Magnification: ×40 (A to D).
In contrast, the viral antigen was first detected in lung tissue from one of the three DkYK10-infected chickens killed at 24 hpi, but the distribution and intensity of M antigen did not increase as dramatically as they did in CkYM7-infected chickens. Slight to moderate amounts of M antigen were detected from 48 to 81 hpi in several organs, although it was found most frequently in lungs (Table 3). There was some variability in number of M antigen-positive cells among individual chickens infected with the DkYK10. These cells appeared to be macrophages in lungs (Fig. 7B) but were not identified in other tissues.
Macrophage invasion of the lungs and spleens was histopathologically observed in chickens infected with either CkYM7 or DkYK10. The amount of viral antigen detected in tissues correlated with the viral titers of the two HPAI virus strains measured from tissue (Table 3 and Fig. 7).
Detection of apoptotic cells.
The apoptotic cells were detected in lungs and spleens of all three chickens inoculated with CkYM7 (Fig. 8A and D) or DkYK10 (Fig. 8B and E). The apoptotic cells were more frequently detected in spleens than in lungs in both virus strains, but there was no difference in intensity in lungs or spleens between the two HPAI virus strains. Most of the apoptotic cells might be macrophages in both tissues in both virus strains. Negligible numbers of apoptotic cells were detected in lungs and spleens of the uninfected chickens (Fig. 8C and F).
FIG. 8.

Detection of apoptotic cells in lungs and spleens of chickens infected with CkYM7 or DkYK10. The apoptotic cells were detected in lung (A) and spleen (D) tissues 36 h after inoculation with CkYM7 and lung (B) and spleen (E) tissues 96 h after inoculation with DkYK10. Lung (C) and spleen (F) tissues derived from uninfected chickens. Magnification: ×40 (A to F).
DISCUSSION
IL-1 and IL-6 produced by lymphocytes and macrophages stimulate vascular endothelial cells to synthesize prostaglandin E2, which binds to the EP3 receptor in neural cells in the hypothalamus. The signal in the hypothalamus is then transferred to peripheral thermoregulatory systems to generate fever. It was unknown whether HPAI virus infection in chickens caused high fever. To our knowledge, the present study is the first to demonstrate that there are two types of fever kinetics in chickens infected with HPAI viruses of different pathogenicities. The extremely virulent strain CkYK7 replicated quickly and efficiently in chickens and caused no or slight fever. The extremely rapid replication of CkYM7 in macrophages and vascular endothelial cells described below might disrupt the thermoregulation system of chickens. This speculation was supported by the fact that fever was not induced in chickens that survived for 57 h after inoculation with minimum amount of CkYM7 (103.0 EID50). In contrast, the less-virulent strain DkYK10 gave rise to high and long fevers in chickens, which declined gradually after the peak. This suggests that although the chicken's thermoregulatory system responded normally for the first 2 days of infection, it was then disrupted, as indicated by the declining fever. The kinetic curves of fevers were preserved in chickens inoculated with different amounts of DkYK10. These results demonstrate that fever is an indicator to determine the pathogenicity of avian influenza virus in chickens. At present, the intravenous pathogenicity test is an authorized test for the determination of pathogenicity of avian influenza viruses in chickens by the World Organization for Animal Health (OIE). In this test, less-objective indicators such as observations of clinical signs, gross lesions, and day of death time were used (34). Actually, the INPIs were not much different between CkYM7 and DkYK10 (2.7 and 2.3, respectively) (Table 2). We also found that pathogenicity levels of two HPAI viruses were more clearly differentiated by the fever and MDT than by the INPI test (see Fig. S1 and S2 and Table 1 in the supplemental material). The wireless thermosensor would be the useful tool to determine the pathogenicity of infectious pathogens in birds and mammals.
Clinical signs and gross lesions are closely associated with pathogenicity of AI viruses in animals, and the distribution and severity of gross lesions and clinical signs vary among HPAI viruses in chickens (1, 3, 12, 15, 27, 28, 31, 50-52, 57). We investigated onset of clinical signs and gross lesions in chickens infected with two HPAI viruses of different pathogenicities. Only severe depression was observed prior to death in chickens infected with CkYM7. In contrast, severe clinical signs and gross lesions were detected in chickens infected with DkYK10 from 72 hpi onward. These observations suggest that onset of clinical signs and gross lesions began at 48 hpi and became obvious by 72 hpi in chickens that survived the peracute phase. Swayne and others have suggested that altered permeability of cardiovascular endothelial cells is responsible for edema and hemorrhage (3, 37, 48, 50, 51). This stage is probably followed by a consumptive coagulopathy with thrombocytopenia (32) and efficient virus replication in multiple organs, resulting in multiple organ failure and death. In our study, these processes were not observed in chickens infected with CkYM7 but were observed in those infected with DkYK10, who survived longer. Variations in the severity of clinical signs and gross lesions among HPAI virus strains appear to be associated with time of death. However, typical clinical signs and gross lesions were not observed in chickens inoculated with 103.0 EID50 of strain CkYM7, which survived for approximately 57 h. Although time of death is a critical factor for producing clinical signs and gross lesions in chickens, our results indicate that these signs and lesions were intrinsically associated with pathogenicity of virus strains, not directly with time of death.
The broad spectrum of pathogenicity in birds and interspecies transmission are main characteristics of the H5N1 HPAI viruses (1, 6, 12, 24, 37, 46, 47), and mammalian transmission raises the major public health concern (7, 13, 36, 44, 49, 55). Cytokine production may play a critical role in the pathogenesis of H5N1 HPAI virus infection in mammals (8, 13, 36). The “cytokine storm” has been hypothesized to the main cause of mortality in mammals infected with H5N1 and also in the human Spanish flu (20). Overproduction or dysregulation of proinflammatory cytokines in primary human macrophages and respiratory epithelial cells following infection with H5N1 HPAI viruses has been reported (4, 13). Although studies using mice for influenza virus pathogenesis and treatment in mammals have been performed, the role of cytokine overproduction in the pathogenesis of H5N1 HPAI virus infections is still in dispute. Dybing et al. reported that decreased levels of transforming growth factor β may contribute to the severe pathological changes observed in infected mice (9). In addition, Szretter et al. suggested that TNF-α may contribute to morbidity in early stages of infection and that IL-1 may be important for clearance of the H5N1 virus (42, 53). However, Salomon et al. demonstrated that mice deficient in TNF-α, IL-6, or CC chemokine ligand 2 were not protected; mortality caused by H5N1 virus was the same as that in wild-type mice (42).
In contrast, the role of cytokines in the pathogenesis of the HPAI viruses in chickens is poorly understood. HPAI viruses replicate systemically in chickens and are highly lethal (52). To understand the basis of HPAI viral pathogenesis in chickens, we compared the expression of antiviral and proinflammatory cytokine mRNAs following inoculation with one of two HPAI viruses or one LPAI virus. Our study showed that two HPAI viruses, CkYM7 and DkYK10, increased quick and intensive production of antiviral and proinflammatory cytokines in chickens compared with the LPAI CkIB1 strain. IFN-α, IL-6, IL-8, IL-15, and IL-18 are produced in macrophages immediately after infection, while IFN-β, IL-6, IL-8, IL-15, and IL-18 are quickly generated in epithelial cells or vascular endothelial cells. IFN-γ, which is induced by IL-12 or IL-18, plays a critical role in cellular immunity, whereas IL-4, which is induced by IL-15, plays an important part in humoral immunity. After inoculation with CkYM7, antiviral (IFN-α and IFN-β) and proinflammatory (IL-6, IL-8, IL-15, and IL-18) cytokine mRNAs were quickly induced at 24 hpi, but 8 h later, mRNA levels became dramatically lower. Since apoptotic cells were detected in macrophages in lung tissues of chickens infected with CkYM7, the acute drop in cytokine mRNA expression in chickens infected with CkYM7 could be caused by the efficient replication of CkYM7 in the cytokine-producing macrophages. In contrast, in chickens infected with DkYK10, mRNA expression of antiviral (IFN-α and IFN-β) and proinflammatory (IL-4, IL-6, IL-8, and IL-15) cytokines decreased at 48 hpi after a transient peak at 24 hpi. The decreased expression levels were comparable to (IFN-α, IFN-β, and IL-6) or lower than (IL-4, IL-8, and IL-15) those observed in chickens infected with CkIB1. The downregulation of cytokine mRNA synthesis in DkYK10 could be caused by apoptosis of cytokine-producing cells, although we did not determine when the apoptosis was elicited in the chickens in this study. It is also possible that the depressed proinflammatory cytokine gene expression of IL-6, IL-15, and IL-18 might have failed to switch to Th1-type (IFN-γ) and Th2-type (IL-4) cytokine mRNA synthesis. Thus, our data do not support the hypothesis that proinflammatory cytokines play a role in the severity of clinical signs and gross lesions observed in chickens infected with DkYK10. Instead, our study favors the idea that rapid and efficient replication of HPAI viruses, particularly in macrophages and/or vascular endothelial cells, overwhelms the innate immune responses of chickens infected with HPAI viruses.
To determine the factors behind the different pathogenicities of the two HPAI virus strains, we sequentially compared viral titers in lungs, kidneys, and spleens and demonstrated a clear difference in the viral titers of chickens infected with the two HPAI viruses. CkYM7 viral titers were more than twofold higher than DkYK10 titers, although there was some variation among chickens infected with DkYK10. CkYM7 replicated rapidly and efficiently in chickens, and viral antigens were detected predominantly in macrophages and vascular endothelial cells, which resulted in apoptosis of macrophages and disruption of innate immune responses. These results suggest that replication of CkYK7 preferentially in vascular endothelial cells and macrophages could account for the increased pathogenicity of recent Asian aH5N1 viruses in chickens. We propose that the preferential replication of CkYM7 in these cytokine-producing cells could result in disruption of the innate immune responses, as well as other important functions of chickens. In contrast, the merely moderately rapid replication of DkYK10 in tissues and induction of the same intensity of apoptosis as those detected in chickens infected with CkYM7 might partly account for downregulation of cytokine expression from 48 hpi onward and death at 87 hpi. Also, it is possible that DkYK10 may replicate more efficiently in other tissues such as heart or brain. These data suggest that pathogenesis may be different between the two HPAI viruses.
The molecular basis of the increased pathogenesis of Asian H5N1 HPAI viruses in chickens remains to be determined. Several critical amino acids contributing to the pathogenicity of HPAI viruses in animals have been identified by other groups (Table 4). Both of the HPAI strains used in this study have multiple basic amino acids at the hemagglutinin (HA) cleavage site, which are required for replication in multiple tissues (16, 52). Five amino acids surrounding the HA receptor binding site that are known to be involved in pathogenicity of H5N1 viruses in chickens (17) are identical between the two viruses. The amino acid at position 627 in PB2 (14) is glutamic acid in both strains. The glutamic acid at position 92 in NS1, which confers resistance to antiviral cytokines in pigs (43, 45), and the alanine at position 149 of NS1, which are critical to high pathogenicity in chicken (23), were present in both viruses. The amino acids at positions 515 in PA and 436 in PB1, which are associated with lethality in ducks (18), were the same in the two viruses (18). Recently, NP, PB1, and PB2 genes were shown to contribute to the altered replication of H5N1 HPAI viruses in chickens (58), but the potential critical amino acids were not related in our case. Whole-genome-sequence comparisons revealed that CkYM7 and DkYK10 differed at 89 amino acids (98% identity) scattered in eight gene segments with a 20-amino-acid deletion in the NA stalk region of DkYK10 (Table 4). It is possible that those amino acids associated with adaptation from ducks to chickens also play a critical role in the increased pathogenicity of CkYM7 in chickens. Because 50% chicken infectious doses of CkYM7 and DkYK10 were the same (102.5 EID50), the HA and NA surface glycoproteins, which are key proteins for binding to or release from infected cells, may not be responsible for the high-growth phenotype of CkYM7 in chickens. It is more likely that viral polymerase proteins are probably involved in the efficient replication of CkYM7 in chickens. We are currently undertaking a molecular genetics approach to explore the genetic basis underlying the increased pathogenicity of Asian H5N1 HPAI viruses in chickens.
TABLE 4.
Comparisons of amino acid sequences between CkYM7 and DkYK10
| Gene product | Amino acid identity (%) | No. of amino acids different/totala | Pathogenesis- related amino acid(s) | Reference | Critical identical amino acid(s) related to pathogenesis in:
|
|
|---|---|---|---|---|---|---|
| CkYM7 | DkYK10 | |||||
| PB2 | 98.7 | 10/759 | 627K | 14 | E | E |
| 701N | 10 | D | D | |||
| 714R | 10 | S | S | |||
| PB1 | 98.8 | 9/757 | 436Y | 18 | Y | Y |
| PA | 98.5 | 11/716 | 515T | 18 | T | T |
| HA | 97.2 | 16/567 | 97, 108, 126, 138, 212 | 17 | E, Y, H, W, T | E, Y, H, W, T |
| Cleavage site | 16 | PQRERRKKR | PQRERRRKKR | |||
| NP | 98.6 | 7/498 | Not identified | 58 | ||
| 319K | 10 | N | N | |||
| NA | 91.7 | 19/469b | Glycosylation site | 17 | ||
| M1 | 99.6 | 1/252 | ||||
| M2 | 97.9 | 2/97 | ||||
| NS1 | 96.4 | 8/225 | 92E | 43 | E | E |
| 149A | 23 | G | G | |||
| NS2 | 95.0 | 6/121 | ||||
CkYM7 virus NA protein contains a 20-amino-acid stalk deletion as well as 19 amino acid differences. Therefore, the total number of amino acids different/total shown represents 89 (+ 20 amino acids deleted)/4,461.
Including the 20-amino-acid stalk deletion.
Supplementary Material
Acknowledgments
We are grateful for the excellent technical assistance provided by M. Kobayashi and M. Shimada in preparing the tissue sections.
This work was supported in part by a grant from the Core Research for Evolutional Science and Technology (CREST) program of the Japan Science and Technology Agency and by a grant from National Agriculture and Food Research Organization (NARO), Japan.
Footnotes
Published ahead of print on 20 May 2009.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Alexander, D. J., G. Parsons, and R. J. Manvell. 1986. Experimental assessment of the pathogenicity of eight avian influenza A viruses of H5 subtype for chickens, turkeys, ducks and quail. Avian Pathol. 15647-662. [DOI] [PubMed] [Google Scholar]
- 2.Amonsin, A., S. Payungporn, A. Theamboonlers, R. Thanawongnuwech, S. Suradhat, N. Pariyothorn, R. Tantilertcharoen, S. Damrongwantanapokin, C. Buranathai, A. Chaisingh, T. Songserm, and Y. Poovorawan. 2006. Genetic characterization of H5N1 influenza A viruses isolated from zoo tigers in Thailand. Virology 344480-491. [DOI] [PubMed] [Google Scholar]
- 3.Brown, C. C., H. J. Olander, and D. A. Senne. 1992. A pathogenesis study of highly pathogenic avian influenza virus H5N2 in chickens, using immunohistochemistry. J. Comp. Pathol. 107341-348. [DOI] [PubMed] [Google Scholar]
- 4.Chan, M. C., C. Y. Cheung, W. H. Chui, S. W. Tsao, J. M. Nicholls, Y. O. Chan, R. W. Chan, H. T. Long, L. L. Poon, Y. Guan, and J. S. Peiris. 2005. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir. Res. 6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, H., G. Deng, Z. Li, G. Tian, Y. Li, P. Jiao, L. Zhang, Z. Liu, R. G. Webster, and K. Yu. 2004. The evolution of H5N1 influenza viruses in ducks in southern China. Proc. Natl. Acad. Sci. USA 10110452-10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, H., G. J. Smith, S. Y. Zhang, K. Qin, J. Wang, K. S. Li, R. G. Webster, J. S. Peiris, and Y. Guan. 2005. Avian flu: H5N1 virus outbreak in migratory waterfowl. Nature 436191-192. [DOI] [PubMed] [Google Scholar]
- 7.de Jong, M. D., V. C. Bach, T. Q. Phan, M. H. Vo, T. T. Tran, B. H. Nguyen, M. Beld, T. P. Le, H. K. Truong, V. V. Nguyen, T. H. Tran, D. Q. Ha, and J. Farrar. 2005. Fatal avian influenza A (H5N1) in a child presenting with diarrhea followed by coma. N. Engl. J. Med. 352686-691. [DOI] [PubMed] [Google Scholar]
- 8.de Jong, M. D., C. P. Simmons, T. T. Thanh, V. M. Hien, G. J. Smith, T. N. Chau, D. M. Hoang, N. V. Chau, T. H. Khanh, V. C. Dong, P. T. Qui, B. V. Cam, D. Q. Ha, Y. Guan, J. S. Peiris, N. T. Chinh, T. T. Hien, and J. Farrar. 2006. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 121203-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dybing, J. K., S. Schultz-Cherry, D. E. Swayne, D. L. Suarez, and M. L. Perdue. 2000. Distinct pathogenesis of Hong Kong-origin H5N1 viruses in mice compared to that of other highly pathogenic H5 avian influenza viruses. J. Virol. 741443-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabriel, G., B. Dauber, T. Wolff, O. Planz, H. D. Klenk, and J. Stech. 2005. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc. Natl. Acad. Sci. USA 10218590-18595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Govorkova, E. A., J. E. Rehg, S. Krauss, H.-L. Yen, Y. Guan, M. Peiris, T. D. Nguyen, T. H. Hanh, P. Puthavathana, H. T. Long, C. Buranathai, W. Lim, R. G. Webster, and E. Hoffmann. 2005. Lethality to ferrets of H5N1 influenza viruses isolated from humans and poultry in 2004. J. Virol. 792191-2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan, Y., J. S. Peiris, A. S. Lipatov, T. M. Ellis, K. C. Dyrting, S. Krauss, L. J. Zhang, R. G. Webster, and K. F. Shortridge. 2002. Emergence of multiple genotypes of H5N1 avian influenza viruses in Hong Kong SAR. Proc. Natl. Acad. Sci. USA 998950-8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guan, Y., L. L. Poon, C. Y. Cheung, T. M. Ellis, W. Lim, A. S. Lipatov, K. H. Chan, K. M. Sturm-Ramirez, C. L. Cheung, Y. H. Leung, K. Y. Yuen, R. G. Webster, and J. S. Peiris. 2004. H5N1 influenza: a protean pandemic threat. Proc. Natl. Acad. Sci. USA 1018156-8161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatta, M., P. Gao, P. Halfmann, and Y. Kawaoka. 2001. Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 2931840-1842. [DOI] [PubMed] [Google Scholar]
- 15.Hooper, P. T., G. W. Russell, P. W. Selleck, and W. L. Stanislawek. 1995. Observations on the relationship in chickens between the virulence of some avian influenza viruses and their pathogenicity for various organs. Avian Dis. 39458-464. [PubMed] [Google Scholar]
- 16.Horimoto, T., and Y. Kawaoka. 1997. Biologic effects of introducing additional basic amino acid residues into the hemagglutinin cleavage site of a virulent avian influenza virus. Virus Res. 5035-40. [DOI] [PubMed] [Google Scholar]
- 17.Hulse, D. J., R. G. Webster, R. J. Russell, and D. R. Perez. 2004. Molecular determinants within the surface proteins involved in the pathogenicity of H5N1 influenza viruses in chickens. J. Virol. 789954-9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hulse-Post, D. J., J. Franks, K. Boyd, R. Salomon, E. Hoffmann, H. L. Yen, R. J. Webby, D. Walker, T. D. Nguyen, and R. G. Webster. 2007. Molecular changes in the polymerase genes (PA and PB1) associated with high pathogenicity of H5N1 influenza virus in mallard ducks. J. Virol. 818515-8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaiser, P., G. Underwood, and F. Davison. 2003. Differential cytokine responses following Marek's disease virus infection of chickens differing in resistance to Marek's disease. J. Virol. 77762-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobasa, D., S. M. Jones, K. Shinya, J. C. Kash, J. Copps, H. Ebihara, Y. Hatta, J. H. Kim, P. Halfmann, M. Hatta, F. Feldmann, J. B. Alimonti, L. Fernando, Y. Li, M. G. Katze, H. Feldmann, and Y. Kawaoka. 2007. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445319-323. [DOI] [PubMed] [Google Scholar]
- 21.Kuiken, T., G. Rimmelzwaan, D. van Riel, G. van Amerongen, M. Baars, R. Fouchier, and A. Osterhaus. 2004. Avian H5N1 influenza in cats. Science 306241. [DOI] [PubMed] [Google Scholar]
- 22.Li, K. S., Y. Guan, J. Wang, G. J. Smith, K. M. Xu, L. Duan, A. P. Rahardjo, P. Puthavathana, C. Buranathai, T. D. Nguyen, A. T. Estoepangestie, A. Chaisingh, P. Auewarakul, H. T. Long, N. T. Hanh, R. J. Webby, L. L. Poon, H. Chen, K. F. Shortridge, K. Y. Yuen, R. G. Webster, and J. S. Peiris. 2004. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature 430209-213. [DOI] [PubMed] [Google Scholar]
- 23.Li, Z., Y. Jiang, P. Jiao, A. Wang, F. Zhao, G. Tian, X. Wang, K. Yu, Z. Bu, and H. Chen. 2006. The NS1 gene contributes to the virulence of H5N1 avian influenza viruses. J. Virol. 8011115-11123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu, J., H. Xiao, F. Lei, Q. Zhu, K. Qin, X. W. Zhang, X. L. Zhang, D. Zhao, G. Wang, Y. Feng, J. Ma, W. Liu, J. Wang, and G. F. Gao. 2005. Highly pathogenic H5N1 influenza virus infection in migratory birds. Science 3091206. [DOI] [PubMed] [Google Scholar]
- 25.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25402-408. [DOI] [PubMed] [Google Scholar]
- 26.Maines, T. R., X. H. Lu, S. M. Erb, L. Edwards, J. Guarner, P. W. Greer, D. C. Nguyen, K. J. Szretter, L.-M. Chen, P. Thawatsupha, M. Chittaganpitch, S. Waicharoen, D. T. Nguyen, T. Nguyen, H. H. T. Nguyen, J.-H. Kim, L. T. Hoang, C. Kang, L. S. Phuong, W. Lim, S. Zaki, R. O. Donis, N. J. Cox, J. M. Katz, and T. M. Tumpey. 2005. Avian influenza (H5N1) viruses isolated from humans in Asia in 2004 exhibit increased virulence in mammals. J. Virol. 7911788-11800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mase, M., M. Eto, N. Tanimura, K. Imai, K. Tsukamoto, T. Horimoto, Y. Kawaoka, and S. Yamaguchi. 2005. Isolation of a genotypically unique H5N1 influenza virus from duck meat imported into Japan from China. Virology 339101-109. [DOI] [PubMed] [Google Scholar]
- 28.Mase, M., T. Imada, K. Nakamura, N. Tanimura, K. Imai, K. Tsukamoto, and S. Yamaguchi. 2005. Experimental assessment of the pathogenicity of H5N1 influenza A viruses isolated in Japan. Avian Dis. 49582-584. [DOI] [PubMed] [Google Scholar]
- 29.Mase, M., N. Tanimura, T. Imada, M. Okamatsu, K. Tsukamoto, and S. Yamaguchi. 2006. Recent H5N1 avian influenza A virus increases rapidly in virulence to mice after a single passage in mice. J. Gen. Virol. 873655-3659. [DOI] [PubMed] [Google Scholar]
- 30.Mase, M., K. Tsukamoto, T. Imada, K. Imai, N. Tanimura, K. Nakamura, Y. Yamamoto, T. Hitomi, T. Kira, T. Nakai, M. Kiso, T. Horimoto, Y. Kawaoka, and S. Yamaguchi. 2005. Characterization of H5N1 influenza A viruses isolated during the 2003-2004 influenza outbreaks in Japan. Virology 332167-176. [DOI] [PubMed] [Google Scholar]
- 31.Mo, I. P., M. Brugh, O. J. Fletcher, G. N. Rowland, and D. E. Swayne. 1997. Comparative pathology of chickens experimentally inoculated with avian influenza viruses of low and high pathogenicity. Avian Dis. 41125-136. [PubMed] [Google Scholar]
- 32.Muramoto, Y., H. Ozaki, A. Takada, C. H. Park, Y. Sunden, T. Umemura, Y. Kawaoka, H. Matsuda, and H. Kida. 2006. Highly pathogenic H5N1 influenza virus causes coagulopathy in chickens. Microbiol. Immunol. 5073-81. [DOI] [PubMed] [Google Scholar]
- 33.Nakamura, K., T. Imada, K. Imai, Y. Yamamoto, N. Tanimura, M. Yamada, M. Mase, K. Tsukamoto, and S. Yamaguchi. 2008. Pathology of specific-pathogen-free chickens inoculated with H5N1 avian influenza viruses isolated in Japan in 2004. Avian Dis. 528-13. [DOI] [PubMed] [Google Scholar]
- 34.OIE. March 2009. Avian influenza. Terrestrial animal health code. OIE—World Organisation for Animal Health, Paris, France. http://www.oie.int/eng/normes/mcode/en_chapitre_2.7.12.htm.
- 35.Okamatsu, M., T. Saito, Y. Yamamoto, M. Mase, S. Tsuduku, K. Nakamura, K. Tsukamoto, and S. Yamaguchi. 2007. Low pathogenicity H5N2 avian influenza outbreak in Japan during the 2005-2006. Vet. Microbiol. 12435-46. [DOI] [PubMed] [Google Scholar]
- 36.Peiris, J. S., W. C. Yu, C. W. Leung, C. Y. Cheung, W. F. Ng, J. M. Nicholls, T. K. Ng, K. H. Chan, S. T. Lai, W. L. Lim, K. Y. Yuen, and Y. Guan. 2004. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet 363617-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perkins, L. E., and D. E. Swayne. 2003. Comparative susceptibility of selected avian and mammalian species to a Hong Kong-origin H5N1 high-pathogenicity avian influenza virus. Avian Dis. 47956-967. [DOI] [PubMed] [Google Scholar]
- 38.Peters, M. A., G. F. Browning, E. A. Washington, B. S. Crabb, and P. Kaiser. 2003. Embryonic age influences the capacity for cytokine induction in chicken thymocytes. Immunology 110358-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reed, L., and H. Muench. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27493-497. [Google Scholar]
- 40.Rimmelzwaan, G. F., T. Kuiken, G. van Amerongen, T. M. Bestebroer, R. A. Fouchier, and A. D. Osterhaus. 2001. Pathogenesis of influenza A (H5N1) virus infection in a primate model. J. Virol. 756687-6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rothwell, L., J. R. Young, R. Zoorob, C. A. Whittaker, P. Hesketh, A. Archer, A. L. Smith, and P. Kaiser. 2004. Cloning and characterization of chicken IL-10 and its role in the immune response to Eimeria maxima. J. Immunol. 1732675-2682. [DOI] [PubMed] [Google Scholar]
- 42.Salomon, R., E. Hoffmann, and R. G. Webster. 2007. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc. Natl. Acad. Sci. USA 10412479-12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seo, S. H., E. Hoffmann, and R. G. Webster. 2002. Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat. Med. 8950-954. [DOI] [PubMed] [Google Scholar]
- 44.Shinya, K., M. Hatta, S. Yamada, A. Takada, S. Watanabe, P. Halfmann, T. Horimoto, G. Neumann, J. H. Kim, W. Lim, Y. Guan, M. Peiris, M. Kiso, T. Suzuki, Y. Suzuki, and Y. Kawaoka. 2005. Characterization of a human H5N1 influenza A virus isolated in 2003. J. Virol. 799926-9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Solórzano, A., R. J. Webby, K. M. Lager, B. H. Janke, A. Garcia-Sastre, and J. A. Richt. 2005. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J. Virol. 797535-7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sturm-Ramirez, K. M., T. Ellis, B. Bousfield, L. Bissett, K. Dyrting, J. E. Rehg, L. Poon, Y. Guan, M. Peiris, and R. G. Webster. 2004. Reemerging H5N1 influenza viruses in Hong Kong in 2002 are highly pathogenic to ducks. J. Virol. 784892-4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sturm-Ramirez, K. M., D. J. Hulse-Post, E. A. Govorkova, J. Humberd, P. Seiler, P. Puthavathana, C. Buranathai, T. D. Nguyen, A. Chaisingh, H. T. Long, T. S. P. Naipospos, H. Chen, T. M. Ellis, Y. Guan, J. S. M. Peiris, and R. G. Webster. 2005. Are ducks contributing to the endemicity of highly pathogenic H5N1 influenza virus in Asia? J. Virol. 7911269-11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suarez, D. L., M. L. Perdue, N. Cox, T. Rowe, C. Bender, J. Huang, and D. E. Swayne. 1998. Comparisons of highly virulent H5N1 influenza A viruses isolated from humans and chickens from Hong Kong. J. Virol. 726678-6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subbarao, K., A. Klimov, J. Katz, H. Regnery, W. Lim, H. Hall, M. Perdue, D. Swayne, C. Bender, J. Huang, M. Hemphill, T. Rowe, M. Shaw, X. Xu, K. Fukuda, and N. Cox. 1998. Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science 279393-396. [DOI] [PubMed] [Google Scholar]
- 50.Swayne, D. E. 1997. Pathobiology of H5N2 Mexican avian influenza virus infections of chickens. Vet. Pathol. 34557-567. [DOI] [PubMed] [Google Scholar]
- 51.Swayne, D. E. 2007. Understanding the complex pathobiology of high pathogenicity avian influenza viruses in birds. Avian Dis. 51242-249. [DOI] [PubMed] [Google Scholar]
- 52.Swayne, D. E., and D. A. Halvorson. 2003. Influenza, p. 135-160. In Y. M. Saif, H. J. Barnes, J. R. Glisson, A. M. Fadly, L. R. McDougald, and D. E. Swayne (ed.), Diseases of poultry, 11th ed. Iowa State University Press, Ames, IA.
- 53.Szretter, K. J., S. Gangappa, X. Lu, C. Smith, W.-J. Shieh, S. R. Zaki, S. Sambhara, T. M. Tumpey, and J. M. Katz. 2007. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J. Virol. 812736-2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanimura, N., K. Tsukamoto, M. Okamatsu, M. Mase, T. Imada, K. Nakamura, M. Kubo, S. Yamaguchi, W. Irishio, M. Hayashi, T. Nakai, A. Yamauchi, M. Nishimura, and K. Imai. 2006. Pathology of fatal highly pathogenic H5N1 avian influenza virus infection in large-billed crows (Corvus macrorhynchos) during the 2004 outbreak in Japan. Vet. Pathol. 43500-509. [DOI] [PubMed] [Google Scholar]
- 55.Tran, T. H., T. L. Nguyen, T. D. Nguyen, T. S. Luong, P. M. Pham, V. C. Nguyen, T. S. Pham, C. D. Vo, T. Q. Le, T. T. Ngo, B. K. Dao, P. P. Le, T. T. Nguyen, T. L. Hoang, V. T. Cao, T. G. Le, D. T. Nguyen, H. N. Le, K. T. Nguyen, H. S. Le, V. T. Le, D. Christiane, T. T. Tran, J. de Menno, C. Schultsz, P. Cheng, W. Lim, P. Horby, and J. Farrar. 2004. Avian influenza A (H5N1) in 10 patients in Vietnam. N. Engl. J. Med. 3501179-1188. [DOI] [PubMed] [Google Scholar]
- 56.Tumpey, T. M., X. Lu, T. Morken, S. R. Zaki, and J. M. Katz. 2000. Depletion of lymphocytes and diminished cytokine production in mice infected with a highly virulent influenza A (H5N1) virus isolated from humans. J. Virol. 746105-6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uys, C. J., and W. B. Becker. 1967. Experimental infection of chickens with influenza A-Tern/South Africa/1961 and Chicken/Scotland/1959 viruses. II. Pathology. J. Comp. Pathol 77167-173. [DOI] [PubMed] [Google Scholar]
- 58.Wasilenko, J. L., C. W. Lee, L. Sarmento, E. Spackman, D. R. Kapczynski, D. L. Suarez, and M. J. Pantin-Jackwood. 2008. NP, PB1, and PB2 viral genes contribute to altered replication of H5N1 avian influenza viruses in chickens. J. Virol. 824544-4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Withanage, G. S. K., P. Wigley, P. Kaiser, P. Mastroeni, H. Brooks, C. Powers, R. Beal, P. Barrow, D. Maskell, and I. McConnell. 2005. Cytokine and chemokine responses associated with clearance of a primary Salmonella enterica serovar Typhimurium infection in the chicken and in protective immunity to rechallenge. Infect. Immun. 735173-5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.