

Abstract
Neutrophil elastase (NE) degrades basal lamina and extracellular matrix molecules, and recruits leukocytes during inflammation; however, a basic understanding of the role of NE in stroke pathology is lacking. We measured an increased number of extravascular NE-positive cells, as well as increased levels of tissue elastase protein and activity, following transient middle cerebral artery occlusion (tMCAo). Both pharmacologic inhibition of NE with ZN200355 (ZN), and genetic deletion of NE, significantly reduced infarct volume, blood-brain barrier disruption, vasogenic edema, and leukocyte-endothelial adherence 24 h after tMCAo. ZN also reduced infarct volume in MMP9-null mice following tMCAo. There were, however, no reductions in infarct volume or vasogenic edema in NE-null mice in two models of permanent middle cerebral artery occlusion. Our findings confirm the involvement of NE in neurovascular stroke pathology, when reperfusion allows neutrophils access to vulnerable brain, with pharmacologic or genetic inhibition of NE being both neuro- and vasculo-protective in this setting.
Keywords: protease, blood-brain barrier, ischemia, transient middle cerebral artery occlusion, vasogenic edema, leukocyte-endothelial adherence
Leukocytes play a critical role in post-ischemic inflammation and secondary neurovascular injury in most tissues, including brain (for review, see: (Man et al. 2007; Wang et al. 2007). Mechanistically, this results from oxidative injury secondary to the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-associated respiratory burst and/or proteolytic injury from the release of destructive enzymes. Both of these events can occur independent of leukocyte transmigration across endothelium into parenchyma (Shapiro 2002). With respect to the protease pathway, the 33-kDa serine proteinase elastase (E.C. 3.4.21.37) is stored in a biologically active form within primary neutrophil granules. Neutrophil elastase (NE) is important in host defense (Belaaouaj et al. 1998), and degranulation stimuli during inflammation can include the paracrine action of platelet activating factor and interleukin-8 released from endothelial cells (Henriksen et al. 2008). NE degrades structural matrix proteins (e.g. elastin, collagens, laminins, and fibronectin), resulting in increased vascular permeability and hemorrhage in non-cerebral tissues (Ishikawa et al. 2003; Houtz et al. 2004).
In conjunction with membrane degradation, NE also regulates leukocyte adhesion and diapedesis/transmigration in peripheral vascular beds in response to pro-inflammatory stimuli (Woodman et al. 1993; Young et al. 2004). NE-mediated degradation of endothelial junction proteins, in addition to the aforementioned proteolysis of matrix proteins, “clears a path” for the transmigrating leukocytes during diapedesis (Hermant et al. 2003; Ionescu et al. 2003). In brain, exogenous delivery of NE, following either intracarotid infusion (Nagy et al. 1998) or intracerebral injection (Armao et al. 1997), increases blood-brain barrier (BBB) permeability, suggesting that BBB tight junctions, basal lamina, and perhaps even cortical endothelial cells are vulnerable to NE-mediated proteolysis during pathological conditions associated with cortical inflammation.
Studies of non-CNS tissues using pharmacologic inhibitors of NE highlight the specific role played by NE in ischemia-reperfusion pathology. NE inhibition reduces leukocyte adherence and extravasation in intestinal and myocardial ischemia (Zimmerman et al. 1990; Tiefenbacher et al. 1997). Post-ischemic vasogenic edema in the rabbit lung (Kishima et al. 1998), as well as in rat hindlimb (Welbourn et al. 1991) and liver (Okajima et al. 2004), can be attenuated by NE inhibitors. Few studies, however, have investigated endogenous NE in stroke, despite evidence of significant neutrophil accumulation following cerebral ischemia (Wang et al. 2007; Gelderblom et al. 2009). One study by Shimakura and colleagues showed that the continuous infusion of an NE inhibitor, ONO-5046, reduced post-ischemic vasogenic edema and infarct volume 24 h following permanent middle cerebral artery occlusion in rats (Shimakura et al. 2000). ONO-5046 has also been shown to protect hippocampal neurons following forebrain ischemia (Matayoshi et al. 2009). While these findings are consistent with NE mediating post-ischemic neurovascular dysfunction, more specific studies on leukocyte dynamics and BBB disruption, particularly in transient focal ischemia models, using both pharmacologic and genetic approaches, are needed to begin to systematically assess the deleterious effects of NE in the ischemic brain.
The present investigation was undertaken, in mice, to identify the potential post-stroke pathophysiologic roles of neutrophil-derived elastase. We hypothesized that vascular and parenchymal exposure to NE would contribute to infarct severity and vasogenic edema, secondary to promoting leukocyte adherence and attendant BBB disruption, and that NE-mediated pathology would only occur in areas of transient ischemia, where reperfusion allows neutrophils access to vulnerable regions. By cross-validating genetic and pharmacologic approaches, we obtained causal data confirming the involvement of NE in neurovascular pathology following cerebral ischemia/reperfusion.
Materials and Methods
Transient and permanent focal cerebral ischemia
The Animal Studies Committee at Washington University School of Medicine approved all experimental methods and animal care procedures in accordance with NIH guidelines. Occlusion of the middle cerebral artery was performed in adult male Swiss-Webster/ND4 (SW/ND4), 129SvEv (wild-type; WT), and NE−/− mice or MMP9 −/− mice on a 129SvEv background, at 10–16 weeks of age, as described previously (Gidday et al. 2005). Pilot studies were conducted in SW/ND4 and WT mice to establish transient middle cerebral artery occlusion (tMCAo) durations that led to roughly similar, moderate-sized infarcts and that maximized 24-h animal viability for subsequent analysis of leukocyte adherence and BBB integrity. Monitoring of relative cerebral blood flow (CBF) changes during and after ischemia in the final experimental groups revealed no potential strain-dependent effects (Majid et al. 2000). Matched protocols were then conducted in a blinded, randomized fashion in drug-treated or mutant mice of the same strain. During all surgical procedures, core body temperatures in mice were monitored by a rectal probe and maintained at 37°C using a thermo-regulated heating pad.
For tMCAo, animals were anesthetized with chloral hydrate (350 mg/kg) and xylazine (4 mg/kg; 0.001 ml/g i.p.), and a blunted 6.0-gauge nylon suture was threaded 9.0–10.5 mm through the left common carotid to the origin of the middle cerebral artery (MCA; (Miller et al. 2001). Interruption of blood flow to the MCA was confirmed using transcranial laser Doppler flowmetry (TSI, Inc.). The MCA was considered successfully occluded with a >80% drop in blood flow versus baseline, and successfully reperfused with a >50% blood flow return versus baseline at 10 min of reperfusion. Following suture withdrawal, mice were kept in a warmed incubator until recovered from anesthesia and fully ambulatory. A sham tMCAo group, in which the animals were anesthetized, and the intraluminal suture was placed anterogradely in the carotid but not advanced to the ostium of the MCA, was also studied. All mice subjected to tMCAo underwent serial neurobehavioral examinations performed in blinded fashion at the end of ischemia and at 24 h after reperfusion (Table 2). Each animal was assigned a neurological deficit score of 0–4, with 0 being no observable deficits and 4 being an inability to walk spontaneously (Gidday et al. 2005).
Table 2.
Relative cortical blood flow and neurological deficit scores for all tMCAo groups during and after ischemia
| ANALYSIS | (n) | duration of tMCAo (min) | CBF after 15 min of Ischemia* (% baseline) | CBF after 15 min of reperfusion† (% baseline) | neurological deficit score at end of Ischemia | neurological deficit score after 15 min of reperfusion | neurological deficit score after 24 h of reperfusion |
|---|---|---|---|---|---|---|---|
| Vasogenic edema | |||||||
| SW + vehicle | 7 | 90 | 9±2 | 78±7 | 1.6±0.1 | 1.7±0.1 | 1.6±0.1 |
| SW + ZN | 8 | 90 | 9±1 | 82±4 | 1.7±0.2 | 1.5±0.1 | 1.1±0.1 |
| WT | 11 | 90 | 10±1 | 92±3 | 1.1±0.1 | 1.1±0.1 | 1.6±0.2 |
| NE−/− | 11 | 90 | 11±2 | 91±3 | 1.6±0.2 | 1.9±0.2 | 1.9±0.2 |
| BBB breakdown | |||||||
| SW + vehicle | 9 | 90 | 8±0 | 78±7 | 1.7±0.1 | 1.8±0.1 | 1.8±0.2 |
| SW + ZN | 13 | 90 | 7±0 | 83±5 | 2.0±0.1 | 1.8±02 | 1.6±0.3 |
| WT | 9 | 90 | 9±1 | 8l±5 | 1.9±0.2 | 1.9±0.3 | 2.2±0.3 |
| NE−/− | 10 | 90 | 10±2 | 87±6 | 1.4±0.2 | 1.4±0.2 | 1.0±0.1 |
| Leukocyte adherence | |||||||
| SW + vehicle (0, 4 h) | 10 | 60 | 5±1 | 93±6 | 2.1±0.2 | 2.1±0.1 | 1.6±0.2 |
| SW + ZN (0, 4 h) | 13 | 60 | 6±0 | 86±6 | 2.0±0.1 | 1.8±0.1 | 1.5±0.1 |
| SW + vehicle (0, 4, 22 h) | 6 | 60 | 6±1 | 103±7 | n/a | 1.8±0.2 | 1.6±0.3 |
| SW + ZN (0, 4, 22 h) | 6 | 60 | 5±0 | 92±6 | n/a | 2.0±0.3 | 1.6±0.2 |
| WT | 16 | 60 | 8±0 | 87±8 | 1.9±0.1 | 1.8±0.1 | 1.3±0.1 |
| NE−/− | 14 | 60 | 7±0 | 80±5 | 1.7±0.2 | 1.3±0.1 | 1.1±0.1 |
| Infarct volume | |||||||
| SW + vehicle | 8 | 90 | 7±2 | 78±5 | 1.6±0.2 | 1..2±0.1 | 1.0±0.0 |
| SW + ZN | 8 | 90 | 9±2 | 85±3 | 1.5±0.2 | 1.4±0.2 | 1.2±0.2 |
| WT | 11 | 120 | 9±1 | 90±7 | 2.0±0.1 | 2.0±0.2 | 2.2±0.3 |
| NE−/− | 11 | 120 | 13±1 | 86±4 | 1.6±0.2 | 1.5±0.2 | 1.4±0.2 |
| NE−/− + vehicle | 7 | 120 | 13±1 | 85±5 | 1.6±0.3 | 1.6±0.2 | 1.7±0.4 |
| NE−/− + ZN | 8 | 120 | 10±1 | 92±3§ | 1.7±0.3 | 1.7±0.3 | 1.5±0.6 |
| WT (MMP9) | 12 | 90 | 6±1 | 92±4 | 2.2±0.2 | 2.2±0.2 | 1.9±0.2 |
| MMP9−/− + vehicle | 14 | 90 | 6±1 | 76±3 | 2.1±0.1 | 1.9±0.2 | 1.8±0.1 |
| MMP9−/− + ZN | 10 | 90 | 8±1 | 84±4 | 1.9±0.0 | 1.5±0.1 | 1.2±0.2 |
Values are mean (SEM); n is animal number; SW Swiss Webster, ZN ZN200355; WT; wild type. NE, Neutrophil elastase. n/a, not available
p<0.05 vs. baseline CBF for all groups
p<0.05 vs. baseline CBF for all groups except [NE−/− + ZN] as denoted by §
Permanent middle cerebral artery occlusion (pMCAo) was performed in two different ways in 129 SvEv WT and matched NE−/− mice. In one group, surgical procedures were identical to the tMCAo model, but after the suture was placed at the origin of the MCA, it was secured tightly in place and remained blocking the ostium of the MCA for the 24-h duration of the experiment. In another group of WT and NE−/− mice, a more distal occlusion of the MCA was performed via a craniotomy, with electrocoagulation of the surface presentation of the MCA as described previously (Majid et al. 2000; Miller et al. 2001). As with tMCAO, consequent reductions of >80% of distal baseline cortical blood flow were confirmed using Doppler flowmetry immediately following occlusion, and animals were excluded from the pMCAo group if they did not meet this criterion. Post-operative recovery procedures in these mice were the same as indicated above.
For experiments assessing the effect of pharmacologic inhibition of NE in SW/ND4 and NE−/− mice, the selective, peptidic, trifluoromethyl ketone-based NE inhibitor ZN200355 (ZN; 10 mg/kg [(Mehta et al. 1994)]; AstraZeneca) was dissolved in physiologic saline for intraperitoneal (ip) injection. We chose to administer 10 mg/kg of ZN at both 0 h and 4 h of reperfusion, and additionally at 22 h of reperfusion in some of the leukocyte-endothelial adherence studies, based on previously published pharmacokinetics (Williams et al. 1991; Mehta et al. 1994; Tiefenbacher et al. 1997).
Elastin-Congo red degradation assay
SW/ND4 mice underwent a 90-min tMCAo and were sacrificed 12, 24, or 48 h following reperfusion. Animals were sacrificed via cervical dislocation and the brain was immediately removed and flash-frozen. Tissue samples of the ipsilateral hemisphere were homogenized in extraction buffer (50mM Tris, pH 7.6, 150 mM NaCl, 5 mM CaCl2, and 0.05% Brij-35; Zhang et al. 1997). Total protein concentration was determined using a DC protein assay kit (BioRad), as per manufacturer’s instructions. Equal amounts of protein were incubated with 1 ml (10 mg/ml) Congo Red-conjugated insoluble elastin (Elastin Products Inc) at 37 °C while rotating for 16 h. The solution was centrifuged to pellet any undigested, insoluble elastin, and the absorbance of cleaved elastin-Congo red by elastolytic activity was measured at 490 nm. Various concentrations of purified NE (Elastin Products Inc; 0–10 μg) were used in parallel to determine the concentration of elastolytic activity in the tissue samples.
Immunoblot analysis for NE
Based on protein concentration, equal amounts of protein from whole-brain cortical homogenates were separated under reducing conditions on a 15% SDS polyacrylamide gel and transferred onto Immobilon-P PVDF transfer membranes (Millipore Corp). After blocking overnight (5% nonfat dried milk, TBS, 0.1% Tween 20) at 4 °C, the membranes were incubated for 2 h with anti-NE antibody (1:6000; courtesy of Barry Starcher, University of Texas Health Sciences Center at Tyler), followed by 1 h of incubation with horseradish peroxidase-conjugated donkey anti-rabbit secondary antibody (1:25000; Jackson ImmunoResearch). After washing, immunoreactive proteins were detected by chemiluminescence using the ECL Plus Western Blotting Detection System (Amersham Pharmacia) and subsequent autoradiography.
Immunohistochemistry and confocal microscopy
Either under naïve conditions or 24 h following tMCAo, animals were transcardiac perfused (20 ml PBS followed by 40 ml 4% paraformaldehyde), and brains were removed, cryoprotected, and sectioned on a cryostat at a 10-μm thickness. At time of histology (McCandless et al. 2008), sections were brought to room temperature, rinsed in dH2O, hydrated in 10 mM phosphate-buffered saline (PBS), and blocked (10% goat serum, 0.1% Triton, 10 mM PBS). Sections were washed and exposed to primary antibody (rat anti-mouse CD31, 1:50, BD Biosciences; rabbit anti-mouse NE, 1:100, courtesy of Dr. Barry Starcher, University of Texas Health Center at Tyler) overnight. The following day, sections were washed (3 X 15min, 0.2% FSG, 10 mM PBS), blocked, and probed with secondary antibody (Alexa 488 goat anti-rat 1:300 and Alexa 568 goat anti-rabbit, 1:300, Molecular Probes) for 1 h. Sections were rinsed, stained for 5 min with ToPro3, and coverslipped with Immu-mount (Thermo Scientific). All images were obtained using a 60X objective on a confocal laser-scanning microscope via standard imaging software. Images were localized to the cortical aspect of the peri-infarct region corresponding to the MCA territory, with control images taken from matched cortical areas in naïve animals.
Quantification of vasogenic edema and blood-brain barrier integrity
For vasogenic edema, animals were sacrificed 24 h after tMCAo or pMCAO by halothane overdose, then transcardially perfused with 24 ml physiological saline (3 ml/min) containing 10 U/ml heparin (Gidday et al. 2005). Vasogenic edema occurring over the initial 24 h of reperfusion was assessed as described in previously-published methods (Tureyen et al. 2008). Briefly, the cerebellum and brainstem were removed and each hemisphere weighed to determine the “wet” weight. The brain was dehydrated in an oven at 105 °C for 24 h, and weighed after a 30 min cooling period to obtain the “dry” weight. Water content was determined for each hemisphere and presented as a percent change of ipsilateral hemisphere relative to the contralateral hemisphere. In another group of animals, hemispheric Evans blue extravasation was quantified to determine the extent of BBB disruption (Gidday et al. 2005). Evans blue was administered through retro-orbital injection (2.5 mL/kg; 0.9% NaCl; 100 μl/animal) 4 h prior to sacrifice. Following transcardiac perfusion, both hemispheres were removed, homogenized in 300 μl N, N-dimethylformaline (Sigma-Aldrich), and incubated for 18 h at 50°C. Standard spectrophotometer techniques quantified the final Evans blue concentration (μg/hemisphere) and results are presented as percent change increase in the ipsilateral hemisphere relative to the contralateral hemisphere.
Epifluorescence videomicroscopy for leukocyte-endothelial adherence
As described in detail previously (Gidday et al. 2005; Altay et al. 2007), 24 h following tMCAo, animals were anesthetized with chloral hydrate (350 mg/kg) and xylazine (4 mg/kg; 0.001 ml/g ip), tracheostomized, and mechanically ventilated (5 ml/kg; 100 breaths/min; Harvard Apparatus, Model 845) prior to femoral artery cannulation. During the procedure, core body temperatures in mice were monitored and maintained at 37°C using a thermo-regulated heating pad. A craniotomy ipsilateral to the occluded MCA exposed an area with heterogenous patterning of secondary and tertiary venules. An epifluorescence microscope and a charge-coupled device camera (Olympus) captured and recorded intravascular (rhodamine 6-G intra-arterial; 0.007% in PBS; 0.25–0.5 ml; 150 μl/min) leukocyte dynamics. Manual counting of leukocytes sticking to pial venules for >10 consecutive seconds was conducted in a blinded fashion over six distinct user-defined microvessel networks that contained secondary and tertiary venules (20–60 μm diameter), and is presented as the mean number of leukocytes per square millimeter of total endothelial vessel surface (Ishikawa et al. 2004; Gidday et al. 2005; Altay et al. 2007). Arterial blood samples were obtained for pH and gas analyses following the adherence measurements in all experimental groups in which this endpoint was quantified (Table 1).
Table 1.
Blood gas and blood pressure values for different groups 24 h after tMCAo
| (n) | pH | PaCO2 (mmHg) | PaO2 (mmHg) | MABP (mmHg) | |
|---|---|---|---|---|---|
| Sham surgery | |||||
| SW | 9 | 7.31 ±0.01 | 33±2 | 123±10 | 79±7 |
| WT | 10 | 7.28±0.02 | 34±2 | 126±11 | 80±6 |
| tMCAo | |||||
| SW + vehicle (0, 4 h) | 10 | 7.30±0.03 | 39±3 | 128±16 | 86±6 |
| SW + ZN (0,4 h) | 14 | 7.32±0.02 | 30±2† | 107±6 | 87±4 |
| SW + vehicle (0, 4, 22 h) | 6 | n/a | 52±8* | 92±6* | 60±10 |
| SW + ZN (0, 4, 22 h) | 6 | n/a | 57±8* | 81±9* | 69±6 |
| WT | 16 | 7.32±0.03 | 30±2 | 116±6 | 96±2* |
| NE−/− | 14 | 7.27±0.03 | 30±2 | 97±5 | 84±3† |
Values are mean (SEM); PaCO2, arterial PCO2; PaO2 arterial PO2; MABP, mean arterial blood pressure; n/a, not available
p<0.05 vs. respective sham
p<0.05 vs. vehicle or WT
Infarct quantification
Animals were sacrificed 24 h after tMCAo or pMCAo and transcardiac perfused as stated above. Upon dissection and gross examination, animals with subarachnoid hemorrhaging at the Circle of Willis, secondary to suture placement, were excluded from the study. The frequency of complications due to suture placement occurred with similar frequency in all experimental and control groups. Brains were removed and sectioned into 1.0-mm thick coronal sections on a brain matrix. 2,3,5-triphenyl tetrazolium chloride (TTC) stain delineated infarct regions, which were quantified by a blinded observer using image analysis software. Infarct volume data were corrected for edema, based on the cross-sectional area of the right hemisphere as control, following standard protocols (Gidday et al. 2005).
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Statistical comparisons were evaluated using Mann-Whitney rank sum test for barrier indices, leukocyte adherence, and infarct volumes, Chi-squared tests for neurological deficit scores, and one-way ANOVA with Tukey post-hoc for physiologic parameters and elastin-Congo red assay. Significance was determined as p<0.05.
Results
Physiologic and hemodynamic parameters
Table 1 depicts representative physiologic data, by experimental group, for the animals in which leukocyte adherence was measured. Transcranial Doppler flowmetry established CBF prior to, and immediately following, tMCAo suture placement to confirm the MCA occlusion. As shown in Table 2, suture placement significantly lowered CBF during ischemia relative to baseline CBF within each experimental group, and CBF at 15 min of reperfusion remained significantly lower than baseline (p<0.05) in all groups but one (NE−/− mice with ZN treatment). There was no significant inter-group difference between the magnitude of CBF changes during ischemia or after reperfusion for all end points presented in this study, despite inter-group differences in both ischemic duration and strain. In addition, the neuromotor recovery of all animals was evaluated at the end of ischemia and at 24 h of reperfusion (Table 2). There was a significant reduction in neurological deficit score (NDS) in NE−/− animals vs. WT, as shown in Figure 1, both at the end of ischemia and at 24 h of reperfusion. In particular, at the end of ischemia, mean scores were elevated in WT mice (2.0±0.1; n=11) over NE−/− mice (1.6±0.2; n=11; p<0.05). These WT mice continued to exhibit elevated neurological deficits 24 h after reperfusion (WT 2.2±0.3 vs. NE−/− 1.4±0.2; p<0.05).
Figure 1.

Neutrophil elastase negatively influences neurobehavioral recovery following transient middle cerebral artery occlusion (tMCAo). Individual neurological deficit scores (denoted by each circle) at both the end of tMCAo, prior to the onset of reperfusion, and also at 24 h following reperfusion, were significantly reduced in NE−/− mice vs. wild-type controls. Bars represent the median value for each group. *p<0.05 vs. wild-type.
Stroke increases tissue neutrophil elastase activity and expression
Elastase enzymatic activity in the ipsilateral hemisphere was evaluated in naïve animals, and at 12, 24, and 48 h following 90-min of tMCAo in SW/ND4 mice. The concentration of enzyme activity, based on the optical density of dissolved elastin, was 3.8±0.2 μg/ml in naïve animals (n=5). Figure 2A shows that elastase activity increased by 70% at 12 h of reperfusion to 6.5±0.3 μg/ml (n=5; p<0.0001), and by 48% at 24 h of reperfusion to 5.7±0.3 μg/ml (n=5; p<0.01), with a return to near naïve expression levels at 48 h (4.4±0.4 μg/ml; n=4). Immunoblot analysis of the tissue homogenates, using an antibody directed against neutrophil elastase (MW = 33 kDa), which does not cross-react with the proform of macrophage-derived elastase (MW = 55 kDa) or its metabolically active forms (MW = 45 and 22 kDa), revealed increases in NE protein at 12 and 24 h of reperfusion, which returned to non-ischemic levels at 48 h of reperfusion (Fig. 2B). These findings confirmed that the aforementioned increase in elastase activity could not solely be the consequence of elastase released from macrophages. Moreover, these data corresponded with an increased presence of NE-immunopositive cells in the peri-infarct region at 24 h following tMCAo, as detected by immunohistochemistry (Figure 3). The majority of these NE-positive cells were clearly localized outside of the peri-vascular space, indicating that substantial neutrophil diapedesis into brain parenchyma occurred within the initial 24 h of reperfusion. Very few NE-positive cells were detected in matched cortical areas of non-ischemic naïve animals.
Figure 2.

Elastase activity following transient middle cerebral artery occlusion (tMCAo). (A) Congo red-conjugated elastin assay was used to determine elastase activity in the ipsilateral hemisphere following a 90-min tMCAo. Elastase activity significantly increased digestion of elastin (μg/ml) at 12 and 24 h following reperfusion. Values shown are mean±SEM. *p<0.05 vs. naïve levels of elastase activity in non-ischemic, naïve mice. (B) Immunoblot analysis of whole-brain homogenates using an NE-specific antibody identifies NE protein (33 kDa) in pairs of samples from naïve animals and mice subjected to tMCAO followed by 12, 24, or 48 h of reperfusion. Image is representative of five mice per condition examined.
Figure 3.

NE-immunoreactive cells in neocortex of non-ischemic, naïve animals and in animals 24 h following transient middle cerebral artery occlusion (tMCAo). Ten micron-thick cortical sections were immunostained for (A, E) endothelial cells (CD31, green), (B, F) neutrophil elastase (NE, red), and (C, G) general nuclei (ToPro3, blue). Merged photomicrographs shows a paucity of NE stain in (D) naïve animals, with a sharp increase in NE-containing cells (H) 24 h following reperfusion. In panel H: arrows denote representative extravascular cells immunoreactive for NE; arrowhead denotes an NE-expressing cell in the peri-vascular space; asterisk denotes a cell with NE in the surrounding tissue. Scale bar = 40μm.
NE contributes to infarct volume following transient MCAo
Post-ischemic administration of the NE inhibitor ZN, at 0 and 4 h of reperfusion, significantly decreased infarct volume by 56% from 61±9 mm3 in vehicle-treated mice (n=8) to 27±3 mm3 in ZN-treated mice (n=8; p<0.01) following tMCAo (Fig. 4A). Assessments of the effect of NE gene deletion on lesion severity following stroke revealed a significant, 37% reduction in infarct volume in NE−/− mice (44±5 mm3; n=11) relative to WT mice (70±7 mm3; n=11; p<0.01) following tMCAo (Fig. 4B). The infarct-sparing phenotype associated with NE gene deletion was also accompanied by significant reductions in ischemic and post-ischemic neurological deficit scores (Table 2 and Figure 1). Of note, when NE−/− mice were administered ZN, no additional reduction in infarct volume was measured (45±8 mm3; n=8) relative to that measured in vehicle-treated NE−/− mice (48±4 mm3; n=7), indicative of the lack of nonspecific protective effects of this elastase inhibitor. ZN was also administered to MMP9 null mice following 90-min of tMCAo to determine if an NE-induced activation of MMP9 contributed to its injurious effects (Fig. 4C). In MMP9−/− mice, infarct volumes were reduced 53% by ZN (24±4 mm3; n=10) relative to vehicle (51±7 mm3; n=14; p<0.05), indicative of MMP9-independent injury mechanisms; note that MMP9−/− mice already exhibited less injury than WT controls (73±9 mm3; n=12; p<0.05) prior to ZN administration, as shown previously by us and others (Gidday et al. 2005).
Figure 4.
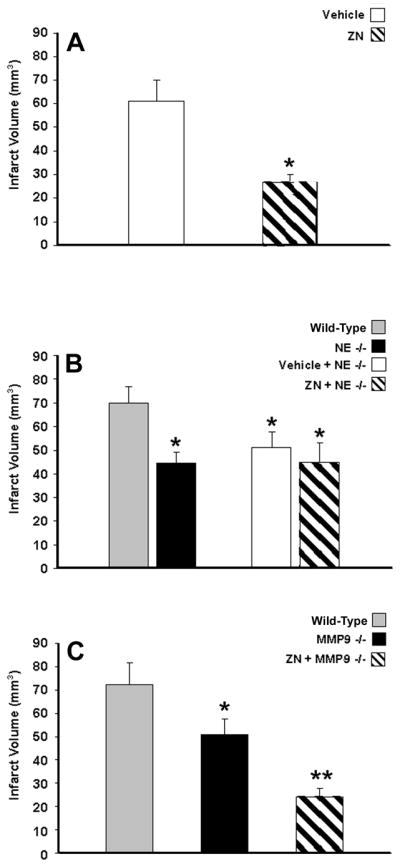
NE contribution to infarct volume following transient middle cerebral artery occlusion (tMCAo). (A) Post-ischemic NE inhibition by ZN200355 (ZN) administration significantly reduced infarct volume in SW/ND4 mice following 90-min tMCAo. (B) Infarct volumes were also significantly lower in NE−/− mice following 2-h tMCAo. ZN did not further reduce infarct volume following tMCAo in NE−/− mice, attesting to the specificity of its protective effects. (C) Administration of ZN to MMP9−/− mice significantly reduced infarct volume following 90-min tMCAo when compared to MMP9−/− mice with vehicle administration. Values shown are mean±SEM. *p<0.05 vs. respective vehicle administration or wild-type (WT) controls; **p<0.05 vs. vehicle-treated MMP9−/− mice.
Although NE gene deletion was protective in the setting of tMCAo, when NE−/− mice were subjected to a permanent occlusion of the MCA, no infarct-sparing effect could be detected in either a distal or proximal occlusion model, even with large sample sizes. When subjected to a distal pMCAo by a craniotomy and subsequent electrocoagulation of the MCA, infarct volumes in WT (27±2 mm3; n=12) and NE−/− (22±2 mm3; n=12) mice were statistically indistinguishable (Fig. 5A). When pMCAo was achieved by the permanent placement of the intraluminal suture, there was also no significant difference in infarct volumes between WT (95±5 mm3; n=15) and NE−/− (99±6 mm3; n=17) mice (Fig. 5A).
Figure 5.

The role of reperfusion in neutrophil elastase (NE)-induced neurovascular pathology. At 24 h following a permanent middle cerebral artery occlusion (pMCAo), neither (A) infarct volume secondary to pMCAo by either electrocoagulation of the MCA via craniotomy, or permanent placement of the intraluminal suture, nor (B) vasogenic edema were affected in NE-null mice compared to wild-type mice. Values shown are mean±SEM.
NE contributes to vasogenic edema and BBB breakdown
Vasogenic edema was evaluated at 24 h after ZN administration following a 90-min tMCAo in SW/ND4 mice, and also 24 h after either a 90-min tMCAo, or pMCAo secondary to cauterization of the distal MCA, in NE−/− and WT mice. As shown in Figure 6A, the increase in ipsilateral water content measured in vehicle-treated mice (4.00±0.38%; n=7) was reduced by 50% in mice given ZN (2.00±0.41%; n=8; p<0.05). A similar, 52% reduction in post-ischemic vasogenic edema occurred in NE-null mice (1.79±0.34%; n=11; p<0.01) when compared to WT control (3.71±0.54%; n=11; Fig. 6B). There was no significant effect of NE gene deletion on post-ischemic water content when the MCA was permanently occluded in WT mice (2.58±0.34%; n=12) relative to NE−/− mice (2.43±0.29%; n=14; Fig. 5B).
Figure 6.

Neutrophil elastase (NE) contribution to post-ischemic vasogenic edema and BBB disruption. (A) ZN200355 (ZN) administration in SW/ND4 mice reduced ipsilateral vasogenic edema at 24 h of reperfusion following 2-h transient middle cerebral artery occlusion (tMCAo). (B) Vasogenic edema was significantly reduced in NE−/− mice versus wild-types following tMCAo. Evans Blue content (μg/hemisphere) 24 h following a 90-min tMCAo, normalized to the contralateral hemisphere, was significantly reduced in (C) SW/ND4 mice given ZN, relative to vehicle, and (D) NE−/− mice versus wild-type controls. Values shown are mean±SEM. *p<0.05 vs. vehicle administration or wild-type.
To differentiate between changes in osmotic water balance and BBB disruption, cerebrovascular protein leakage was evaluated by Evans blue extravasation following 90-min tMCAo. Figure 6C shows that inhibition of NE with ZN completely abrogated increases in ipsilateral hemispheric Evans blue content from 156±57% in vehicle-treated mice (n=9) to -2±10% in ZN-treated mice (n=13; p<0.05). Genetic inhibition of NE reduced Evans blue extravasation by 86%, from 562±166% in WT mice (n=9) to 80±64% in NE-null mice (n=10; p<0.01; Fig. 6D).
NE contributes to post-ischemic leukocyte adherence
Sham tMCAo studies in SW/ND4 mice established a baseline of 86±23 adherent leukocytes per mm2 of venular endothelium (n=9). As shown in Fig. 7A, 24 h after tMCAo, the number of leukocytes adherent to cerebral endothelium was increased by 256% (p<0.01) in vehicle-treated animals (220±44 leukocytes/mm2; n=10). This post-ischemic elevation in adherence was not significantly reduced when ZN was administered at 0 and 4 h of reperfusion (186±28 leukocytes/mm2; n=13). To test the possibility that this lack of reduction in leukocyte adherence was due to a loss of pharmacologic activity of ZN, additional groups of ischemic animals underwent a third administration of ZN, or vehicle control, at 22 h of reperfusion. The number of adherent leukocytes at 24 h of reperfusion was unchanged in vehicle-treated mice (302±48 leukocytes/mm2; n=6; p<0.01), but a 47% reduction in the number of adherent leukocytes was evidenced in animals with the additional ZN treatment (163±61; n=6; p<0.05 vs. vehicle at 22 h). Similarly, the tMCAo-induced increase in adherent leukocytes measured in WT mice (190±23 leukocytes/mm2; n=16; p<0.05) was significantly abrogated, by 94%, in NE-null mice, to levels (116±23 leukocytes/mm2; n=14) not different from WT shams (111±26 leukocytes/mm2; n=10; Fig. 7B). Representative photomicrographs of post-ischemic leukocyte adherence in the venular microcirculation of these two genotypes are shown in Figs. 7C and D.
Figure 7.

Neutrophil elastase (NE) contribution to post-ischemic leukocyte-endothelial adherence. (A) Increases in leukocyte adherence to cortical venular endothelium observed 24 h following a 60-min tMCAo relative to strain-matched, nonischemic sham mice were not significantly decreased by ZN200355 (ZN) treatment at 0 and 4 h of reperfusion, but was significantly reduced when an additional dose of ZN was administered at 22 h of reperfusion. (B) NE gene deletion significantly attenuated leukocyte adherence relative to that measured in wild-type mice 24 h following tMCAo. (C and D) Leukocyte adherence in representative video photomicrographs of surface cortical venules in (C) wild-type (WT) and (D) NE−/− mice 24 h following tMCAo. Scale bar = 100 μm; values shown are mean±SEM. *p<0.05 vs. sham; #p<0.05 vs. vehicle administration or wild-type.
Discussion
In many pathologic conditions, leukocyte-derived proteases, such as NE, degrade the basement membrane to allow leukocyte extravasation into the surrounding tissue, jeopardizing BBB integrity in the process (Nagy et al. 1998; Man et al. 2007). Once present in the parenchymal extracellular space, NE may also degrade additional extracellular matrix substrates (e.g. collagens, laminins) that threaten neuronal and glial viability. In the present study, we confirmed the presence of extravascular cells immunoreactive for NE in the peri-infarct region, with a concomitant increase in tissue NE expression levels and enzymatic activity, following tMCAo. We also identified several specific, and detrimental, roles for NE in neurovascular unit pathology following focal stroke with reperfusion. Both pharmacologic inhibition and genetic deletion of NE resulted in significantly reduced vasogenic edema, BBB disruption, and leukocyte-endothelial adherence to cortical venules, ultimately leading to a reduction in infarct volume after tMCAo.
The extent to which neutrophil-dependent and neutrophil-independent mechanisms participate in BBB disruption, in different types of stroke, as well as the relationship between post-stroke barrier integrity and ultimate lesion volume, still require considerable elucidation. We contend that the reduction in vasogenic edema and Evans blue leak across the BBB, and the reductions in leukocyte adherence to venular endothelium resulting from both pharmacologic inhibition and genetic deletion of NE, strongly implicate NE in neurovascular unit injury, secondary to its ability to degrade basal lamina and extracellular matrix (Ishikawa et al. 2003; Houtz et al. 2004; Henriksen et al. 2008). In future studies, it will be important to establish whether neutrophil protease release requires adherence or extravasation, or if it can occur in non-adherent, slowly circulating neutrophils as they pass through the cerebral microcirculation.
In our study, post-ischemic leukocyte adherence to reperfused cerebrovenular endothelium was reduced in mutant mice lacking the NE gene. In addition, the NE inhibitor ZN reduced leukocyte adherence, but only when ZN was administered 2 h prior to adherence measurements. This likely relates to the timing of the inflammatory response being measured relative to the administration of the inhibitor, as our intravital microscopy-based measurement of leukocyte-endothelial adherence provides a snapshot of a dynamic process occurring at the time of measurement. At 24 h of reperfusion, though, the extent of NE inhibition by ZN, given 20 and 24 hours earlier, is likely to be considerably diminished based upon previous pharmacokinetic studies (Williams et al. 1991; Mehta et al. 1994). A large portion of vasogenic edema and Evans blue leak, however, may occur during early reperfusion, the cumulative effects of which are measured at 24 h post-ischemia. Thus, in contrast to leukocyte adherence, these latter responses can be strongly influenced by ZN administered early during reperfusion. That ZN-mediated neurovascular protection resulted from NE inhibition, and not other nonspecific actions, is supported by our finding that no additional infarct-sparing effects were observed when ZN was administered to NE-null mice. However, additional investigations should address the neurovascular protective efficacy of delayed treatment with ZN, or other elastase inhibitors, and confirm that the beneficial effects of NE inhibition are not transitory, but rather provide long-term functional benefits.
The role of NE in leukocyte adherence and diapedesis includes the ability of NE to mediate the expression of intracellular adhesion molecule-1 (ICAM-1) on endothelial cells (Yamaguchi et al. 1998) and CD11/CD18 on neutrophils (Woodman et al. 1993). Despite this, a recent study showed no reduction in leukotriene B4-induced leukocyte adherence to skeletal muscle venules in NE−/− mice (Young et al. 2007). The reduction in leukocyte-endothelial adherence in NE−/− mice in our model of focal ischemia/reperfusion may result from differences in inflammatory stimuli and/or an uncharacterized immune privilege feature of the BBB. Activation of parallel pathways may be sufficient to promote leukocyte adherence in the systemic vasculature of NE-null mice, whereas higher levels of activation of these alternative adherence-promoting mechanisms are required to affect similar changes at the level of the BBB following stroke.
NE-mediated injury of neurovascular unit cells can also be indirect, given that NE can upregulate the expression of other proteases capable of recruiting additional leukocytes to the lesion and/or degrading the extracellular matrix; one such protease is matrix metalloproteinase (MMP) gelatinase MMP9 (Itoh et al. 1995). We previously showed that MMP9−/− mice, and chimeras lacking MMP9 in circulating leukocytes, exhibited reduced leukocyte adherence, BBB disruption, and infarct size following tMCAo (Gidday et al. 2005). However, we observed additional and significant neuroprotective effects of pharmacologic NE inhibition when ZN was administered to MMP9-null mice. These findings indicate that NE worsens stroke outcome by mechanisms that are independent of its potential ability to activate this particular protease.
No protective effects of NE gene deletion on vasogenic edema or infarct volume were noted in two models of pMCAo. These findings indicate that vascular and neuronal exposure to circulating, adherent, and/or infiltrating neutrophils, as occurs with reperfusion, is necessary for NE-mediated injury. Activating stimuli for neutrophil degranulation (Henriksen et al. 2008) may vary between permanently ischemic and reperfused tissue; therefore, future pMCAo studies utilizing penumbral-based measures might actually confirm modest neuroprotective effects of NE inhibition in peri-infarct regions with limited reperfusion.
Clinical studies document that proteases, including both NE (Iwatsuki et al. 1998; Cojocaru et al. 2006) and MMP9 (Rosell et al. 2008), are upregulated within the first days following cerebral ischemia, and can be prognosticators of secondary complications following stroke. In light of these and our present results, it would seem prudent to pursue additional preclinical studies to further address the proximal signaling pathways responsible for neutrophil degranulation in ischemic brain, as well as the molecular basis of NE-mediated neurovascular pathology. Given the predominance of evidence indicating that neutrophil recruitment is delayed following focal stroke (Gelderblom et al. 2009), as is the elevation of plasma NE levels in clinical stroke (Iwatsuki et al. 1998), inhibiting the injurious effects by NE could prove invaluable in the treatment of reperfused stroke.
Acknowledgments
The authors thank Steven S. Shapiro for the breeder pairs of NE−/− mice, and Lena Martensson at AstraZeneca (Wilmington, Delaware) for kindly providing the ZN200355. The authors would like to recognize NIH RO1 NS21045 (TSP), RO1 HL079278 (JMG), PO1 NS032636 (JMG), P01 HL29594 (TLAK), and the Spastic Paralysis Research Foundation of the Illinois-Eastern Iowa District of Kiwanis International (TSP), for funding this research. Ann Stowe is a Hope Center Fellow supported by the Hope Center for Neurological Disorders, Washington University School of Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altay T, McLaughlin B, Wu JY, Park TS, Gidday JM. Slit modulates cerebrovascular inflammation and mediates neuroprotection against global cerebral ischemia. Exp Neurol. 2007;207:186–94. doi: 10.1016/j.expneurol.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armao D, Kornfeld M, Estrada EY, Grossetete M, Rosenberg GA. Neutral proteases and disruption of the blood-brain barrier in rat. Brain Res. 1997;767:259–64. doi: 10.1016/s0006-8993(97)00567-2. [DOI] [PubMed] [Google Scholar]
- Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, et al. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med. 1998;4:615–8. doi: 10.1038/nm0598-615. [DOI] [PubMed] [Google Scholar]
- Cojocaru IM, Cojocaru M, Burcin C. Evaluation of granulocyte elastase as a sensitive diagnostic parameter of inflammation in first ischemic stroke. Rom J Intern Med. 2006;44:317–21. [PubMed] [Google Scholar]
- Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and Spatial Dynamics of Cerebral Immune Cell Accumulation in Stroke. Stroke. 2009 doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, et al. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–68. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Henriksen PA, Sallenave JM. Human neutrophil elastase: mediator and therapeutic target in atherosclerosis. Int J Biochem Cell Biol. 2008;40:1095–100. doi: 10.1016/j.biocel.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Hermant B, Bibert S, Concord E, Dublet B, Weidenhaupt M, Vernet T, et al. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J Biol Chem. 2003;278:14002–12. doi: 10.1074/jbc.M300351200. [DOI] [PubMed] [Google Scholar]
- Houtz PK, Jones PD, Aronson NE, Richardson LM, Lai-Fook SJ. Effect of pancreatic and leukocyte elastase on hydraulic conductivity in lung interstitial segments. J Appl Physiol. 2004;97:2139–47. doi: 10.1152/japplphysiol.00567.2004. [DOI] [PubMed] [Google Scholar]
- Ionescu CV, Cepinskas G, Savickiene J, Sandig M, Kvietys PR. Neutrophils induce sequential focal changes in endothelial adherens junction components: role of elastase. Microcirculation. 2003;10:205–20. doi: 10.1038/sj.mn.7800185. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Stokes KY, Zhang JH, Nanda A, Granger DN. Cerebral microvascular responses to hypercholesterolemia: roles of NADPH oxidase and P-selectin. Circ Res. 2004;94:239–44. doi: 10.1161/01.RES.0000111524.05779.60. [DOI] [PubMed] [Google Scholar]
- Ishikawa N, Oda M, Kawaguchi M, Tsunezuka Y, Watanabe G. The effects of a specific neutrophil elastase inhibitor (ONO-5046) in pulmonary ischemia-reperfusion injury. Transpl Int. 2003;16:341–6. doi: 10.1007/s00147-003-0556-8. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Nagase H. Preferential inactivation of tissue inhibitor of metalloproteinases-1 that is bound to the precursor of matrix metalloproteinase 9 (progelatinase B) by human neutrophil elastase. J Biol Chem. 1995;270:16518–21. doi: 10.1074/jbc.270.28.16518. [DOI] [PubMed] [Google Scholar]
- Iwatsuki K, Kumura E, Yoshimine T, Yamamoto K, Sato M, Hayakawa T. Increase in jugular levels of polymorphonuclear neutrophil elastase in patients with acute cerebral infarction. Neurol Res. 1998;20:397–402. doi: 10.1080/01616412.1998.11740537. [DOI] [PubMed] [Google Scholar]
- Kishima H, Takeda S, Miyoshi S, Matsumura A, Minami M, Utsumi T, et al. Microvascular permeability of the non-heart-beating rabbit lung after warm ischemia and reperfusion: role of neutrophil elastase. Ann Thorac Surg. 1998;65:913–8. doi: 10.1016/s0003-4975(98)00076-9. [DOI] [PubMed] [Google Scholar]
- Majid A, He YY, Gidday JM, Kaplan SS, Gonzales ER, Park TS, et al. Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke. 2000;31:2707–14. doi: 10.1161/01.str.31.11.2707. [DOI] [PubMed] [Google Scholar]
- Man S, Ubogu EE, Ransohoff RM. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain Pathol. 2007;17:243–50. doi: 10.1111/j.1750-3639.2007.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matayoshi H, Hirata T, Yamashita S, Ishida K, Mizukami Y, Gondo T, et al. Neutrophil elastase inhibitor attenuates hippocampal neuronal damage after transient forebrain ischemia in rats. Brain Res. 2009;1259:98–106. doi: 10.1016/j.brainres.2008.12.070. [DOI] [PubMed] [Google Scholar]
- McCandless EE, Piccio L, Woerner BM, Schmidt RE, Rubin JB, Cross AH, et al. Pathological expression of CXCL12 at the blood-brain barrier correlates with severity of multiple sclerosis. Am J Pathol. 2008;172:799–808. doi: 10.2353/ajpath.2008.070918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta JL, Nichols WW, Nicolini FA, Hendricks J, Donnelly WH, Saldeen TG. Neutrophil elastase inhibitor ICI 200,880 protects against attenuation of coronary flow reserve and myocardial dysfunction following temporary coronary artery occlusion in the dog. Cardiovasc Res. 1994;28:947–56. doi: 10.1093/cvr/28.7.947. [DOI] [PubMed] [Google Scholar]
- Miller BA, Perez RS, Shah AR, Gonzales ER, Park TS, Gidday JM. Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia-reperfusion. Neuroreport. 2001;12:1663–9. doi: 10.1097/00001756-200106130-00030. [DOI] [PubMed] [Google Scholar]
- Nagy Z, Kolev K, Csonka E, Vastag M, Machovich R. Perturbation of the integrity of the blood-brain barrier by fibrinolytic enzymes. Blood Coagul Fibrinolysis. 1998;9:471–8. doi: 10.1097/00001721-199809000-00003. [DOI] [PubMed] [Google Scholar]
- Okajima K, Harada N, Uchiba M, Mori M. Neutrophil elastase contributes to the development of ischemia-reperfusion-induced liver injury by decreasing endothelial production of prostacyclin in rats. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1116–23. doi: 10.1152/ajpgi.00061.2004. [DOI] [PubMed] [Google Scholar]
- Rosell A, Cuadrado E, Ortega-Aznar A, Hernandez-Guillamon M, Lo EH, Montaner J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke. 2008;39:1121–6. doi: 10.1161/STROKEAHA.107.500868. [DOI] [PubMed] [Google Scholar]
- Shapiro SD. Neutrophil elastase: path clearer, pathogen killer, or just pathologic? Am J Respir Cell Mol Biol. 2002;26:266–8. doi: 10.1165/ajrcmb.26.3.f233. [DOI] [PubMed] [Google Scholar]
- Shimakura A, Kamanaka Y, Ikeda Y, Kondo K, Suzuki Y, Umemura K. Neutrophil elastase inhibition reduces cerebral ischemic damage in the middle cerebral artery occlusion. Brain Res. 2000;858:55–60. doi: 10.1016/s0006-8993(99)02431-2. [DOI] [PubMed] [Google Scholar]
- Tiefenbacher CP, Ebert M, Niroomand F, Batkai S, Tillmanns H, Zimmermann R, et al. Inhibition of elastase improves myocardial function after repetitive ischaemia and myocardial infarction in the rat heart. Pflugers Arch. 1997;433:563–70. doi: 10.1007/s004240050315. [DOI] [PubMed] [Google Scholar]
- Tureyen K, Brooks N, Bowen K, Svaren J, Vemuganti R. Transcription factor early growth response-1 induction mediates inflammatory gene expression and brain damage following transient focal ischemia. J Neurochem. 2008;105:1313–24. doi: 10.1111/j.1471-4159.2008.05233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welbourn CR, Goldman G, Paterson IS, Valeri CR, Shepro D, Hechtman HB. Neutrophil elastase and oxygen radicals: synergism in lung injury after hindlimb ischemia. Am J Physiol. 1991;260:H1852–6. doi: 10.1152/ajpheart.1991.260.6.H1852. [DOI] [PubMed] [Google Scholar]
- Williams JC, Stein RL, Giles RE, Krell RD. Biochemistry and pharmacology of ICI 200,880, a synthetic peptide inhibitor of human neutrophil elastase. Ann N Y Acad Sci. 1991;624:230–43. doi: 10.1111/j.1749-6632.1991.tb17022.x. [DOI] [PubMed] [Google Scholar]
- Woodman RC, Reinhardt PH, Kanwar S, Johnston FL, Kubes P. Effects of human neutrophil elastase (HNE) on neutrophil function in vitro and in inflamed microvessels. Blood. 1993;82:2188–95. [PubMed] [Google Scholar]
- Yamaguchi Y, Matsumura F, Wang FS, Akizuki E, Liang J, Matsuda T, et al. Neutrophil elastase enhances intercellular adhesion molecule-1 expression. Transplantation. 1998;65:1622–8. doi: 10.1097/00007890-199806270-00014. [DOI] [PubMed] [Google Scholar]
- Young RE, Thompson RD, Larbi KY, La M, Roberts CE, Shapiro SD, et al. Neutrophil elastase (NE)-deficient mice demonstrate a nonredundant role for NE in neutrophil migration, generation of proinflammatory mediators, and phagocytosis in response to zymosan particles in vivo. J Immunol. 2004;172:4493–502. doi: 10.4049/jimmunol.172.7.4493. [DOI] [PubMed] [Google Scholar]
- Young RE, Voisin MB, Wang S, Dangerfield J, Nourshargh S. Role of neutrophil elastase in LTB4-induced neutrophil transmigration in vivo assessed with a specific inhibitor and neutrophil elastase deficient mice. Br J Pharmacol. 2007;151:628–37. doi: 10.1038/sj.bjp.0707267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JW, Gottschall PE. Zymographic measurement of gelatinase activity in brain tissue after detergent extraction and affinity-support purification. J Neurosci Methods. 1997;76:15–20. doi: 10.1016/s0165-0270(97)00065-4. [DOI] [PubMed] [Google Scholar]
- Zimmerman BJ, Granger DN. Reperfusion-induced leukocyte infiltration: role of elastase. Am J Physiol. 1990;259:H390–4. doi: 10.1152/ajpheart.1990.259.2.H390. [DOI] [PubMed] [Google Scholar]