

Abstract
Split inteins are parasitic genetic elements frequently found inserted into reading frames of essential proteins. Their association and excision restore host protein function through a protein self-splicing reaction. They have gained an increasingly important role in the chemical modification of proteins to create cyclical, segmentally labeled, and fluorescently tagged proteins. Ideally, inteins would seamlessly splice polypeptides together with no remnant sequences and at high efficiency. Here, we describe experiments that identify the branched intermediate, a transient step in the overall splicing reaction, as a key determinant of the splicing efficiency at different splice-site junctions. To alter intein specificity, we developed a cell-based selection scheme to evolve split inteins that splice with high efficiency at different splice junctions and at higher temperatures. Mutations within these evolved inteins occur at sites distant from the active site. We present a hypothesis that a network of conserved coevolving amino acids in inteins mediates these long-range effects.
Keywords: directed evolution, protein ligation, Crk-II, SCA, coevolution
Protein function is modulated by a variety of posttranslational modifications, such as phosphorylation, ubiquitylation, and methylation (1). One of the most dramatic posttranslational modifications is protein splicing, an autocatalytic process in which an intervening polypeptide sequence, termed an intein, is excised from a precursor protein with concomitant splicing of the franking sequences, known as exteins. Protein splicing has proven to be a highly versatile process for methods development and forms the hub of several protein engineering technologies (2). Inteins have been successfully used in protein semisynthesis to create posttranslationally modified proteins (2, 3), in the cyclization of peptides to create small molecule toxins (4), in plant biotechnology to reconstruct proteins in vivo (5), in the segmental labeling of proteins for NMR studies (6), and in protein semisynthesis in living cells (7).
Inteins come in 2 flavors—cis splicing inteins are single polypeptides that are embedded in a host protein, whereas trans-splicing inteins (herein called split inteins) are separate polypeptides that mediate protein splicing after the intein pieces and their protein cargo associate (8, 9) (Fig. 1A). Despite the many applications of split inteins in chemical biology and protein chemistry, they are plagued with various idiosyncratic parameters that limit their more general use. Most importantly, the 2 parts of the naturally split inteins associate and typically splice at a C-terminal junction containing the canonical “CFN” tripeptide sequence (10, 11), which are the first 3 amino acids of the C-extein sequence (Fig. 1A). This tripeptide remains in the product after splicing, meaning that it will most often be a mutant protein. It is currently unclear why this “CFN” sequence is required for efficient protein trans-splicing. In contrast, work from several laboratories, including our own, indicates that the N-terminal splice junction is much more tolerant to noncanonical sequences (6, 11–13).
Fig. 1.

The mechanism of protein trans-splicing. (A) Schematic showing the basic steps in the intein-mediated protein trans-splicing reaction. IntN and IntC refer to the N- and C-terminal parts of the split intein, N- and C-extein refer to the N- and C-terminal parts of the spliced protein, and “CFN” refers to the amino-acid sequence in the C-extein at the IntC boundary. (B) Schematic showing the bond rearrangements catalyzed by the intein during protein splicing.
In this study, we begin by identifying a key catalytic step in the splicing reaction that is sensitive to the identity of the amino acids at the C-terminal splice junction. To alter this rate-limiting step, we describe a general cell-based selection scheme that allows the directed evolution of mutant split inteins that can splice at noncanonical C-terminal splice junctions. We demonstrate that these mutant inteins can be used for traceless protein splicing by creating the Crk-II proto-oncogenic adaptor protein from peptide fragments without the need to introduce mutations into the final protein. In addition, this evolution scheme was used to generate mutant split inteins that support efficient splicing at 37 °C and can be used in mammalian cell applications. Intriguingly, the location of the mutated amino acids in each case is distant from the active site and likely does not interact directly with extein amino acids. We present a hypothesis for how these distant sites might affect the function of amino acids in the active site.
Results
The Rate-Limiting Step of Splicing Using DnaE Split Inteins.
We first set out to understand why protein trans-splicing is so inefficient when noncanonical sequences are placed at the C-terminal splice junction. Split inteins catalyze a remarkable series of chemical rearrangements that require the intein to be properly assembled and folded (9) (Fig. 1B). The first step in splicing involves an N-S acyl shift in which the N-extein polypeptide is transferred to the side chain of the first residue of the intein. This is then followed by a trans-(thio)esterification reaction in which this acyl unit is transferred to the first residue of the C-extein (which is either serine, threonine, or cysteine) to form a branched intermediate. In the penultimate step of the process, this branched intermediate is cleaved from the intein by a transamidation reaction involving the C-terminal asparagine residue of the intein. This then sets up the final step of the process involving an S-N acyl transfer to create a normal peptide bond between the 2 exteins. We monitored the overall protein splicing reaction using an in vitro protein trans-splicing assay with purified proteins. Constructs were generated in which model N- and C-exteins were fused to the fragments of the prototypical DnaE split intein from Synechocytis sp. PCC6803 (herein abbreviated to Ssp). In initial studies, we reacted an N-terminal construct (H6-Ub-SspN) with a C-terminal construct (SspC-“CFN”-HA) containing the canonical “CFN” sequence at the C-terminal splice junction (see Table 1 for a description of all constructs used in this study). As expected, splicing was found to be highly efficient as read out by Western blotting against the HA tag [supporting information (SI) Fig. S1]. Next, we replaced the “CFN” motif with a noncanonical sequence. For this, we chose the “SGV” tripeptide, which is derived from a linker sequence in the multidomain adaptor protein Crk-II; we previously assembled this protein using the Ssp intein, albeit using a “CFN” mutant sequence in place of the native “SGV” motif (12). Consistent with our previous results (12), we observed no spliced product upon reaction of H6-Ub-SspN with SspC-“SGV”-HA (Fig. 2 A and B). However, a 16-kDa band accumulated, which is consistent in size with the expected branched intermediate in which ubiquitin is linked to the SspC-“SGV”-HA peptide (expected molecular weight of 15.7 kDa). Only trace amounts of this intermediate were observed when the canonical “CFN” junction was used (Fig. S1). Therefore, the rate-limiting step in Ssp splicing at this noncanonical site appears to be the resolution of the branched intermediate into the spliced product.
Table 1.
Description of all constructs used in this study
| Protein | Molecular weight | Description |
|---|---|---|
| Ssp intein | 14.0 kDa (N-term), 3.9 kDa (C-term) | N- and C-terminal inteins from Ssp DnaE |
| Npu* intein | 11.9 kDa (N-term), 3.9 kDa (C-term) | N-terminal intein from Npu DnaE with C-terminal intein from Ssp DnaE |
| mNpu* intein | 11.9 kDa (N-term), 3.9 kDa (C-term) | Npu* intein evolved with “SGV” (E15D, L25I, and L92M mutations in NpuN; D23Y mutation in SspC) |
| mNpu37* intein | 11.9 kDa (N-term), 3.9 kDa (C-term) | Npu* intein evolved at 37°C (L25S and P21RC mutations) |
| H6-Ub-SspN | 24.4 kDa | His6–tagged ubiquitin fused to the N-terminal intein from Ssp DnaE |
| H6-Ub-NpuN | 22.3 kDa | His6–tagged ubiquitin fused to the N-terminal intein from Npu DnaE |
| SspC-“CFN”-HA | 5.2 kDa | C-terminal intein from Ssp DnaE with “CFN” sequence preceding the HA tag |
| SspC-“SGV”-HA | 5.2 kDa | C-terminal intein from Ssp DnaE with “SGV” sequence preceding the HA tag |
| CrkII | 38.3 kDa | Full-length protein with domain structure of SH2-“SGV”-SH3-SH3 |
| Flag-SH2-IntN-Ub | 35.4 kDa | Flag-tagged CrkII SH2 domain followed by the N-terminal intein domain and ubiquitin |
| MBP-IntC-“SGV”-SH3-SH3-H6 | 68.2 kDa | His-tagged CrkII SH3 domains preceded by MBP and the C-terminal intein domain |
Fig. 2.

In vitro protein trans-splicing of Ssp and Npu* DnaE inteins. (A) Schematic showing the starting materials, branched intermediate, and expected product of the splicing reaction. The molecular weights shown are for the Npu* reaction. IntN = SspN or NpuN. (B) Time course of in vitro splicing reactions employing either Ssp or Npu* split inteins using an “SGV” splice junction. Reaction mixtures were resolved by nonreducing SDS-PAGE and blotted using an anti-HA antibody. BI, branched intermediate (expected: 15.7 kDa); SP, spliced product (expected: 11.8 kDa). (C) Plot showing the buildup of BI (squares) and SP (circles) over time for the Ssp (Left) and Npu* (Right) reactions. Data points are derived from densitometry analysis of Western blots of the type shown in B. The results of 3 independent in vitro trans-splicing experiments are shown with the data fit to a 2-step sequential kinetic model. (D) Sequence alignment of Ssp and Npu DnaE inteins. The N-extein residues are “AEY”, whereas the C-extein residues are “CFN”.
Ssp is a member of a family of naturally split DnaE inteins, all of which use the canonical “CFN” splice junction but whose splicing activities differ from each other (10, 11, 14). Intriguingly, the activity of the chimeric split intein composed of the N-terminal Nostoc punctiforme DnaE intein and C-terminal Ssp intein was previously shown to have broader sequence specificity than WT Ssp despite nearly 70% sequence identity (14) (Fig. 2D). We used the same in vitro splicing assay to characterize the functional differences between Ssp and this chimeric intein, termed Npu* (Fig. 2B). Unlike in the Ssp splicing reaction, the Npu* reaction led to the appearance of both the spliced product and the 16-kDa band, which was confirmed as the branched intermediate by mass spectrometry (Fig. S2). We determined the rate constants for the branched intermediate formation as well as its conversion to the spliced product for both Ssp and Npu* by quantifying the bands on the gel and fitting their values to the differential equations describing a sequential kinetic reaction model (Fig. 2C). The rate of branched intermediate formation was similar between Ssp (0.32 ± 0.02 h−1) and Npu* (0.38 ± 0.03 h−1), although the resolution to the spliced product was slower in the case of Npu* (0.14 ± 0.04 h−1) and not measurable in the case of the Ssp intein. These data suggest that the resolution of the branched intermediate is rate-limiting for both inteins and that the primary difference between these inteins is the rate of this step.
A Selection System to Evolve Intein Function.
Although Npu* is superior to Ssp at splicing the “SGV” junction, it would be desirable to have an intein that splices this junction and, by extension, other noncanonical sequences even better. One approach to this problem is to mutate all the residues in and around the intein active site. Although many structure-activity analyses have been performed on inteins, there is currently no structural information on the branched intermediate, and, by extension, very little is known about how the intein catalyzes the final steps in protein splicing. Thus, we concluded that a rational approach to engineering an improved split intein would not be straightforward. We instead asked whether we could evolve Npu* to splice the “SGV” junction better. A kanamycin resistance-based intein selection system was developed to evolve split intein function rapidly. Genticin resistance has been used to select for cis-splicing intein function in yeast (15), so we anticipated that this system could be adapted to our purposes and would provide kanamycin resistance in Escherichia coli. The Npu* intein was fused within the coding region of the aminoglycoside transferase gene (KanR) such that protein splicing assembles an active enzyme and so endows kanamycin resistance to E. coli cells (Fig. 3A and SI Text). Initially, 4 different splice site junctions in KanR were evaluated for activity in E. coli selected on kanamycin plates (Fig. S3); in addition to the site previously used (15), 3 new sites were chosen that are surface-exposed, contain a serine, and are present in a loop. All 4 junctions were evaluated based on the following criteria: (i) growth with “CFN” replacing the native KanR sequence to ensure that enzyme function is not compromised by the mutations, (ii) growth with active Npu* intein inserted into the KanR gene, and (iii) no growth with a catalytically inactive Npu* intein to eliminate splicing-independent resistance. The KanR splice site with the sequence “SPD” satisfied all these criteria (Fig. 3B), whereas the splice site used in a previous yeast-based study (with the sequence “SGE”) showed kanamycin resistance with the catalytically inactive intein, suggesting that this site is not a good reporter of Npu*-mediated trans-splicing in E. coli. The other 2 sites (with sequences “SVA” and “SDR”) were nonfunctional. For these reasons, the SPD junction site was chosen for all subsequent experiments.
Fig. 3.

Kanamycin resistance-based split intein selection assay. (A) Schematic of the KanR-Npu* selection vector construct and principle behind the selection method. (B) An LB agar plate grown at 30 °C that contains 100 μM ampicillin and 100 μM kanamycin streaked with test plasmids containing pBSK (vector alone), KanR (resistance gene alone), KanR-Npu* fusion, or KanR-Npu*(C1A) fusion (inactive intein).
The KanR-intein selection system was used to evolve Npu* to splice the “SGV” junction with increased activity at 30 °C (Fig. 4A). Each round of selection began by creating a library of PCR-based random mutants using oligonucleotides that preserve the N- and C-terminal catalytic residues (Fig. S4). This library was transformed into E. coli and plated on LB agar containing kanamycin to select for split intein function. Colonies that grew were then further tested in a 96-well plate assay to quantify intein activity based on the level of kanamycin resistance. In this assay, the growth of E. coli containing the mutant intein is measured in different concentrations of kanamycin. The mean and standard error of the mean of the OD of the best intein from each round are plotted in Fig. 4B. Three rounds of selection were sufficient to isolate an evolved split intein (termed mNpu*) with mutations E15D, L25I, and L92M in the N-terminus and D23Y in the C-terminus (Fig. 4C). The mNpu* intein has an IC50[Kan] that is 6-fold higher than WT Npu*.
Fig. 4.

Evolution of split inteins with an “SGV” splice junction. (A) Schematic of the directed evolution experiment designed to isolate split Npu* inteins with increased activity at an “SGV” C-terminal splice junction. (B) Growth of transformed E. coli cells after 22 h at 30 °C in LB containing different concentrations of kanamycin. Each curve is the growth profile for cells expressing the Npu*-KanR mutant with the highest activity (as determined by this assay) from each round of evolution. The WT Npu* starting point is shown for comparison. Errors = SEM (n = 3). (C) Mutations in the final selected Npu* intein (mNpu*) are mapped onto the Ssp intein structure (pdb1DE3) (16). The blue- and light blue-colored ribbons indicate the IntN and IntC portions of the intein, respectively. Essential catalytic residues are rendered as red sticks, and mutated amino acids are shown as a purple space-filling representation.
We wondered if the higher IC50[Kan] of mNpu* was specific to the “SGV” splice junction or if the overall activity of the intein simply increased. This was addressed by determining if mNpu* had the same increase in activity with different splice site junctions. The IC50[Kan] of Npu* and mNpu* inteins was measured with 3 additional splice site junctions (“CFN”, “SMD”, and “SPD”); “CFN” is the canonical junction for Npu*, “SMD” is a sequence between the MH1 and MH2 domains of SMAD2, and “SPD” is the sequence of the WT KanR gene. We observed a maximum 2.5-fold increase in mNpu* activity with these alternate splice sites, significantly less than that seen with the “SGV” site (Fig. S5). The larger increase in activity observed for the “SGV” site compared with these other sites suggests that much of the improved activity of the mNpu* is specific for the “SGV” splice junction, the site to which evolution was directed. In addition, these results demonstrate the utility of this selection method to screen for suitable splice sites rapidly for use in protein splicing applications.
Traceless Splicing of the Multidomain Crk-II Protein.
The ultimate goal was to evolve split inteins with activity toward particular splice sites within multidomain proteins. With this in mind, the split Npu* or mNpu* was inserted into the “SGV” sequence between the SH2 and SH3 domains of Crk-II to create 2 intein-fused polypeptide constructs (Fig. 5A). The ability of these proteins to generate native full-length Crk-II was assayed using 2 different conditions. In the first, the 2 fragments were coexpressed in E. coli from the same plasmid and their in vivo splicing activity was monitored following the induction of protein expression at 30 °C. Western blotting was used to monitor the progress of the trans-splicing reaction over time (Fig. 5 B and C). The mNpu* intein spliced the native “SGV” site ≈5-fold better than Npu*, consistent with the 6-fold difference in IC50s[Kan] from the 96-well growth assay.
Fig. 5.

Traceless splicing of Crk-II using evolved inteins. (A) Schematic showing the starting materials and expected Crk-II product of the splicing reaction. (B) Time course of in vivo splicing reactions employing either Npu* or mNpu* split inteins using a native “SGV” splice junction. Cells were lysed at the indicated time point postinduction, resolved by nonreducing SDS-PAGE, and blotted using an anti-Flag antibody. (C) Plot showing the buildup of Crk-II over time for the Npu* (diamonds) and mNpu* (squares) reactions. Data points are derived from densitometry analysis of Western blots of the type shown in B. The ratio of the slope of mNpu* to Npu* is 4.8 and is the rate enhancement of the evolution experiment. (D) Time course of in vitro splicing reactions in which lysates containing the individually expressed split intein-fused Crk-II constructs were mixed and analyzed as in B. BI, branched intermediate (expected: 81.0 kDa); SP, Crk-II spliced product (expected: 38.3 kDa); N, N-terminal intein precursor (expected: 35.4 kDa). Note that the band annotated as SP also reacts with an anti-His antibody (data not shown).
The second condition tests the utility of this system for in vitro splicing of individually expressed and combined protein fragments. The IntN- and IntC-containing fragments were individually transformed into E. coli and expressed in separate flasks. The resulting cell lysates were mixed and incubated at 30 °C, and the trans-splicing reaction was monitored over time (Fig. 5D). After 27 h, the vast majority of the mNpu* precursors had spliced with no branched intermediate remaining, indicating that the reaction was near complete. Although spliced product was observed with Npu*, a significant fraction was still found as a branched intermediate. Thus, mNpu* resolves the branched intermediate better than Npu*. These results, together with the selection experiment described above, demonstrate the versatility of this evolution system to generate split inteins that work in different protein systems (Crk-II and KanR), both in vivo and in vitro.
Evolution of a Mammalian Cell-Compatible Intein.
The power of this E. coli-based selection system is in its utility to select for protein function using different selective pressures, such as temperature. We next asked whether the split intein selection system can be used to evolve Npu* to higher activity at 37 °C, a property that might facilitate its use in mammalian cells. Two rounds of selection were performed on Npu*, which has a low level (and therefore a selectable level) of activity at 37 °C (Fig. S6). This led to the isolation of an intein containing an N-terminal L25S mutation and a C-terminal P21R mutation (termed mNpu37*). This intein has a 4-fold higher splicing activity in E. coli at 37 °C (Fig. S6). We next compared the activity of the Ssp, Npu*, and mNpu37* inteins in cultured HeLa cells. Maltose-binding protein (MBP) fused to the N-terminal portion of the respective intein was cotransfected in the HeLa cells, with GFP fused to the C-terminal portion of the intein (Fig. S7A). Cells were collected and lysed, and the spliced product was analyzed 8 h posttransfection by Western blot analysis against MBP and GFP (Fig. S7B). No spliced product was detected for Ssp, even though both reactants were well expressed. In contrast, Npu* and mNpu37* both supported trans-splicing to yield the expected MBP-GFP product. The relative levels of spliced product to unspliced precursors were higher for the evolved mNpu37* than for Npu*, consistent with the higher activity in the selection assay. Together, these experiments demonstrate the versatility of this selection system to evolve inteins using a different selective pressure, temperature.
Discussion
In summary, we have developed a protein evolution system that can be used to isolate split inteins with novel functions. In particular, we have shown that the evolved split inteins generated by this method can be used to perform traceless protein splicing both in vivo and in vitro, thereby addressing one of the critical limitations of protein trans-splicing in chemical biology applications. This study also sheds light on the fundamental way that inteins are built to perform specific functions. In particular, we examined the consequences of replacing the invariant “CFN” C-extein motif found in DnaE split inteins with a noncanonical sequence. Surprisingly, we find that the identity of the C-extein amino acids dramatically affects the rate of branched intermediate resolution, although having a minimal effect on its formation (Fig. 2). Perhaps mutation of the C-extein amino acids would be a good strategy to trap an intein in its branched intermediate state for structural studies, because little is known about the structure of the branched intermediate itself.
The crystal structure of the Ssp DnaE intein, which is nearly 70% identical to Npu* and a close structural model for the Npu* intein, suggests a possible mechanism for the reduced rate of branched intermediate resolution in the noncanonical “SGV”-containing substrates. The phenylalanine (F+2) side chain in the “CFN”-containing extein stacks with the histidine side chain (H24C) that coordinates a bound water molecule in the active site and is believed to have a critical role in asparagine (N36C) cyclization and resolution of the branched intermediate (16) (Fig. 6A). Although 2 other crystal structures containing C-extein residues reveal different specific amino-acid interactions with their +2 side chain (17, 18), the particular structural features of the Ssp intein suggest that mutation of “CFN” to “SGV” at the splice junction may disrupt efficient asparagine cyclization in this intein, in part by displacing the critical H24C amino acid. Intriguingly, H24C is physically next to the D23YC mutation discovered in the evolved mNpu* intein and may account for some of the increased rate of branched intermediate resolution in this evolved intein; the aspartate-to-tyrosine mutation undoubtedly changes the local packing interactions, which could reposition H24C and the bound water molecule to favor asparagine cyclization (Fig. 6A). Although it is currently impossible to predict accurately the energetic effects of a mutation from a structure alone, these structural observations lead us to hypothesize that the D23YC mutation partially compensates for the loss of the phenylalanine through repositioning of its normal stacking partner H24C, effectively coupling D23YC to the splice junction 8–10 Å away.
Fig. 6.

Proposed role of mutations in the evolved inteins. (A) Packing interactions within the C-terminal splice junction of the Ssp DnaE intein. Shown in red are the locations of the cysteine within the C-extein (C+1) and the C-terminal asparagine (N36C) of the intein; N36C is mutated to an alanine in the Ssp structure. The phenylalanine (F+2) of the C-extein, the catalytic histidine in the intein (H24C), and the adjacent residue (D23, which is mutated in mNpu*) are shown as a spaced-filled representation. (B) The SCA network of coevolving residues (tan surface) is mapped onto the Ssp DnaE intein structure. The essential catalytic intein residues (C1, T79, H82, and C + 1) are in red, whereas sites of mutation from the “SGV” splice junction and 37 °C evolution experiments are shown in purple.
The mechanism by which the other 3 amino acids affect intein function is not obvious from the Ssp crystal structure alone. In fact, the mutations seem to be scattered over the whole structure, including the opposite face from the active site (Fig. 4C, Fig. S6, and Movie S1). How can these mutations outside of the active site affect protein splicing? Protein mutagenesis and NMR dynamics, as well as the evolutionary analyses of protein families, suggest that networks of amino acids connect perturbations at distant sites to changes in the functional properties within active sites (19). One approach to identify these networks is to assume that coevolving residues in a protein family reveal functionally interacting amino acids independent of their location in the 3-D structure. Such an approach, utilizing the statistical coupling analysis (SCA) (20, 21), has been successfully used to identify distant points of allosteric regulation in proteins such as G-protein coupled receptors and globins (21) and was even used as constraints to create synthetic proteins that function like their natural counterparts (22, 23). We performed SCA on the intein protein family using sequences from InBase (24) and discovered a network of spatially contiguous amino acids formed by residues within and distant from the active site (Fig. 6B and Fig. S8). In the evolved mNpu* and mNpu37* inteins, 2 of the mutated amino acids (E15D and P21RC) are part of the network of coevolving amino acids, whereas the other 4 mutated amino acids are physically juxtaposed to network amino acids (Fig. 6B and Fig. S8). The proximity of these amino acids to the coupled network in inteins suggests a conduit by which these mutations could alter protein function. A similar trend is found when mutations from cis-intein evolution experiments are mapped onto the intein structure (15, 25, 26) (Fig. S9). It is important to note that not all amino acid mutations in these evolution experiments appear within or juxtaposed to the network of coevolving residues, suggesting that other mechanisms may also account for changes in protein function. Nevertheless, this network of covarying residues is likely a part of the conduit by which many of these amino acids alter splicing, which suggests that SCA results may be useful in rationalizing the results of other protein evolution experiments.
This study describes a method to evolve split inteins for practical applications, while also addressing basic features of intein catalytic mechanism and splice-site specificity. Several groups have independently evolved new function into cis-splicing inteins (15, 25–27). However, directed protein evolution has not been applied to split inteins or, to our knowledge, any other protein complementation pair. We show that a KanR-based selection system can be used to coevolve mutant pairs of split inteins in E. coli. This system was used to evolve a split intein that allows the efficient and traceless assembly of a multidomain protein from component fragments, the first of its kind. One obvious utility of this technology is segmental labeling of multidomain proteins, which can now be accomplished without mutating the protein of interest. In principle, this technology should also facilitate the evolution of cysteine-less inteins that splice in oxidizing environments, such as the endoplasmic reticulum or extracellular matrix, and that would efficiently cyclize peptides to target intracellular processes in mammalian cells. Thus, the use of inteins for traceless-protein splicing holds great potential as a way to study native proteins and their roles in cellular processes.
Methods
Protein Expression and Purification.
E. coli BL21 (DE3) cells were transformed with the H6-Ub-IntN expression plasmid. Transformed cells were grown to OD = 0.6 at 37 °C and induced by addition of isopropyl-β-D-thiogalactopyranoside (IPTG) to a concentration of 500 μM for 3 h. Cells were collected, resuspended in lysis buffer (50 mM Tris, 150 NaCl, pH 7.5) with protease inhibitors and lysed by sonication. H6-Ub-IntN was purified from the soluble fraction using Ni-NTA (Qiagen), followed by separation on a Superdex S75 column. Purified protein was dialyzed against splicing buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 7.2).
Protein Trans-Splicing Reactions.
Splicing reactions were initiated by mixing equal volumes of the appropriate IntN (2 μM) and IntC (4 μM) constructs in splicing buffer at 30 °C for a total volume of 100 μL (SI Text and Fig. S10). Aliquots were removed at specific time points, mixed with SDS-PAGE loading buffer [80 mM Tris (pH 6.8), 2% (w/v) SDS, 10% (v/v) glycerol, 0.02% (w/v) bromophenol blue], and flash frozen at −80 °C. The reaction mixtures were analyzed by SDS-PAGE (under nonreducing conditions), followed by immunoblotting with an anti-HA antibody.
Kinetic Analysis.
The intensity of the branched intermediate and spliced product was quantified using an Odyssey Imaging System (Li-Cor Biosciences) with dye-conjugated secondary antibodies. The integrated bands were fit to the sequential model:
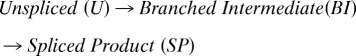 |
Raw densitometry data for 3 independent experiments were normalized and fit collectively to the following solutions for the differential equations that describe the overall reaction:
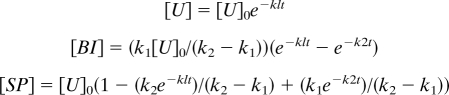 |
where [U]0 is the concentration of initial unspliced reactants, [BI] is the branched intermediate concentration, [SP] is the spliced product concentration, k1 is the rate of U→BI, k2 is the rate of BI→SP, and t is time from starting the reaction.
Crk-II Trans-Splicing Reactions.
For in vivo splicing reactions, the coexpression plasmid for Crk-II was transformed into E. coli BL21 (DE3), grown to an OD = 0.6 at 37 °C, and induced with 500 μm of IPTG at 30 °C (t = 0 min). After 15 min, cells were lysed with B-PER reagent (Thermo Scientific) plus additives [final 0.5 mM EDTA, 100 mM NaCl, 50 mM Tris (pH 7.2), 1 mM Tris(2-Carboxyethyl)Phosphine Hydrochloride] and aliquots were frozen in SDS-PAGE loading buffer at −80 °C at indicated time points.
For in vitro splicing reactions, the Crk-II N-terminal-intein and C-terminal intein expression plasmids were individually expressed in E. coli BL21 (DE3), grown to an OD = 0.6 at 37 °C, and induced by 500 μM IPTG for 20 min. Cells expressing each construct were lysed with B-PER plus additives (as above) and clarified by centrifugation, and the splicing reactions were initiated by mixing equal volumes of lysates at 30 °C. All reactions were monitored by SDS-PAGE, followed by immunoblotting with anti-Flag antibody.
Selection Assay.
Point mutations were introduced into Npu* using a Stratagene GeneMorphII kit with a mutation rate of ≈1%. The start codons, stop codons, and splice junctions (including the Cys1 and penultimate asparagine amino acids of the split intein) were preserved with oligonucleotides that extended through these amino acids (see Fig. S4 and SI Text). The WT Npu* was used in the first round of mutagenesis, whereas a pool of DNA from all selected colonies from the previous round was used in each subsequent round of selection. Each round involved cloning a new library of point mutants into the KanR-Npu* selection vector using NsiI/NcoI sites within the KanR gene, transforming this library into E. coli DH5α cells and plating the cells onto LB agar containing 100 μg/mL ampicillin and various concentrations of kanamycin; the concentration of kanamycin was increased from an initial concentration of 20 μg/mL to 150 μg/mL during the final round of selection. Plates were incubated at 30 °C or 37 °C overnight, and colonies were sequenced.
Ninety-Six–Well Plate Assay.
E. coli DH5α cells expressing KanR-Npu* WT and mutant proteins were grown in 150 μL LB media containing 100 μg/mL ampicillin and increasing concentrations of kanamycin. The plate was incubated with shaking at 30 °C for 22 h for the “SGV” splice site selection and at 37 °C for 16 h for the temperature selection. The final OD was determined using a Molecular Devices VersaMax microplate reader.
Expression in HeLa Cells.
HeLa cells were cultured in DMEM (Gibco) supplemented with 10% FBS (Sigma) at 37 °C and CO2 according to standard procedures. Cells were cotransfected with IntN and IntC plasmids (0.5 μg) using FuGENE-6 (Roche Applied Science). Cells were incubated for 8 h posttransfection; at that point, the medium was aspirated and ice-cold PBS was added to resuspend the cells. Cells were lysed by boiling in SDS-PAGE buffer. Splicing was detected by SDS-PAGE, followed by immunoblotting with anti-MBP and anti-GFP antibodies.
Statistical Coupling Analysis (SCA).
An alignment of 357 intein sequences was created from sequences deposited in New England Biolab's InBase (24) using HMMER (28, 29) from a profile created using published intein structures (available on request). The statistical coupling between positions was calculated as described previously (20). The network of coevolving positions was determined from a hierarchical clustering analysis to group self-consistent positions that statistically covary with each other (21) (Fig. S8). Inteins have 1 self-consistent cluster consisting of 29 aa (19% of protein), which is mapped onto the Ssp DnaE intein structure using PyMol (30) in Fig. 5B (see also Movie S1).
Supplementary Material
Acknowledgments.
We thank Miquel Vila-Perello for the SspC-“CFN”-HA peptide and for help with peptide synthesis and mass spectrometry, Kyle Chiang for help with HeLa cells, Matt Pratt for eukaryotic expression constructs pEB4 and pMPG04, and Fran Perler for access to InBase intein sequences. We also thank Champak Chatterjee for helpful discussions. This work is supported by National Institutes of Health Grants GM086868 and GM55843 (to T.W.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0902964106/DCSupplemental.
References
- 1.Walsh CT, Garneau-Tsodikova S, Gatto GJ., Jr Protein posttranslational modifications: The chemistry of proteome diversifications. Angew Chem Int Ed. 2005;44:7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 2.Muralidharan V, Muir TW. Protein ligation: An enabling technology for the biophysical analysis of proteins. Nat Methods. 2006;3:429–438. doi: 10.1038/nmeth886. [DOI] [PubMed] [Google Scholar]
- 3.Lew BM, Mills KV, Paulus H. Protein splicing in vitro with a semisynthetic two-component minimal intein. J Biol Chem. 1998;273:15887–15890. doi: 10.1074/jbc.273.26.15887. [DOI] [PubMed] [Google Scholar]
- 4.Tavassoli A, Benkovic SJ. Split-intein mediated circular ligation used in the synthesis of cyclic peptide libraries in E. coli. Nat Protoc. 2007;2:1126–1133. doi: 10.1038/nprot.2007.152. [DOI] [PubMed] [Google Scholar]
- 5.Chin HG, et al. Protein trans-splicing in transgenic plant chloroplast: Reconstruction of herbicide resistance from split genes. Proc Natl Acad Sci USA. 2003;100:4510–4515. doi: 10.1073/pnas.0736538100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muona M, Aranko AS, Iwai H. Segmental isotopic labelling of a multidomain protein by protein ligation by protein trans-splicing. Chembiochem. 2008;9:2958–2961. doi: 10.1002/cbic.200800604. [DOI] [PubMed] [Google Scholar]
- 7.Giriat I, Muir TW. Protein semi-synthesis in living cells. J Am Chem Soc. 2003;125:7180–7181. doi: 10.1021/ja034736i. [DOI] [PubMed] [Google Scholar]
- 8.Paulus H. Protein splicing and related forms of protein autoprocessing. Annu Rev Biochem. 2000;69:447–496. doi: 10.1146/annurev.biochem.69.1.447. [DOI] [PubMed] [Google Scholar]
- 9.Saleh L, Perler FB. Protein splicing in cis and in trans. Chem Rec. 2006;6:183–193. doi: 10.1002/tcr.20082. [DOI] [PubMed] [Google Scholar]
- 10.Dassa B, Amitai G, Caspi J, Schueler-Furman O, Pietrokovski S. Trans protein splicing of cyanobacterial split inteins in endogenous and exogenous combinations. Biochemistry. 2007;46:322–330. doi: 10.1021/bi0611762. [DOI] [PubMed] [Google Scholar]
- 11.Zettler J, Schutz V, Mootz HD. The naturally split Npu DnaE intein exhibits an extraordinarily high rate in the protein trans-splicing reaction. FEBS Lett. 2009;583:909–914. doi: 10.1016/j.febslet.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Shi J, Muir TW. Development of a tandem protein trans-splicing system based on native and engineered split inteins. J Am Chem Soc. 2005;127:6198–6206. doi: 10.1021/ja042287w. [DOI] [PubMed] [Google Scholar]
- 13.Zuger S, Iwai H. Intein-based biosynthetic incorporation of unlabeled protein tags into isotopically labeled proteins for NMR studies. Nat Biotechnol. 2005;23:736–740. doi: 10.1038/nbt1097. [DOI] [PubMed] [Google Scholar]
- 14.Iwai H, Zuger S, Jin J, Tam PH. Highly efficient protein trans-splicing by a naturally split DnaE intein from Nostoc punctiforme. FEBS Lett. 2006;580:1853–1858. doi: 10.1016/j.febslet.2006.02.045. [DOI] [PubMed] [Google Scholar]
- 15.Buskirk AR, Ong YC, Gartner ZJ, Liu DR. Directed evolution of ligand dependence: Small-molecule-activated protein splicing. Proc Natl Acad Sci USA. 2004;101:10505–10510. doi: 10.1073/pnas.0402762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun P, et al. Crystal structures of an intein from the split DnaE gene of Synechocystis sp. PCC6803 reveal the catalytic model without the penultimate histidine and the mechanism of zinc ion inhibition of protein splicing. J Mol Biol. 2005;353:1093–1105. doi: 10.1016/j.jmb.2005.09.039. [DOI] [PubMed] [Google Scholar]
- 17.Ding Y, et al. Crystal structure of a mini-intein reveals a conserved catalytic module involved in side chain cyclization of asparagine during protein splicing. J Biol Chem. 2003;278:39133–39142. doi: 10.1074/jbc.M306197200. [DOI] [PubMed] [Google Scholar]
- 18.Mizutani R, Anraku Y, Satow Y. Protein splicing of yeast VMA1-derived endonuclease via thiazolidine intermediates. J Synchrotron Radiat. 2004;11(Pt 1):109–112. doi: 10.1107/s0909049503023495. [DOI] [PubMed] [Google Scholar]
- 19.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 20.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 21.Suel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 22.Russ WP, Lowery DM, Mishra P, Yaffe MB, Ranganathan R. Natural-like function in artificial WW domains. Nature. 2005;437:579–583. doi: 10.1038/nature03990. [DOI] [PubMed] [Google Scholar]
- 23.Socolich M, et al. Evolutionary information for specifying a protein fold. Nature. 2005;437:512–518. doi: 10.1038/nature03991. [DOI] [PubMed] [Google Scholar]
- 24.Perler FB. InBase: The Intein Database. Nucleic Acids Res. 2002;30:383–384. doi: 10.1093/nar/30.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wood DW, Wu W, Belfort G, Derbyshire V, Belfort M. A genetic system yields self-cleaving inteins for bioseparations. Nat Biotechnol. 1999;17:889–892. doi: 10.1038/12879. [DOI] [PubMed] [Google Scholar]
- 26.Adam E, Perler FB. Development of a positive genetic selection system for inhibition of protein splicing using mycobacterial inteins in Escherichia coli DNA gyrase subunit A. J Mol Microbiol Biotechnol. 2002;4:479–487. [PubMed] [Google Scholar]
- 27.Lew BM, Paulus H. An in vivo screening system against protein splicing useful for the isolation of non-splicing mutants or inhibitors of the RecA intein of Mycobacterium tuberculosis. Gene. 2002;282:169–177. doi: 10.1016/s0378-1119(01)00836-8. [DOI] [PubMed] [Google Scholar]
- 28.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 29.Krogh A, Brown M, Mian IS, Sjolander K, Haussler D. Hidden Markov models in computational biology. Applications to protein modeling. J Mol Biol. 1994;235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- 30.DeLano WL. PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}