

Abstract
Scaffold proteins contribute to the spatiotemporal control of MAPK signaling and KSR1 is an ERK cascade scaffold that localizes to the plasma membrane in response to growth factor treatment. To better understand the molecular mechanisms of KSR1 function, we examined the interaction of KSR1 with each of the ERK cascade components, Raf, MEK, and ERK. Here, we identify a hydrophobic motif within the proline-rich sequence (PRS) of MEK1 and MEK2 that is required for constitutive binding to KSR1 and find that MEK binding and residues in the KSR1 CA1 region enable KSR1 to form a ternary complex with B-Raf and MEK following growth factor treatment that enhances MEK activation. We also find that docking of active ERK to the KSR1 scaffold allows ERK to phosphorylate KSR1 and B-Raf on feedback S/TP sites. Strikingly, feedback phosphorylation of KSR1 and B-Raf promote their dissociation and result in the release of KSR1 from the plasma membrane. Together, these findings provide unique insight into the signaling dynamics of the KSR1 scaffold and reveal that through regulated interactions with Raf and ERK, KSR1 acts to both potentiate and attenuate ERK cascade activation, thus regulating the intensity and duration of ERK cascade signaling emanating from the plasma membrane during growth factor signaling.
Keywords: ERK cascade, protein scaffolds, signal tranduction
Cells respond to a diverse array of signal inputs and ensuring that the correct biological response is achieved requires that the appropriate signaling pathways are engaged and that mechanisms are in place to regulate the strength, duration, and location of pathway activation. Receptor tyrosine kinases (RTKs) serve as entry points for many extracellular cues, relaying signals that control cell proliferation and differentiation, promote cell migration and survival, and modulate cellular metabolism. RTK pathways are intricate signaling networks, containing modules of multiprotein complexes that assemble at various intracellular compartments to process, integrate, and transmit information that will ultimately specify a particular biological response (1).
An essential effector cascade required for most RTK function is the ERK cascade composed of the Raf, MEK, and ERK kinases (2). The critical link that allows RTKs to signal to ERK is the RasGTPase. Through the recruitment of guanine nucleotide exchange factors to the cell surface, activated RTKs stimulate the GTP-loading of Ras, which allows Ras to interact directly with its target effectors, one of which is the Raf kinase family. Binding to Ras recruits the cytosolic Raf kinases to the plasma membrane where they are activated, thus initiating the phosphorylation cascade that results in MEK and ERK activation.
In addition to the enzymatic components of the ERK cascade, protein scaffolds have also been found to play an important role in ERK cascade activation and signaling (3, 4). The Ste5 protein in yeast is the prototypical scaffold for MAPK signaling cascades, and studies characterizing Ste5 have revealed that by interacting with the kinase components of a particular MAPK cascade, a scaffold can increase the efficiency and specificity of MAPK activation. Moreover, scaffolds can regulate the localization of the cascade components, and as a result, can target signaling to specific cellular compartments and/or substrates (5, 6).
Several proteins with proposed scaffolding functions in the ERK cascade have been identified in metazoans, including members of the KSR family, MP1 IQGAP, Sef, and Paxillin (3,4). Of these, the KSR family has been most extensively studied. KSR proteins were initially identified as positive regulators of Ras pathway signaling through genetic screens performed in Drosophila melanogaster and Caenorhabditis elegans (7–9). Subsequently, the mammalian KSR1 protein has been shown to contribute to many aspects of Ras-dependent signal transduction including cell proliferation, adipogenesis, cell transformation, and survival signaling (10–13). KSR1 interacts with all of the ERK cascade kinases and the CK2 kinase, which can facilitate Raf kinase activation (14–16). The KSR1 scaffold contains the 5 conserved domains (CA1–5) characteristic of KSR proteins (9), and its intracellular localization is dynamic, residing in the cytosol of quiescent cells and translocating to the cell surface in response to RTK and Ras activation (17). Notably, of all of the ERK scaffolds engaged in mammalian RTK signaling pathways, KSR1 is the only one whose localization changes in response to signal activation.
To gain a better understanding of how KSR1 functions to regulate ERK cascade signaling, we analyzed the interaction of KSR1 with each of the ERK cascade kinases. Here, we find that in response to growth factor treatment and RTK activation, KSR1 first acts as a signal enhancer to potentiate MEK activation through ternary interactions with B-Raf and MEK. Subsequently, through the docking of activated ERK, KSR1 serves as a signal attenuator, facilitating the ERK-dependent feedback phosphorylation of KSR1 and B-Raf on sites that promote their dissociation and result in the release of KSR1 from the plasma membrane. These findings demonstrate an important role for KSR1 in the spatiotemporal regulation of ERK cascade activation.
Results
Identification of Residues in the MEK1/2 PRS That Mediate Binding to KSR1.
MEK1 and MEK2 contain a proline-rich sequence (PRS) located between kinase subdomains IX and X that is not found in other mammalian MAP kinase kinase (MKK) family members, but is conserved in MEK homologs from Xenopus, Drosophila, and C. elegans (Fig. 1A). Previous studies have shown that the PRS of MEK1 is required for the direct binding of MEK1 to MP1, an ERK scaffold that localizes to endosomes (18, 19). Therefore, we examined whether the PRS might also contribute to the direct binding of MEK1 to the KSR1 scaffold. KSR1−/− MEFs that stably express Pyo-tagged WT-KSR1 at close to endogenous levels (WT-KSR1 MEFs) were transfected with constructs encoding HA-tagged WT- or ΔPRS-MEK1 proteins, following which the interaction between KSR1 and MEK1 was examined. For comparison, binding of the MEK1 proteins to the MP1 scaffold was also evaluated in CCL39 cells. As shown in Fig. 1B, not only did deletion of the PRS disrupt binding of MEK1 to MP1, it also prevented binding between MEK1 and KSR1, indicating that the PRS is required for MEK1 to interact with both of these ERK scaffolds.
Fig. 1.

Identification of PRS residues in MEK1/2 required for KSR1 and B-Raf binding. (A) Alignment of PRS sequences from rat MEK1 and MEK2 and MEK homologs in Xenopus, Drosophila, and C. elegans. Human MKK6, which lacks the PRS, is also shown. ●, indicates conserved PRS residues examined in this study; *, indicates residues required for the MEK1/MP1 interaction. (B) WT-KSR1 MEFs were transfected with HA-WT- or ΔPRS-MEK1 (deletes residues 270–307) constructs. CCL39 cells were cotransfected with constructs encoding Flag-MP1 and HA-WT-, ΔPRS-, L274S-MEK1 or vector control. The KSR1 and MP1 scaffolds were immunoprecipitated and examined for MEK1 binding by immunoblot analysis. (C) Various HA-MEK1 or MEK2 proteins were expressed in WT-KSR1 MEFs, and the MEK/KSR1 and MEK/B-Raf interactions were detected by coimmunoprecipitation and immunoblot analysis.
Previous reports have found that the MP1 scaffold binds MEK1 but not MEK2 and that certain residues unique to the MEK1 PRS are required for the MEK1/MP1 interaction (20). In contrast, yeast 2-hybrid studies and biochemical analyses have demonstrated that the KSR1 scaffold interacts with both MEK1 and MEK2 (14, 17), suggesting that PRS residues common between MEK1 and MEK2 might contribute to KSR1 binding. Moreover, because the interaction with MEK is required for KSR function in flies and worms (21, 22), it might also be expected that residues involved in KSR1 binding would be present in the PRS of Drosophila and C. elegans MEK proteins. To investigate this possibility, we generated a panel of MEK1 mutants containing alanine substitutions at various conserved residues in the PRS and a mutant containing the L274S mutation, which has been shown to disrupt the MEK1/MP1 interaction (ref. 20, Fig. 1B). As shown in Fig. 1C, the L274S mutation did not perturb the MEK1/KSR1 interaction, nor did mutation of the conserved F275 or D303 residues. In contrast, mutation of two hydrophobic residues (M308A and I310A) at the 3′ end of the MEK1 PRS did disrupt binding to KSR1, as did mutation of the equivalent residues (M316A and I318A) in MEK2, identifying these residues (hereafter referred to as the MAI motif) as necessary for the binding of MEK1 and MEK2 to the KSR1 scaffold.
The MAI Motif Contributes to the MEK/B-Raf Interaction.
A previous study has also indicated that the PRS contributes to the interaction of the MEK proteins with members of the Raf kinase family (23). Consistent with this report, we found that deletion of the PRS disrupted binding of HA-tagged MEK1 to endogenous B-Raf in the WT-KSR1 MEFs, as did mutation of the MAI motifs in MEK1 and MEK2 (Fig. 1C). Mutation of the MEK1/2 MAI motifs also inhibited binding of B-Raf to the HA-tagged MEK proteins in MEFs lacking KSR1 and in those expressing a KSR1 mutant defective in MEK binding (C809Y-KSR1) (supporting information (SI) Fig. S1A), indicating that when exogenously expressed, the MEK proteins can interact with B-Raf in the absence of KSR1 and that the MAI motif is critical for this interaction. In regard to the binding of the MEKs to C-Raf, we were unable to detect an interaction between endogenous C-Raf and the HA-tagged MEK proteins in the WT-KSR1 MEFs. However, when both C-Raf and the MEKs were transiently overexpressed (Fig. S1C), Flag-tagged C-Raf was found to interact with HA-tagged MEK1 and MEK2 in a manner dependent on the MEK MAI motif (Fig. S1C).
For all KSR family members mutation of a conserved cysteine residue in the KSR CA5 domain (C809 in KSR1) disrupts the MEK/KSR interaction (21, 22, 24). Even though the Raf kinases contain a cysteine residue at a position analogous to the C809 residue of KSR1, we found that in cotransfection experiments, Flag-tagged B-Raf and C-Raf proteins containing a mutation in this conserved cysteine (C696Y-B-Raf and C588Y-C-Raf) were still competent to bind HA-tagged MEK proteins (Fig. S1 B and C). Together, these data indicate that although the MEK1/2 MAI motif is critical for both the KSR1/MEK and Raf/MEK interactions, there are differences in the determinants by which KSR1 and Raf recognize MEK.
KSR1 Facilitates the MEK/B-Raf Interaction.
To further investigate the MEK/KSR1 and MEK/Raf interactions, we compared the ability of exogenously expressed KSR1, B-Raf, and C-Raf to bind endogenous MEK in cycling CCL39 cells. Using proteins that all contain the Flag-epitope tag, we found that even when KSR1 was expressed at levels equivalent to or less than either Raf protein, the amount of endogenous MEK bound to Flag-KSR1 was much greater than that observed for Flag-B-Raf and that little-to-no binding was detected with Flag-C-Raf (Fig. 2A). As evidence that the Flag-epitope tag was not influencing the binding interactions, similar results were observed for proteins containing the Pyo-epitope tag (Fig. 2A). These findings indicate that MEK preferentially associates with the KSR1 scaffold, and taken together with the preceding observations, suggest that binding of MEK to the Raf proteins is significantly influenced by protein expression levels.
Fig. 2.
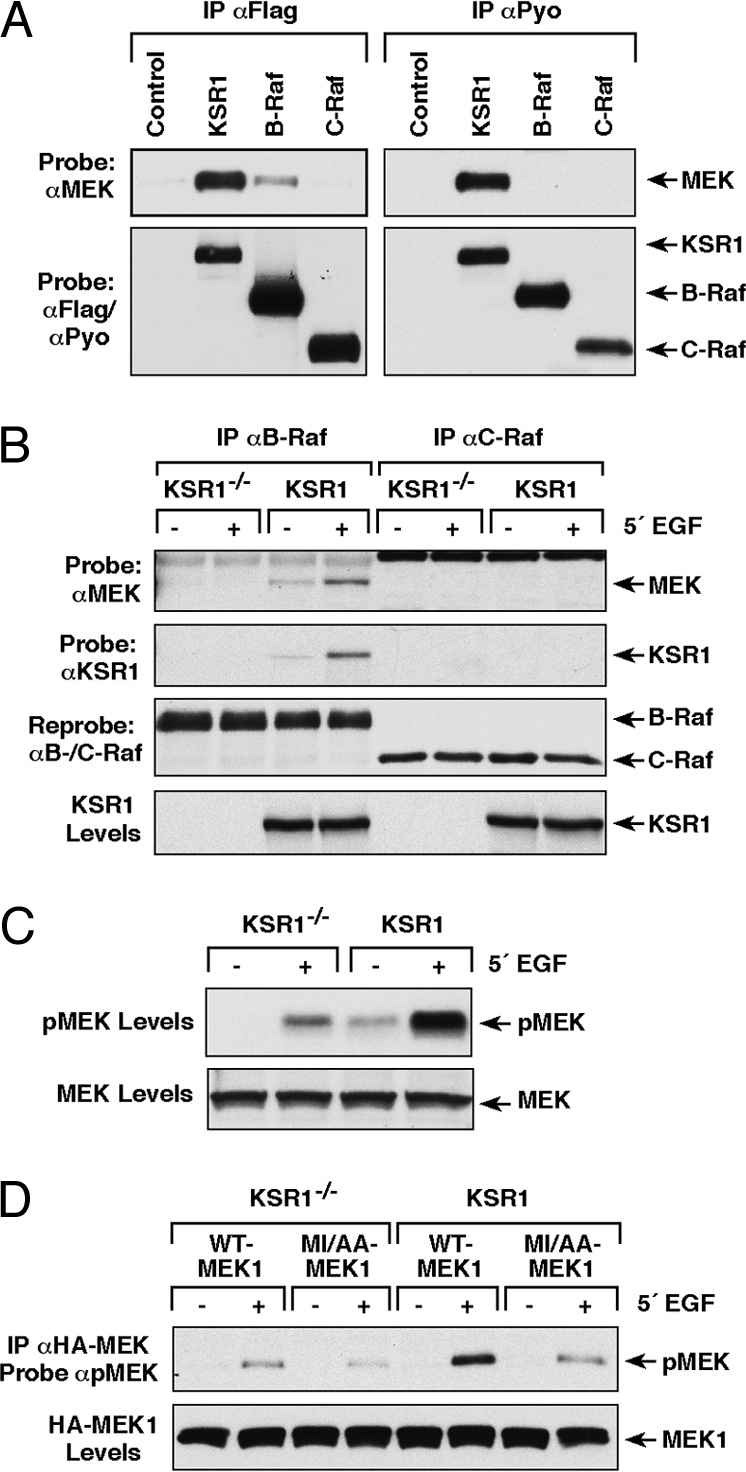
KSR1 facilitates the MEK/B-Raf interaction. (A) CCL39 cells were transfected with plasmids encoding Flag- or Pyo-KSR1, B-Raf, and C-Raf. Flag or Pyo-tagged proteins were immunoprecipitated and the amount of endogenous MEK present in each complex was determined by immuoblot analysis. (B and C) KSR1−/− or WT-KSR1 MEFs were serum starved and treated with EGF. Endogenous B-Raf or C-Raf proteins were immunoprecipitated and examined for KSR1 and endogenous MEK binding (B). Lysates were examined for the phosphorylation of MEK on activating sites using pMEK antibodies (C). (D) KSR1−/− or WT-KSR1 MEFs expressing HA-WT- or MI/AA-MEK1 were serum starved and treated with EGF. HA-MEK1 proteins were immunoprecipitated and examined for phosphorylation on activating sites using pMEK antibodies.
The KSR1/MEK interaction is constitutive, and given that KSR1, like the Raf proteins, translocates to the plasma membrane in response to RTK and Ras activation (14, 15, 25, 26), we next examined whether KSR1 might facilitate the Raf/MEK interaction under endogenous signaling conditions. For these experiments, we assessed the ability of endogenous Raf proteins to interact with endogenous MEK in KSR1−/− or WT-KSR1 MEFs (Fig. 2B). In WT-KSR1 MEFs, binding of endogenous MEK to endogenous B-Raf was observed; however, binding to endogenous C-Raf was not detected. The interaction between the endogenous MEK and B-Raf proteins increased upon growth factor treatment and the increased binding correlated with the growth factor-induced association of B-Raf and KSR1. Notably, in cells lacking KSR1, we were unable to detect an interaction between the endogenous MEK and B-Raf proteins by coimmunoprecipitation. When the activation state of endogenous MEK was examined, we found that the Raf-dependent phosphorylation of MEK on activating sites was significantly reduced in cells lacking KSR1 (Fig. 2C). These data indicate that under endogenous signaling conditions, KSR1 facilitates the association of MEK and B-Raf and thus promotes MEK activation.
Next, to evaluate whether the MEK MAI motif is required for the positive effect that KSR1 has on MEK activation, HA-tagged WT- or MI/AA-MEK1 proteins were expressed in the KSR1−/− and WT-KSR1 MEFs, following which their activation state was examined. In growth factor treated-WT-KSR1 MEFs, the activation state of the MI/AA mutant was significantly reduced in comparison to WT-MEK1 and was comparable to that observed for both MEK proteins in KSR1−/− MEFs (Fig. 2D). Consistent with their reduced activation state, the MI/AA mutant also exhibited reduced kinase activity when compared to WT-MEK in immune complex kinase assays using ERK as an exogenous substrate (Fig. S2). These findings demonstrate that the MAI motif plays a critical role in mediating the interactions required for KSR1 to facilitate MEK activation.
Complex Formation Between KSR1, MEK, and B-Raf.
As observed above, the association of B-Raf and KSR1 increases upon growth factor stimulation; however, the requirements for this interaction have not been well characterized in mammalian cells. To address this issue, various KSR1 mutant proteins were stably expressed in the KSR1−/− MEFs at close to physiological levels, following which the mutants were examined for their ability to interact with endogenous B-Raf (Fig. 3A). As controls, the mutant proteins were also evaluated for binding to other known KSR1 interactors and for their localization to the plasma membrane following growth factor treatment (Fig. 3A, Table S1). Consistent with studies examining the Raf/KSR interaction in Drosophila (22), we found that mutation of conserved leucine and arginine residues in the CA1 region of KSR1 (L56G and R57S) disrupted binding of mammalian KSR1 to endogenous B-Raf. In addition, we found that N′- and C809Y-KSR1 proteins unable to interact with MEK were also defective in B-Raf binding despite having intact CA1 regions. However, as demonstrated by analysis of L56G/R57S- and C′-KSR1, binding to MEK alone was not sufficient for the B-Raf/KSR1 interaction. These findings reveal that both MEK binding and an intact CA1 domain are required for KSR1 to stably interact with B-Raf and further support a model whereby ternary interactions between KSR1, MEK, and B-Raf facilitate signal transmission from B-Raf to MEK.
Fig. 3.

Binding of KSR1 to MEK and ERK regulates the KSR1/B-Raf interaction. (A) KSR1−/− MEFs stably expressing the indicated KSR1 proteins were serum starved and treated with EGF. Pyo-KSR1 proteins were immunoprecipitated and examined for binding of endogenous B-Raf and MEK proteins. Tyrosine phosphorylation of EGFr indicates activation of EGFr pathway signaling. (B) Serum-starved WT- or AxAP-KSR1 MEFs were treated as indicated using EGF and U0126. Pyo-KSR1 proteins were immunoprecipitated and examined for binding of endogenous B-Raf, MEK, and pERK. Lysates were probed for pERK levels to confirm that U0126 blocked ERK activation.
KSR1-Bound ERK Disrupts KSR1 Scaffold Complexes via Feedback Phosphorylation.
An interesting observation from the KSR1 mutant analysis was that the KSR1 protein containing mutations in the DEF docking motif (27) for activated ERK (AxAP-KSR1), exhibited increased binding to endogenous B-Raf (Fig. 3A, Table S1). When the B-Raf/KSR1 interaction was examined further in WT-KSR1 MEFs, we found that the association of B-Raf with WT-KSR1 was transient, peaking at 5 min of growth factor treatment and declining to almost basal levels at the 20-min time point (Fig. 3B). In comparison, complex formation between AxAP-KSR1 and B-Raf was elevated and prolonged, suggesting that the docking of activated ERK to the KSR1 scaffold negatively regulates the interaction between KSR1 and B-Raf. In support of this model, blocking ERK activation by treating WT-KSR1 MEFs with the MEK inhibitor U0126 also led to an increased and prolonged association of B-Raf and WT-KSR1 (Fig. 3B). Of note, the constitutive MEK/KSR1 interaction was not altered by growth factor treatment and was comparable for both WT- and AxAP-KSR1 (Fig. 3B).
Both KSR1 and B-Raf have been shown to be substrates of activated ERK (25, 28–30), and to begin to investigate why the docking of activated ERK to the FxFP site influences the KSR1/B-Raf interaction, we performed metabolic labeling experiments and used phosphoS/TP antibodies to characterize the phosphorylation state of KSR1 and B-Raf in WT-KSR1 MEFs. HPLC and mass spectrometry analysis of the labeled WT-KSR1 and endogenous B-Raf proteins revealed that growth factor treatment for 20 min induced the phosphorylation of 4 S/TP sites on each protein: T260, T274, S320, and S443 for KSR1 and S151, T401, S750, and S753 for B-Raf (Fig. S3A). As evidence that these residues are ERK targets, purified active ERK was found to phosphorylate purified KSR1 and a catalytically inactive B-Raf protein (KD-B-Raf) on their respective S/TP sites in vitro (Fig. S3A) and the phosphorylation of these sites in vivo could be blocked by U0126 treatment (Fig. 4A, Fig. S3A). Thus, these findings raise the possibility that ERK-dependent feedback phosphorylation might be responsible for the negative effect that activated ERK has on B-Raf/KSR1 complex stability.
Fig. 4.

KSR1-bound ERK disrupts KSR1 signaling complexes through feedback phosphorylation. (A) Indicated on the left are the ERK-dependent feedback phosphorylation sites identified for KSR1 and B-Raf in Fig. S3A. Serum-starved KSR1−/− MEFs expressing WT- or feedback phosphorylation defective KSR1 and B-Raf proteins (FBm) were treated as indicated using EGF and U0126. B-Raf and KSR1 proteins were immunoprecipitated and probed for pS/TP levels. (B) Serum-starved KSR1−/−, WT-, or AxAP-KSR1 MEFs were treated as indicated with EGF. KSR1 or endogenous B-Raf proteins were immunoprecipated and probed for pS/TP levels. (C) Serum-starved MEFs from WT mice were treated as indicated with EGF. Endogenous KSR1 proteins were immuoprecipiated and examined for binding of endogenous MEK, B-Raf, and pERK (Left panel). Endogenous KSR1 and B-Raf were immuoprecipitated and probed for pS/TP levels (Right panel). (D) Serum-starved KSR1−/− MEFs stably coexpressing WT-KSR1 and WT-B-Raf or FBm-KSR1 and FBm-B-Raf were treated as indicated with EGF. Flag-B-Raf proteins were immunoprecipitated and examined for binding of Pyo-KSR1. Lysates were also examined for activated pMEK. (E) The localization of Pyo-tagged WT-KSR1 and FBm-KSR1 was determined by cell fractionation (Top) and immuofluorescent staining (Bottom). Percentage of cells expressing membrane-localized KSR1 represents 2 independent experiments.
To further investigate this idea, we examined the phosphorylation state of B-Raf and KSR1 in MEFs that lack KSR1 or express WT- or AxAP-KSR1 using the phosphoS/TP antibodies (Fig. 4B). In WT-KSR1 MEFs, phosphorylation of B-Raf on S/TP sites reached a maximal level at 10 min of growth factor treatment and was sustained following 20 min of treatment, whereas in KSR1−/− MEFs, phosphorylation was slightly elevated at the 10-min time point and did not reach maximal levels until 20 min. Interestingly, at 10 min of treatment, phosphorylation of B-Raf in AxAP-KSR1 MEFs was the same as that observed in KSR1−/− MEFs and was significantly decreased in comparison to cells expressing WT-KSR1. Phosphorylation of WT-KSR1 on S/TP sites was also found to peak at 10 min of EGF treatment, and in comparison to WT-KSR1, the phosphorylation of AxAP-KSR1 on S/TP sites was greatly reduced, suggesting that binding of activated ERK to the KSR1 scaffold facilitates the feedback phosphorylation of KSR1 and B-Raf. To further validate these findings, we used MEFs isolated from WT mice to examine the S/TP phosphorylation state of endogenous KSR1 and to monitor its interaction with endogenous B-Raf. Consistent with results observed in WT-KSR1 MEFs, we found that binding between endogenous KSR1 and endogenous B-Raf was inducible and transient (Fig. 4C). Moreover, S/TP phosphorylation of both the endogenous KSR1 and B-Raf proteins peaked following 10 min of EGF treatment (Fig. 4C), confirming the feedback phosphorylation of these proteins under physiological signaling conditions.
Next, to address the effect of feedback phosphorylation on KSR1/B-Raf complex formation, we generated MEF lines that stably coexpress Pyo-tagged WT-KSR1 and Flag-tagged WT-B-Raf or mutant proteins in which the ERK-dependent S/TP sites have been mutated to alanine (FBm-KSR1 and FBm-B-Raf). The FBm proteins were no longer recognized by the phospho S/TP antibody in growth factor treated cells (Fig. 4A), and as shown in Fig. 4D, coexpression of FBm-KSR1 and FBm-B-Raf resulted in an increased and prolonged B-Raf/KSR1 interaction similar to that observed in AxAP-KSR1 MEFs. Moreover, MEK activation was elevated and sustained in cells where feedback phosphorylation of B-Raf and KSR1 was prevented (Fig. 4D). Because previous studies have indicated that feedback phosphorylation of B-Raf on the S750 and T753 sites has a negative regulatory effect on B-Raf signaling in PC12 cells (29), we further examined the effect of specific B-Raf phosphorylation sites on MEK activation. As shown in Fig. S3B, MEK activation following 5 min of EGF treatment was elevated in WT-KSR1 MEFs expressing either S151A-, S750A/T753A-, or FBm-B-Raf; however, sustained MEK activation and sustained interaction with KSR1 was observed only in cells expressing FBm-B-Raf, suggesting that phosphorylation of all of the feedback sites contributes to the disruption of the KSR1/B-Raf complex. Finally, when we examined the effect of feedback phosphorylation on the membrane localization of KSR1, we found that in both biochemical fractionation assays and immunoflourescent staining experiments, the membrane localization of FBm-KSR1 was increased and prolonged in comparison to WT-KSR1 (Fig. 4E). Together these data indicate that ERK-dependent feedback phosphorylation disrupts the association of KSR1 and B-Raf and promotes the release of KSR1 from the plasma membrane, thus downregulating signal transmission.
Discussion
To further understand the signaling dynamics of the KSR1 scaffold complex, we began our study by investigating the interaction of KSR1 with each of the ERK cascade components. Previous studies have shown that the KSR1/MEK interaction is constitutive (14, 15, 21, 22, 25), and although residues of KSR1 required for binding to MEK were identified in the genetic screens that led to the discovery of KSR1 (8, 21), the sequence determinants on MEK for binding to KSR1 have remained unknown. Through mutational analysis, we have identified a hydrophobic motif (MAI motif) in the PRS of MEK1 and MEK2 that is required for KSR1 binding. Interestingly, KSR1 now represents the third protein family that uses the PRS as an interaction site with MEK. The PRS has previously been shown to be required for binding to the MP1 scaffold and the Raf kinases (18, 23).
The PRS is found in all MEK homologs but is not present in other MKK family members, and our findings highlight the importance of the PRS in regulating MEK function, serving as a docking site for MEK-specific upstream activators and scaffold regulators. MP1 is known to associate exclusively with MEK1 and binding is mediated by residues unique to MEK1 that are located near the 5′ end of the PRS. In contrast, both KSR1 and the Raf kinases interact with MEK1 and MEK2, and we find that these interactions are dependent on an intact MAI motif located at the 3′ end of the PRS. Nevertheless, despite the similarities in the MEK residues required for the interactions with KSR1 and Raf, there appear to be differences in the determinants by which KSR1 and the Rafs recognize MEK. For example, mutation of a cysteine residue conserved in the KSR and Raf family members has no effect on the MEK/Raf interaction (Fig. S1 B and C), but completely abolishes the ability of all KSR protein to bind MEK (21, 22, 24). In addition, when these interactions were evaluated in cells using proteins that contain the same epitope tag, we found that endogenous MEK bound preferentially to the KSR1 scaffold, indicating that there are other residues or factors that play a role in determining the relative affinity of MEK for KSR1 and the Raf proteins.
An important observation from our experiments was that the ability to detect the MEK/Raf interaction was significantly impacted by protein expression levels. For example, to detect the MEK/C-Raf interaction by coimmunopreciptation, we found that both proteins needed to be exogenously overexpressed. In regard to the MEK/B-Raf interaction, we found that when MEK was exogenously overexpressed, binding to endogenous B-Raf was observed and was not dependent on KSR1. However, under endogenous expression conditions, binding of MEK to B-Raf was only detected in cells expressing KSR1. These findings highlight the concern of relying solely on overexpression studies to draw conclusions regarding these interactions and suggest that KSR1 plays an important role in facilitating the MEK/Raf interaction under endogenous signaling conditions. In support of this model, we found that binding between endogenous MEK and B-Raf increased in response to growth factor treatment and that the increased binding correlated with the induced association of KSR1 and B-Raf. Interestingly, when we examined the sequence determinants needed for KSR1 to interact with B-Raf, we found that MEK binding and residues in the CA1 region were required for KSR1 to stably associate with B-Raf. Although the CA1 region has also been reported to contribute to the Raf/KSR interaction in Drosophila, it is currently unknown whether the CA1 region contacts the Raf proteins directly or interacts with another molecule that is critical for the interaction. The requirement for MEK binding, however, indicates that growth factor stimulation induces the formation of a stable protein complex that contains KSR1, MEK, and B-Raf. Moreover, the finding that under endogenous signaling conditions, MEK activation is increased ≈10 fold in cells expressing KSR1 supports a model in which ternary interactions between KSR1, MEK, and B-Raf facilitate signal transmission from B-Raf to MEK, resulting in increased MEK activation.
Strikingly, our findings also reveal a role for KSR1 in attenuating signal transmission from B-Raf to MEK through the docking of activated ERK. The binding of ERK to the KSR1 scaffold is greatly increased by growth factor treatment and is mediated primarily by a DEF motif (FxFP) located in the KSR1 CA4 region (25, 27). Structural studies examining the binding of ERK to known docking motifs have revealed that the binding site on ERK for the DEF motif is exposed upon phosphorylation of the ERK activation loop, and as a result, DEF motifs preferentially bind activated ERK (31). Here, we find that WT-KSR1 but not AxAP-KSR1, binds phosphorylated ERK in a growth factor inducible manner, indicating that the DEF motif localizes activated ERK to the KSR1 scaffold complex. Both KSR1 and B-Raf have been reported to be substrates of activated ERK, and we find that in WT-KSR1 MEFs, growth factor treatment induces the phosphorylation of KSR1 and B-Raf on 4 S/TP sites (T260, T274, S320, and S443 for KSR1 and S151, T401, S750, and S753 for B-Raf). Moreover, we find that the docking of activated ERK to the KSR1 complex accelerates the phosphorylation of these sites in response to growth factor treatment. As a functional consequence, phosphorylation of B-Raf and KSR1 on the S/TP sites disrupts the B-Raf/KSR1 interaction and promotes the release of KSR1 from the plasma membrane. Thus, the docking of activated ERK downregulates KSR1's scaffold activity and its ability to potentiate signal transmission from B-Raf to MEK.
In conclusion, our findings reveal unique insight into signaling dynamics of the KSR1 scaffold complex. From the results presented here and previous studies, it is clear that KSR1 functions in multiple ways to modulate ERK cascade signaling (Fig. 5). Not only does KSR1 colocalize the kinase components of the cascade and thus increase the efficiency and specificity of cascade signaling, it also targets cascade signaling to a specific subcellular location—the plasma membrane. In addition, we find that KSR1 can modulate the intensity and duration of ERK cascade signaling through its interactions with the core kinase components of the ERK cascade. These findings are consistent with recent studies of engineered MAPK scaffolds in yeast and demonstrate the importance of scaffold proteins in regulating the dynamics of MAPK signaling (32).
Fig. 5.

KSR1 regulates the intensity and duration of ERK cascade activation. In quiescent cells, KSR1 prevents improper ERK cascade activation by sequestering MEK away from Raf (A). In signaling cells, KSR1 first potentiates signal transmission from Raf to MEK by facilitating the Raf/MEK interaction (B) and then attenuates signaling by docking activated ERK, thus facilitating the disruption of the KSR1 scaffold complex via feedback phosphorylation (C and D).
Methods
Cell Culture, Transfection, and Generation of Cell Lines.
MEF, HeLa, NIH 3T3, and CCL39 cells were cultured in DMEM supplemented with 10% FBS. Cell transfections were performed using the FuGENE 6 reagent (Invitrogen). To generate stable cell lines, KSR1−/− MEFs (11) were infected with recombinant pBabe-puro retroviruses encoding the indicated KSR1 or B-Raf proteins and selected using puromycin (2.5 μg/mL).
Serum-Starvation, EGF Stimulation, and U0126 Inhibition.
All cells were serum starved for 16–18 h in serum-free DMEM before stimulation with 100 ng/mL EGF (Invitrogen). For U0126 inhibition, cells were treated with 10 μM U0126 (Cell Signaling Technology) for 1 h before and during EGF stimulation. Note, in all experiments, activation of the EGF receptor (EGFr)/Ras/ERK pathway was confirmed by monitoring cell lysates for either pY-EGFr or for activated pERK levels by immunoblot analysis. Inhibition of MEK activity by U0126 treatment was confirmed by monitoring cell lysates for activated pERK levels.
Coimmunoprecipiation Assays.
Cells were lysed in Nonidet P-40 lysis buffer [20 mm Tris (pH 8.0), 137 mM NaCl, 10% glycerol, 1% Nonidet P-40, 0.15 units/mL aprotinin, 1 mM PMSF, 20 μM leupeptin, 5 mM sodium vanadate], and lysates were clarified by centrifugation. Lysates were incubated with the appropriate antibody plus protein G Sepharose beads or with anti-Flag M2 affinity agarose (Sigma-Aldrich) for 3 h at 4 °C. Immune complexes were washed and analyzed by immunoblotting.
Immunofluoresence and Cell Fractionation.
Pyo-WT- and FBm-KSR1 proteins were transiently expressed in NIH 3T3 cells grown on glass coverslips. Cells were serum starved for 16–18 h, treated with PDGF (50 ng/mL) for 20 min, and the subcellular localization of KSR1 proteins determined by immunofluorescent staining (26). Isolation of cellular membranes from WT- and FBm-KSR1 MEFs was performed as previously described (33).
See SI Methods for descriptions of additional experimental procedures and reagents.
Supplementary Material
Acknowledgments.
We thank Dr. Michael Weber for generously providing valuable reagents and Suzanne Specht for excellent technical assistance. This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0901590106/DCSupplemental.
References
- 1.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26:3113–3121. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- 2.Shaul YD, Seger R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 4.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 5.Brown MD, Sacks DB. Compartmentalised MAPK pathways. Handb Exp Pharmacol. 2008;186:205–235. doi: 10.1007/978-3-540-72843-6_9. [DOI] [PubMed] [Google Scholar]
- 6.Casar B, et al. Ras subcellular localization defines ERK1/2 substrate specificity through distinct utilization of scaffold proteins. Mol Cell Biol. 2008;29:1338–1353. doi: 10.1128/MCB.01359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kornfeld K, Hom DB, Horvitz HR. The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell. 1995;83:903–913. doi: 10.1016/0092-8674(95)90206-6. [DOI] [PubMed] [Google Scholar]
- 8.Sundaram M, Han M. The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell. 1995;83:889–901. doi: 10.1016/0092-8674(95)90205-8. [DOI] [PubMed] [Google Scholar]
- 9.Therrien M, et al. KSR, a novel protein kinase required for RAS signal transduction. Cell. 1995;83:879–888. doi: 10.1016/0092-8674(95)90204-x. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen A, et al. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol Cell Biol. 2002;22:3035–3045. doi: 10.1128/MCB.22.9.3035-3045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kortum RL, Lewis RE. The molecular scaffold KSR1 regulates the proliferative and oncogenic potential of cells. Mol Cell Biol. 2004;24:4407–4416. doi: 10.1128/MCB.24.10.4407-4416.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kortum RL, et al. The molecular scaffold kinase suppressor of Ras 1 (KSR1) regulates adipogenesis. Mol Cell Biol. 2005;25:7592–7604. doi: 10.1128/MCB.25.17.7592-7604.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKay MM, Morrison DK. Caspase-dependent cleavage disrupts the ERK cascade scaffolding function of KSR1. J Biol Chem. 2007;282:26225–26234. doi: 10.1074/jbc.M702692200. [DOI] [PubMed] [Google Scholar]
- 14.Denouel-Galy A, et al. Murine Ksr interacts with MEK and inhibits Ras-induced transformation. Curr Biol. 1998;8:46–55. doi: 10.1016/s0960-9822(98)70019-3. [DOI] [PubMed] [Google Scholar]
- 15.Yu W, Fantl WJ, Harrowe G, Williams LT. Regulation of the MAP kinase pathway by mammalian Ksr through direct interaction with MEK and ERK. Curr Biol. 1998;8:56–64. doi: 10.1016/s0960-9822(98)70020-x. [DOI] [PubMed] [Google Scholar]
- 16.Ritt DA, et al. CK2 Is a component of the KSR1 scaffold complex that contributes to Raf kinase activation. Curr Biol. 2007;17:179–184. doi: 10.1016/j.cub.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 17.Ory S, et al. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14–3-3 binding sites. Curr Biol. 2003;13:1356–1364. doi: 10.1016/s0960-9822(03)00535-9. [DOI] [PubMed] [Google Scholar]
- 18.Schaeffer HJ, et al. MP1: A MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998;281:1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- 19.Teis D, Wunderlich W, Huber LA. Localization of the MP1-MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev Cell. 2002;3:803–814. doi: 10.1016/s1534-5807(02)00364-7. [DOI] [PubMed] [Google Scholar]
- 20.Pullikuth A, McKinnon E, Schaeffer HJ, Catling AD. The MEK1 scaffolding protein MP1 regulates cell spreading by integrating PAK1 and Rho signals. Mol Cell Biol. 2005;25:5119–5133. doi: 10.1128/MCB.25.12.5119-5133.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stewart S, et al. Kinase suppressor of Ras forms a multiprotein signaling complex and modulates MEK localization. Mol Cell Biol. 1999;19:5523–5534. doi: 10.1128/mcb.19.8.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy F, et al. KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev. 2002;16:427–438. doi: 10.1101/gad.962902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Catling AD, et al. A proline-rich sequence unique to MEK1 and MEK2 is required for raf binding and regulates MEK function. Mol Cell Biol. 1995;15:5214–5225. doi: 10.1128/mcb.15.10.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller J, et al. Identification of B-KSR1, a novel brain-specific isoform of KSR1 that functions in neuronal signaling. Mol Cell Biol. 2000;20:5529–5539. doi: 10.1128/mcb.20.15.5529-5539.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cacace AM, et al. Identification of constitutive and ras-inducible phosphorylation sites of KSR: Implications for 14–3-3 binding, mitogen-activated protein kinase binding, and KSR overexpression. Mol Cell Biol. 1999;19:229–240. doi: 10.1128/mcb.19.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller J, et al. C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol Cell. 2001;8:983–993. doi: 10.1016/s1097-2765(01)00383-5. [DOI] [PubMed] [Google Scholar]
- 27.Jacobs D, et al. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 1999;13:163–175. [PMC free article] [PubMed] [Google Scholar]
- 28.Volle DJ, et al. Phosphorylation of the kinase suppressor of ras by associated kinases. Biochemistry. 1999;38:5130–5137. doi: 10.1021/bi983050d. [DOI] [PubMed] [Google Scholar]
- 29.Brummer T, Naegele H, Reth M, Misawa Y. Identification of novel ERK-mediated feedback phosphorylation sites at the C-terminus of B-Raf. Oncogene. 2003;22:8823–8834. doi: 10.1038/sj.onc.1207185. [DOI] [PubMed] [Google Scholar]
- 30.Wissing J, et al. Proteomics analysis of protein kinases by target class-selective prefractionation and tandem mass spectrometry. Mol Cell Proteomics. 2007;6:537–547. doi: 10.1074/mcp.T600062-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Lee T, et al. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell. 2004;14:43–55. doi: 10.1016/s1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- 32.Bashor CJ, Helman NC, Yan S, Lim WA. Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science. 2008;319:1539–1543. doi: 10.1126/science.1151153. [DOI] [PubMed] [Google Scholar]
- 33.Stokoe D, McCormick F. Activation of c-Raf-1 by Ras and Src through different mechanisms: Activation in vivo and in vitro. EMBO J. 1997;16:2384–2396. doi: 10.1093/emboj/16.9.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.