

Abstract
Myofibroblasts are contractile cells that are characterized by the expression of α-smooth muscle actin and mediate the closure of wounds and the formation of collagen-rich scars. Their presence in organs such as lungs, liver, and kidney has long been established as a marker of progressive fibrosis. The transforming growth factor beta1-driven differentiation of fibroblasts is a major source of myofibroblasts, and recent data have shown that hyaluronan is a major modulator of this process. This study examines this differentiation mechanism in more detail. Transforming growth factor beta1-dependent differentiation to the myofibroblastic phenotype was antagonized by the inhibition of hyaluronan synthesis, confirming that hyaluronan was necessary for differentiation. This response, however, was not reproduced by simply adding hyaluronan to fibroblasts, as the results implicated hyaladherins, as well as the macromolecular assembly of de novo hyaluronan, as essential in this process. We previously suggested that there is a relocalization of lipid-raft components during myofibroblastic differentiation. The present study demonstrates that the hyaluronan receptor CD44, the hyaluronidase HYAL 2, and the transforming growth factor beta1-receptor ALK5 all relocalized from raft to non-raft locations, which was reversed by the addition of exogenous hyaluronan. These data highlight a role for endogenous hyaluronan in the mediation of myofibroblastic differentiation. While hyaluronan synthesis was both essential and necessary for differentiation, exogenously provided hyaluronan antagonized differentiation, underscoring a pathological role for hyaluronan in such cell fate processes.
Fibrosis can be considered disordered wound healing in which there is progression rather than resolution of scarring. The excessive accumulation of extracellular matrix, causing the disruption of normal tissue structure and function characteristic of fibrosis is mediated by a specific phenotype of fibroblast—the myofibroblast. These are contractile cells, characterized by the expression of α-smooth muscle actin (α-SMA).1 They mediate closure of wounds and the formation of collagen-rich scars, and their presence in organs such as lungs, liver, and kidney has long been established as a marker of progressive disease.2,3 The cytokine transforming growth factor-β1 (TGFβ1) is recognized as a mediator of both wound healing and fibrotic progression.3,4,5,6,7,8,9 In addition, to its direct effect on extracellular matrix turnover, it up-regulates α-SMA in fibroblasts both in vitro and in vivo and drives fibroblast to myofibroblast differentiation.3,10,11
As a consequence of myofibroblastic differentiation, recent studies12 described the accumulation of hyaluronan (HA) in the pericellular matrix surrounding the differentiated cells. HA is a ubiquitous connective tissue glycosaminoglycan synthesized by HA synthase (HAS) enzymes of which three vertebrate genes have been isolated and characterized: HAS1, HAS2, and HAS3.13,14 In normal tissues, HA has a role in maintaining extracellular matrix stability and tissue hydration. The increased expression of HA and its cell-surface receptor (CD44), however, have been detected in numerous fibrotic conditions associated with organ dysfunction.15,16,17,18,19 HA normally exists as a high molecular weight molecule that is involved in homeostasis, matrix stability, and tissue hydration. It plays a major role in regulating cell adhesion, migration, differentiation, and proliferation,15,20,21,22,23,24,25,26,27,28 and therefore is strongly implicated in tumor growth and metastasis, as well as in tissue remodeling and wound healing. HA is also involved in mediating cellular responses to TGFβ1. Epithelial cells, for example, have altered TGFβ1 signaling following HA interactions with its receptor, CD44.25,26
We have previously demonstrated that phenotypic conversion of fibroblasts to myofibroblasts is associated with major changes in the accumulation and metabolism of endogenous HA.12 Subsequently, this accumulation of HA has been shown to facilitate the TGFβ1-driven differentiation to myofibroblasts and the effects of TGFβ1 on proliferation.29,30 In the present report, the mechanisms involved in the effects of HA on TGFβ1-dependent responses were investigated and shown to be dependent on HA synthesis. Addition of exogenous HA to fibroblasts, however, antagonized cell responses to TGFβ1, suggesting that these effects were not happening at the level of TGFβ1-receptor. Furthermore, exogenous HA addition did not completely mimic synthesis of endogenous HA, strongly implicating HA assembly, rather than simply HA presence, in the control of TGFβ1-driven differentiation.
Materials and Methods
Reagents
All general reagents were purchased from Sigma-Aldrich, Poole, Dorset, UK, unless stated otherwise. For exogenous addition to the cells (Lot No. F1750762), hyaluronan, in the form of freeze dried white powder, was kindly provided by Denki Kagaku Kogyo, Kabushiki Kaisha, Japan, and was diluted and prepared as described previously.25
Culture of Human Lung Fibroblasts
Primary human lung fibroblasts (AG02262) were purchased from Coriell Cell Repositories (Coriell Institute for Medical Research, NJ). Cells were cultured in a 1:1 (v/v) mix of Dulbecco’s modified Eagle’s medium:F-12 containing 2 mmol/L l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin, supplemented with 10% (v/v) fetal calf serum (Biosera, East Sussex, UK). Cells (up to passage 10) were maintained at 37°C in a 5% CO2 humidified atmosphere. Before experimentation all cells were growth-arrested in serum-free medium for 48 hours to allow cell cycle synchronization.
Myofibroblastic Differentiation
Fibroblasts were differentiated to myofibroblasts by stimulation with TGFβ1 (10 ng/ml) for 72 hours and then maintained at 37°C in a 5% CO2 humidified atmosphere. Resultant myofibroblasts were then washed thoroughly with calcium/magnesium-free PBS, pH 7.3. All experiments, unless otherwise stated, were performed in serum-free medium to avoid any influence of serum factors.
4-Methyl umbelliferone31 and exogenous HA (prepared as described previously25,26 were added to fibroblasts at the same time as the TGFβ1. 4-Methyl umbelliferone was used at a final concentration of 0.5 mmol/L. Exogenous HA (MW 1.73 × 106 Da) was used at a final concentration of 25 μg/ml. The Alk-5 specific inhibitor, SB431542, was used to inhibit TGFβ1 signaling, and the Erk specific inhibitor, PD98059, was used to inhibit mitogen-activated kinase kinase signaling. Both, when used, were used at a final concentration of 10 μmol/L.
Determination of HA Concentration
Cells were grown to confluence in 35-mm dishes and the HA concentration in the cell culture supernatant was determined using a commercially available enzyme-linked HA binding protein assay, according to the manufacturer’s instructions (Hyaluronic Acid Test Kit; Corgenix, Petersborough, UK).
Metabolic Labeling of HA
To metabolically radiolabel HA, confluent monolayers of cells cultured in T75 culture flasks were incubated in serum-free medium containing 20 μCi/ml D-[3H]-glucosamine hydrochloride (GE Health care, Bucks, UK) for 24 hours. The medium was removed and cells washed with PBS. The medium and wash were combined to form the conditioned medium extract. The conditioned medium extract was then treated with an equal volume of 200 μg/ml pronase type XIV (Sigma-Aldrich) for 24 hours. The remaining HA, surface/CD44-associated HA and matrix-associated HA, were removed together by incubating cells with 100 μg/ml pronase for 24 hours. This was decanted and designated the cellular extract.
Each extract was passed over diethylaminoethyl cellulose-Sephacel ion exchange columns (GE Health care) equilibrated with 8 mol/L urea buffer. The columns were washed with several bed volumes of 8 mol/L urea buffer, this removed any low molecular weight peptides and unincorporated radiolabel. Radiolabeled HA was eluted in 8 mol/L urea buffer containing 0.3 mol/L NaCl. Each extract was split into two, and the HA was precipitated by incubation with three times the volume of 1% (w/v) potassium acetate in 95% (v/v) ethanol in the presence of 50 μg/ml of unlabeled HA, heparin, and chondroitin sulfate as co-precipitants, overnight at 4°C. Then extracts were centrifuged at 500 × g for 10 minutes. Supernatants were decanted, the pellets washed in 6 ml of 95% (v/v) ethanol and then centrifuged again at 500 × g for 10 minutes. Following the removal of the supernatants, the first half of each extract was then analyzed by size-exclusion chromatography on a Sephacryl S-500 column (150 cm × 0.6 cm) (GE Health care) equilibrated with 4 M/L guanidine buffer, flow rate 2.4 ml/h (0.6 ml/fraction). The second half of each extract was digested with Streptomyces hyalurolyticus hyaluronidase (ICN Pharmaceuticals Ltd, Hampshire, UK) (1U) overnight at 37°C. The extract was then mixed with 200 μl of 4 mol/L guanidine buffer and analyzed as above by size-exclusion chromatography on the same Sephacryl S-500 column equilibrated with 4 mol/L guanidine buffer. To produce the [3H]-HA elution profiles, the hyaluronidase-resistant counts in the second half of each extract were subtracted from those of the untreated half of the extract and corrected for dilution factors.
Isolation of Collagen
Confluent mono-layers of cells were grown in T25 culture flasks and arrested in serum-free conditions for 48 hours. They were then stimulated as necessary and labeled simultaneously with 20 μCi/ml [3H]-proline, for up to 72 hours. Supernatants (conditioned medium extract) were collected by decanting the flasks. Cell extracts were extracted following incubation at 37°C for 15 minutes with 4 ml extraction buffer: 0.2% (v/v) triton X-100 in 25 mmol/L Tris-HCl, pH7.5 containing proteinase inhibitors (benzamidine hydrochloride, N-ethylmaleimide, and phenylmethylsulfonylfluoride). This was then collected and the flask was washed with a further 2 ml of extraction buffer, and combined with the cell extract. Matrix extracts were extracted overnight at −20°C with 4 ml of 4 mol/L guanidine in 1% (v/v) triton X-100 in 25 mmol/L Tris-HCl pH7.5 containing proteinase inhibitors. The extract was collected and the flask was washed with a further 2 ml at room temperature for 30 minutes. This was combined with the matrix extract and stored at −20°C.
Extract Purification
Each extract was purified by ion exchange chromatography over diethylaminoethyl cellulose columns equilibrated with 10 mmol/L Tris (pH 7.4). Samples were eluted from the columns using 1 mmol/L NaCl2 in 10 mmol/L Tris (pH 7.4), and then dialyzed against H2O overnight. Conditioned medium extract, cellular extract, and matrix extract samples were further purified by gel-filtration through PD10 columns equilibrated in 0.2 mol/L NH3HCO3 and 2 ml fractions were collected. Fractions with radioactivity detectable above background level were bulked together, and the volumes were recorded. Collagenase digests were performed on half of each sample to confirm [3H]-proline incorporation was an accurate measure of collagen production and the collagenase-resistant counts in the second half of each extract were subtracted from those of the undigested half of the extract to give the collagen-specific incorporation.
Immunocytochemistry
Indirect immunofluorescent identification of α-SMA and visualization of filamentous actin (F-actin) were used as confirmation of myofibroblastic differentiation. Cells were grown to confluence in 8-well permanox chamber slides (Nunc), growth arrested for 48 hours, and then stimulated under serum-free conditions for up to 7 days. Following stimulation, cells were fixed in cold 1:1 acetone/methanol for 10 minutes and then washed thoroughly in calcium/magnesium-free PBS. Nonspecific sites on the cells were blocked with 1% (w/v) bovine serum albumin (BSA) in hanks balanced salt solution (HBSS) (Sigma-Aldrich) for 1 hour at 4°C. The cells were washed thoroughly in 0.1% (w/v) BSA in HBSS. Cells were incubated with the appropriate primary antibody (Table 1), diluted in 0.1% (w/v) BSA in HBSS, for 2 hours at room temperature or at 4°C overnight. The cells were washed repeatedly in 0.1% (w/v) BSA in HBSS and then incubated with fluorescein isothiocyanate-conjugated anti-mouse IgG (DakoCytomation), diluted in 0.1% (w/v) BSA in HBSS, for 1 hour at room temperature. The cells were washed extensively with 0.1% (w/v) BSA in HBSS, mounted in Vectashield fluorescent mountant (Vecta Laboratories, Peterborough, UK) and examined under UV-light on a Leica Dialux ×20 fluorescent microscope (Leica Microsystems Ltd, Milton Keynes, UK).
Table 1.
Primary Antibodies for Immunofluorescence
| Antibody | Cat. no | Host | Type | Dilution |
|---|---|---|---|---|
| Anti-Human vimentin (DakoCytomation) | M0725 | Mouse | Monoclonal IgG1 | 1:50 |
| Anti-Human cytokeratin (DakoCytomation) | M0717 | Mouse | Monoclonal IgG1 | 1:50 |
| Anti-Human α-SM actin (Sigma) | A5691 | Mouse | Monoclonal IgG2a | 1:30 |
| Anti-Human HYAL2 a kind gift of Dr R Stern, UCSF | N/A | Rabbit | Polyclonal | 1:200 |
| Anti-Human CD44s (Calbiochem, Merck Biosciences Ltd., Nottingham, UK) | Rat | Monoclonal IgG2b | 1:200 |
Cellular Protein Extraction
Confluent monolayers of cells were washed with PBS. Cells were then lysed using RIPA lysis buffer (Santa Cruz biotechnology inc, Germany); to which 1% (v/v) protease inhibitor cocktail, and freshly prepared 1% (v/v) phenymethylsulphonyl fluoride and 1% (v/v) sodium orthovanadate were added and incubated on ice for 15 minutes. The samples were scraped, collected and centrifuged at 2500 × g at 4°C for 10 minutes. The supernatants were collected and stored at −80°C until analyzed by SDS-polyacrylamide gel electrophoresis (PAGE).
Protein Separation
Each sample equating to 30 μg protein (protein concentration was determined by Bio-Rad protein assay, Bio-Rad Laboratories) was mixed with half that volume of 3× reducing PAGE loading buffer and incubated at 95°C for 5 minutes. SDS-PAGE was performed using Bio-Rad Mini-PROTEAN apparatus. Thirty micrograms protein samples were loaded onto a 12% polyacrylamide gel containing 0.1% SDS (Table 2). Electrophoresis was performed in running buffer at 150 V until the bromophenol blue tracking dye had migrated down 90% of the gel.
Table 2.
Semi-Quantitative PCR Primers
| Primer | Sequence | Product size (bp) | Cycle number | Ascension number |
|---|---|---|---|---|
| β-actin | ||||
| Sense | 5′-CCTTCCTGGGCATGGAGTCCT-3′ | 204 | 25 | NM_001101 |
| Anti-sense | 5′-GGAGCAATGATCTTGATCTT-3′ | |||
| ED-A fibronectin | ||||
| Sense | 5′-ACATTGATCGCCCTAAA-3′ | 274 | 25 | NM_212482 |
| Anti-sense | 5′-ATAGCTGTGGACTGGGT-3′ | |||
Protein Transfer and Detection
The separated proteins were transferred to a nitrocellulose membrane (GE Health care), by Western blot, using the BioRad Mini Blot II apparatus (Bio-Rad Laboratories) in transfer buffer at 150 V for 90 minutes on ice. The nitrocellulose membrane was blocked to prevent non-specific binding by incubating with 5% (w/v) nonfat powdered milk, 0.01% (v/v) Tween-20 in Tris-buffered saline (TBS) for 1 hour. The nitrocellulose membrane was incubated with the appropriate primary antibody (anti-human phospho-Smad 2 [Ser 465/467], anti-human phospho-Smad3 [Ser423/425] or anti-human glyceraldehyde-3-phosphate dehydrogenase, Abcam plc, Cambridge, UK) as loading control. The nitrocellulose membrane was washed repeatedly with 0.01% (v/v) Tween-20 in TBS and incubated with an appropriate horseradish peroxidase-conjugated secondary antibody, diluted as necessary, 5% (w/v) nonfat powdered milk, 0.01% (v/v) Tween-20 in TBS at room temperature for 2 hours. Antibody binding was visualized by ECL-Plus (GE Health care) according to the manufacturer’s protocol. The intensity of the bands visualized were quantified by densitometry (Chemi Doc; Bio Rad) and corrected for the expression of the glyceraldehyde-3-phosphate dehydrogenase housekeeping band for each sample.
Fluorescein Isothiocyanate-Phalloidin Staining
Following stimulation, cells were fixed in 4% (w/v) paraformaldehyde for 15 minutes at room temperature. Cell were then left to air dry, and then permeabilized in 0.1% (v/v) Tween-20 in PBS for 5 minutes at room temperature. Non-specific sites on the cells were blocked with 5% (w/v) BSA in PBS for 20 minutes at room temperature. The cells were washed thoroughly in 0.1% (w/v) BSA in PBS. Cells were incubated with Phalloidin fluorescein isothiocyanate -Conjugate (Fluka, Sigma-Aldrich), diluted in 0.1% (w/v) BSA in PBS, for 1 hour at room temperature, avoiding light. The cells were washed repeatedly in 0.1% (w/v) BSA in PBS and then mounted in Vectashield fluorescent mountant and examined under UV-light on a Leica Dialux 20 fluorescent microscope.
RNA Extraction
Confluent monolayers of cells were cultured in 6-well plates. Cell monolayers were washed once with PBS and then cells lysed in 1 ml Tri-Reagent (Sigma-Aldrich). Chloroform (200 μl) was added to the sample and agitated by inversion until completely emulsified. Samples were then incubated at 4°C for 5 minutes to allow separation of the aqueous and phenol phases, and then centrifuged at 16,000 × g for 20 minutes at 4°C. The aqueous layer was removed and mixed with an equal volume of ice-cold isopropanol. The mixture was incubated at 4°C for 24 hours then the precipitate was pelleted by centrifugation at 16,000 × g for 20 minutes at 4°C. The supernatant was removed and the pellet was washed with 70% (v/v) ethanol by inversion then centrifuged at 16,000 × g for 20 minutes at 4°C. The RNA pellet was air dried at room temperature and then dissolved in 20 μl of H2O.
Reverse Transcription
Reverse transcription (RT) was performed using the random primer method. The RT was performed in a final volume of 20 μl per reaction, containing 1 μg of RNA sample, 2 μl of 10× RT random primers, 2 μl of 10× RT buffer, 0.8 μl of 100 mmol/L dNTPs (deoxynucleotide triphosphate) (mixed nucleotides: dATP, dCTP, dGTP, and dTTP), 1 μl of Multiscribe reverse transcriptase, and 1 μl of RNase inhibitor. All reagents used were supplied as a high-capacity cDNA reverse transcriptase kit (Applied Biosystems). The RT was performed using an Applied Biosystems “Gene Amp PCR System 9700” thermocycler. A negative control (−RT) was prepared with H2O substituted for the Multiscribe reverse transcriptase. The solution was incubated at 25°C for 10 minutes to allow the random hexamer primers to anneal to the RNA. The primers were then extended using reverse transcriptase in the presence of the four dNTPs by heating the solution to 37°C for 2 hours, thus generating cDNA. The final heating step was to heat the solution to 85°C for 5 seconds. The resulting single stranded complementary DNA (cDNA) was stored at −20°C.
Semiquantitative PCR
The PCR was performed in a final volume of 50 μl per reaction, containing 2 μl of cDNA, 20 pmol each of the sense and anti-sense primers (Table 2), 3 μl of 10 mmol/L dNTPs (Sigma-Aldrich), 5 μl PCR buffer (10×) (Applied Biosystems), 0.25 μl (5 U/μl) of AmpliTaq Gold (Applied Biosystems), and 37.25 μl of H2O. A negative control (−PCR) was prepared with H2O substituted for the cDNA. The PCR amplification was performed in a GeneAmp PCR system 9700 Thermocycler (Applied Biosystems). The solution was heated to 94°C for 2 minutes to activate the Taq polymerase, then the necessary number of cycles (Table 2) of 94°C for 30 s, 55°C for 32 s, 72°C for 1 minute, and then 72°C for a further 15 minutes.
Products were separated by electrophoresis on 2% (w/v) agarose gel (Ultrapure agarose, GIBCO) in TAE buffer containing 0.5 mg/ml ethidium bromide. They were visualized and photographed under UV light using a Chemi Doc Gel Documentation system (Bio-Rad Laboratories, Hemel Hempstead, UK).
Quantitative PCR
The quantitative (Q)PCR was performed in a final volume of 20 μl per reaction, containing 1 μl of cDNA, 10 μl of TaqMan Fast Universal Master Mix (20×) (Applied Biosystems), 8 μl of H2O, and 1 μl of a TaqMan gene expression assay primer and probe mix (Applied Biosystems). A negative control (−PCR) was prepared with H2O substituted for the cDNA. The QPCR amplification was performed in 7900HT Fast Real-Time PCR System thermocycler (Applied Biosystems). Amplification was performed using a cycle of 95°C for 1 s, and 60°C for 20 s, for 40 cycles.
The comparative CT method was used for relative quantification of gene expression. The CT (threshold cycle where amplification is in the linear range of the amplification curve) for the standard reference gene (ribosomal RNA) was subtracted from the target gene CT to obtain the ΔCT. The mean ΔCT values for similar samples were then calculated. The expression of the target gene in experimental samples relative to expression in control samples was calculated:
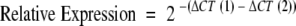 |
where ΔCT (1) is the mean ΔCT value calculated for the experimental samples, and ΔCT (2) is the mean ΔCT value calculated for the control samples. Data were analyzed using RQ Manager software from Applied Biosystems UK, Ltd.
siRNA Transfection of Fibroblasts and Myofibroblasts
Transient transfection of fibroblasts with specific siRNA oligonucleotides (HAS2, Smad2 and Smad3) was performed using Lipofectamine 2000 transfection reagent (Invitrogen) in accordance with the manufacturer’s protocol. Briefly, 2 μl of transfection reagent was diluted in 98 μl Opti-MEM reduced-growth medium (Gibco) and left to incubate at room temperature for 5 minutes. siRNA oligonucleotides were diluted in Opti-MEM reduced growth medium to give a final concentration of 1.2 μmol/L in a total volume of 100 μl. The transfection reagent mix and the siRNA mix were then combined and incubated at room temperature for a further 20 minutes. The newly formed transfection complexes (200 μl) were dispersed into empty wells of a 12 well culture plate. To each well, 9 × 104 cells were then added, so that the total volume in each well was 1.2 ml and the final siRNA concentration was 100 nmol/L. The plate was then rocked gently to ensure adequate mixing of the complexes, and then incubated at 37°C with 5% CO2 for 24 hours. After this time the medium was aspirated and replaced with serum-free Dulbecco’s modified Eagle’s medium:F12 growth medium with no antibiotics. This was incubated at 37°C with 5% CO2 for as long as was necessary to obtain maximum transfection efficiency, before cell stimulation. The success of knockdown by each siRNA used was confirmed by QPCR using primers specific for each gene targeted.
Localization of TGFβ1 Receptor26
Cells were grown to approximately 70% confluence and then growth arrested in serum-free medium for 48 hours. Medium was then replaced with either serum-free medium alone, serum-free medium containing TGFβ1 (10 ng/ml), or serum-free medium containing TGFβ1 (10 ng/ml) with HA (25 μg/ml) for 72 hours. Monolayers were washed twice with ice-cold PBS. Fibroblasts from two confluent dishes were then scraped into 2 ml of 500 mmol/L sodium carbonate, pH 11.0. Homogenization was performed with 10 strokes of a tight-fitting Dounce homogenizer followed by three 20 seconds bursts of Ultrasonic disintegrator (Soniprep 150; Fisher Scientific) to disrupt cellular membranes. The homogenates were adjusted to 45% sucrose by addition of 2 ml of 90% sucrose prepared in (2-[N-morpholino] ethanesulfonic acid-buffered saline; prepared from 25 mmol/L 2-[N-morpholino] ethanesulfonic acid, pH 6.5, with 0.15 M/L NaCl) and placed at the bottom of an ultracentrifuge tube. A discontinuous sucrose gradient (4 ml of 35% sucrose, 3 ml of 5% sucrose, both prepared in 2-[N-morpholino]) was formed above and centrifuged at 39,000 rpm for 20 hours in an SW40 TI rotor (Beckman Instruments, Palo Alto, CA). A light-scattering band was observed at the 5% to 35% sucrose interface. Eleven 1 ml fractions were collected from the top of the tubes.
Western Blot Analysis of Membrane Fractions
Sixty microliters of each fraction were mixed with 15 μl of 5× reducing SDS buffer and boiled for 5 minutes at 95°C before loading onto 10% SDS-PAGE gels. Electrophoresis was performed under reducing conditions at 150 volts for 1 hour and the separated proteins were then transferred at 150 volts over 90 minutes to a nitrocellulose membrane (GE Health care, Buckinghamshire, UK) as described above. The membrane was blocked with TBS containing 5% nonfat powdered milk for 1 hour and then incubated with the primary antibody (anti-TGFβ receptor 1, Santa Cruz Biotechnology Inc, Germany, diluted 1:200 in TBS containing 5% nonfat powdered milk) at 4°C overnight. The blots were subsequently washed with TBS containing 1% Tween and then incubated with the secondary antibody for 1 hour at room temperature (anti-rabbit IgG- horseradish peroxidase, 1:10,000 dilution in TBS containing 5% nonfat powdered milk). Proteins were visualized using enhanced chemiluminescence (GE Health care) according to the manufacturer’s instructions.
The presence of caveolin-1 and EEA-1 were used to determine whether the fraction represented lipid-raft or non-raft proteins respectively. Following transfer of proteins to the nitrocellulose membrane, the membrane was blocked with TBS containing 5% nonfat powdered milk for 1 hour and then incubated with the primary antibody (anti-caveolin 1 or anti-EEA1, diluted 1:2500 in TBS containing 5% BSA; Cell Signaling Technology, Danvers, MA) at 4°C overnight. The blots were subsequently washed with TBS containing 1% Tween and then incubated with the secondary antibody for 1 hour at room temperature (Anti-mouse IgG- horseradish peroxidase, 1:10000 dilution in TBS containing 5% nonfat powdered milk; Santa Cruz Biotechnology Inc, Germany). Proteins were visualized using enhanced chemiluminescence (GE Health Care) according to the manufacturer’s instructions and quantified by densitometry (Chemi Doc; BioRad). Data were expressed as percentage of the total TGFβ-receptor for that experiment in each fraction.
Statistical Analysis
Paired Student’s t-tests were performed for experiments with only one variable. For experiments with multiple variables One-way analysis of variance was used to identify statistical differences, followed by Tukey’s Honest Significant Difference (HSD) method to paired data. The Tukey’s HSD method applied includes corrections for multiple testing and an adjustment for unbalanced designs. The results are expressed as the mean ± SE. All data were analyzed using software (SPSS 14.0 Chicago, IL) and P < 0.05 was considered significant.
Results
TGFβ1-Induction of Myofibroblast Phenotype
Using TGFβ1 to trigger differentiation in fibroblasts led to a time-dependent induction of α-SMA mRNA that peaked around 48 to 72 hours (Figure 1A). This was accompanied by an increase in the expression of α-SMA protein, its incorporation into actin stress fibers and a change in cell morphology (Figure 1, B and C). The induction of phenotypic change was shown to be linked to the Smad signaling pathway, by transfecting fibroblasts with Smad2 or 3 siRNA before incubating them with TGFβ1. Compared with transfection with a scrambled control there was a reduction in α-SMA levels of 80% in the presence of Smad2 siRNA and 60% in the presence of Smad3 siRNA (Figure 2, A–D).
Figure 1.

Induction of the myofibroblast phenotype. Fibroblasts were grown to near confluency and then growth-arrested for 48 hours in serum-free medium. Medium was aspirated and replaced either with fresh serum-free medium (control) or serum-free medium containing 10 ng/ml TGFβ1 for times up to 72 hours. A: mRNA was extracted as described in Materials and Methods and α-SMA expression quantified by QPCR. Ribosomal RNA expression was used as an endogenous control and gene expression was assayed relative to control samples. The comparative CT method was used for relative quantification of gene expression, as described in Materials and Methods and the results are expressed as the mean ± SEM of four independent experiments. Statistical analysis was performed using the paired Student’s t-test and statistical significance was taken as P < 0.05. B: Cells were fixed after 72 hours as described and immunostained for vimentin or α-SMA. Results are representative of four individual experiments. C: Cells fixed after 72 hours were permeabilized in 0.1% (v/v) Tween-20 in PBS and then stained for F-actin with fluorescein isothiocyanate-phalloidin as described. Results are representative of four individual experiments.
Figure 2.

Myofibroblastic induction is dependent on Smad-related TGFβ signaling. Fibroblasts were transfected with siRNA specific to Smad2, Smad3, or a scrambled control for 48 hours in serum-free medium. The medium was changed and the cells stimulated with TGFβ1 (10 ng/ml) for a further 72 hours. mRNA was extracted as described in Materials and Methods. Following transfection with Smad 2-specific siRNA (A and B) or Smad 3-specific siRNA (C and D), the expression of Smad 2 (A), Smad 3 (C), and α-SMA (B and D) were quantified by QPCR. Ribosomal RNA expression was used as an endogenous control and gene expression was assayed relative to control samples. The comparative CT method was used for relative quantification of gene expression and the results are expressed as the mean ± SEM of four independent experiments. Statistical analysis was performed using the one-way analysis of variance test (P < 0.001) followed by Tukey’s HSD posthoc test and statistical significance was taken as P < 0.05.
Associated with the phenotypic differentiation of the cells was the increased expression of type I collagen (Figure 3A), the EDA isoform of fibronectin (Figure 3B) and HA (Figure 3, C and D).
Figure 3.

Phenotype-specific changes in extracellular matrix components. Fibroblasts were grown to near confluency and then growth-arrested for 48 hours in serum-free medium. Medium was aspirated and replaced either with fresh serum-free medium (control) or serum-free medium containing 10 ng/ml TGFβ1 for times up to 72 hours. A: For collagen quantitation, cells were simultaneously metabolically labeled with 20 μCi/ml [3H]-proline and the collagen in each extract purified as described. Results are expressed as mean ± SEM (three independent experiments) for the total labeled collagen of the combined extracts after 72 hours under serum-free conditions (control) or in the presence of 10 ng/ml TGFβ1. Statistical analysis was performed using the paired Student’s t-test and statistical significance was taken as P < 0.05. B: mRNA was extracted from cells after 72 hours in serum-free medium (control) or in serum-free medium containing 10 ng/ml TGFβ1 as described in Materials and Methods and RT-PCR performed for the ED-A isoform of fibronectin. Products were separated on a 1% agarose gel and visualized under UV after ethidium bromide staining. Results show products from four individual experiments. HA in the medium of cells incubated in the presence or absence (control) of 10 ng/ml TGFβ1 was analyzed by enzyme-linked immunosorbent assay for times up to 72 hours (C) (statistical analysis was performed using the paired Student’s t-test and statistical significance was taken as P < 0.05) or metabolic labeling and gel filtration chromatography (D) as described.
Hyaluronan Synthesis Is Essential for Myofibroblastic Differentiation
In previous studies, a link has been described between the accumulation of hyaluronan and the initiation of myofibroblastic differentiation.12,29,30 To investigate in more detail whether HA synthesis was important in this process, 4-mU was used to deplete the cytoplasmic pool of UDP-glucuronic acid, essential for HA chain elongation,31 and siRNA to the inducible HAS2 isoform was used to inhibit production of the synthase. HA enzyme-linked immunosorbent assay was used to confirm HA inhibition (Figure 4A), which was optimal at 0.5 mmol/L (higher concentrations were cytotoxic, data not shown). There was a concentration-dependent inhibition of α-SMA induction following addition of 4-mU to fibroblasts incubated with TGFβ1 (Figure 4B). This was maximal (>95%) at 0.5 mmol/L and above suggesting that HA was an important contributor to the induction of the myofibroblastic phenotype. This was not, however, mediated through an effect on the R-Smads as 4-mU had no effect on the phosphorylation of Smad2 or Smad3 (Figure 4C). siRNA to HAS2 was used to confirm the effect of the inhibitor. The knockdown effect of HAS2 siRNA was confirmed by QPCR (Figure 4D) with 100 nmol/L producing between 50% and 70% knockdown. This was linked to a concomitant reduction in α-SMA (Figure 4E), confirming the link between HA synthesis and differentiation.
Figure 4.

Effect of inhibition of HA chain elongation on differentiation. Fibroblasts were growth-arrested for 48 hours. The medium was then changed and incubations continued in the absence (control) or presence of TGFβ1 for 72 hours. The inhibitor of HA chain elongation, 4-mU (or vehicle, 0.1% dimethyl sulfoxide) was added at various concentrations as shown. A: Dose-response of inhibition of TGFβ1-stimulated HA synthesis in response to 4-mU. Results are expressed as the mean ± SEM of four independent experiments. Statistical analysis was performed using the one-way analysis of variance test (P < 0.001) followed by Tukey’s HSD posthoc test and statistical significance was taken as P < 0.05. B: Effect of optimal dose of 4-mU on α-SMA expression, assessed by QPCR. Ribosomal RNA expression was used as an endogenous control and gene expression was assayed relative to control samples. The comparative CT method was used for relative quantification of gene expression and the results are expressed as the mean ± SEM of four independent experiments. Statistical analysis was performed using the one-way analysis of variance test (P < 0.001) followed by Tukey’s HSD posthoc test and statistical significance was taken as P < 0.05. C: Effect of optimal dose of 4-mU on pSmad 2 and 3 induction. Cells were growth arrested for 48 hours, then incubated with or without TGFβ1 in the presence or absence of 4-mU for 60 minutes. Cell protein was extracted and SDS-PAGE and Western blotting for phosphorylated Smads 2 and 3 performed as described. Results are representative of four independent experiments. D and E: Fibroblasts were transfected with HAS2-specific siRNA or a scrambled control for 48 hours in serum-free medium. The medium was changed and the cells stimulated with TGFβ1 (10 ng/ml) for a further 72 hours. mRNA was extracted and the expression of HAS2 (D) and α-SMA (E) were quantified by QPCR. Ribosomal RNA expression was used as an endogenous control and gene expression was assayed relative to control samples. The comparative CT method was used for relative quantification of gene expression and the results are expressed as the mean ± SEM of four independent experiments. Statistical analysis was performed using the one-way analysis of variance test (P < 0.001) followed by Tukey’s HSD posthoc test and statistical significance was taken as P < 0.05.
Role of Pericellular HA Assembly
The pericellular HA matrix contains a number of hyaladherins that aid in its assembly. In the present study, the expression of the IαI heavy chain 3 (HC3) and TSG-6, were increased by fivefold and 20-fold respectively in fibroblasts incubated with TGFβ1 for 72 hours (Figure 5, A and B). Blockade of the Alk 5 receptor with SB431542 inhibited this increase by 90%, confirming the involvement of the TGFβ1 receptor. The induction of TSG-6 and HC3 was not, however, mediated through either Smad2 or Smad3 as siRNA for either, individually or in combination, had no effect on the mRNA levels for either TSG-6 (Figure 6, A and B) or HC3 (data not shown). TGFβ1 also activates mitogen-activated protein kinase signaling during differentiation32,33,34,35,36,37,38,39 and in the presence of the mitogen-activated protein kinase kinase inhibitor PD98059 there was a 40% decrease in TSG-6 expression (Figure 6C). This suggests that there are at least two signaling pathways (Smad-dependent and ERK-dependent) triggered by the differentiation signal.
Figure 5.

Effect of ALK 5 inhibition on HC3 and TSG-6 expression. Fibroblasts were growth-arrested for 48 hours. The medium was then changed and incubations continued in the absence (control) or presence of TGFβ1 for 72 hours with or without ALK 5 inhibition (10 μmol/L SB431542). mRNA was extracted and QPCR performed for HC3 (A) and TSG-6 (B) as described. Ribosomal RNA expression was used as an endogenous control and gene expression was assayed relative to control samples. The comparative CT method was used for relative quantification of gene expression and the results are expressed as the mean ± SEM of four independent experiments. Statistical analysis was performed using the one-way analysis of variance test (P < 0.001) followed by Tukey’s HSD posthoc test and statistical significance was taken as P < 0.05.
Figure 6.

Effect of Smad and ERK inhibition on TSG-6 expression. Fibroblasts were transfected with (A) Smad2 or (B) Smad3-specific siRNA or a scrambled control for 48 hours in serum-free medium. The medium was changed and the cells stimulated with TGFβ1 (10 ng/ml) for a further 72 hours. Untransfected cells (C) underwent TGFβ1 stimulation for 72 hours in the presence or absence of 10 μmol/L PD98059 (or vehicle, 0.1% dimethyl sulfoxide). Cells were then extracted and QPCR performed for TSG-6 as described. Results represent the mean of four independent experiments. Ribosomal RNA expression was used as an endogenous control and gene expression was assayed relative to control samples. The comparative CT method was used for relative quantification of gene expression and the results are expressed as the mean ± SEM of four independent experiments. Statistical analysis was performed using the one-way analysis of variance test followed by Tukey’s HSD posthoc test and statistical significance was taken as P < 0.05.
The Effect of Exogenously Applied HA on Myofibroblastic Differentiation
To examine whether an increased concentration of HA per se directly affected fibroblast transition to a myofibroblast phenotype, purified HA was added simultaneously with TGFβ1 to the medium of growth-arrested fibroblasts. There was a significant inhibition of the induction of α-SMA in response to TGFβ1 (Figure 7, A and B). This inhibition was shown to be specific to the differentiation process since addition of exogenous HA to myofibroblasts that were terminally differentiated, made no difference to either their resting or TGF-β1-stimulated α-SMA expression (Figure 8).
Figure 7.

Effect of exogenous HA on α-sma induction. Fibroblasts were grown to near confluency and then growth-arrested for 48 hours in serum-free medium. Medium was aspirated and replaced with serum-free medium (control) or in serum-free medium containing 10 ng/ml TGFβ1 ± HA for 72 hours. A: mRNA was extracted from cells as described in Materials and Methods and QPCR performed for α-SMA. Ribosomal RNA expression was used as an endogenous control and gene expression was assayed relative to control samples. The comparative CT method was used for relative quantification of gene expression and the results are expressed as the mean ± SEM of four independent experiments. Statistical analysis was performed using the One-way analysis of variance (P < 0.001) test followed by Tukey’s HSD posthoc test and statistical significance was taken as P < 0.05. B: Corresponding expression of α-SMA protein was confirmed by fixing cells after 72 hours as described and immunostained for vimentin or α-SMA. Results are representative of three independent experiments.
Figure 8.

Effect of HA on induction of α-SMA in myofibroblasts. Myofibroblasts were grown to near confluency and then growth-arrested for 48 hours in serum-free medium. Medium was aspirated and replaced either with fresh serum-free medium (Control) or serum-free medium containing 25 μg/ml HA, 10 ng/ml TGFβ1, or 10 ng/ml TGFβ1 in the presence of 25 μg/ml HA for 72 hours. mRNA was extracted and α-sma QPCR performed as described. Ribosomal RNA expression was used as an endogenous control and gene expression was assayed relative to control samples. The comparative CT method was used for relative quantification of gene expression and the results are expressed as the mean ± SEM of 5 independent experiments. Statistical analysis was performed using the One-way ANOVA (P = 0.00183) test followed by Tukey’s HSD Post-hoc Test and statistical significance was taken as P < 0.05.
Previous work from this laboratory has established that a major effect of exogenous HA in epithelial cells is as a modifier of TGFβ1 signaling.26 Incubating fibroblasts with TGFβ1, in the presence of HA, significantly inhibited the TGFβ1-dependent phosphorylation of Smad 3 (Figure 9A). Taken together with its effect on endogenous HA, these results strongly suggest that there is an HA-dependence to the induction of differentiation that occurs at the level of the TGFβ1 receptor but that this is not reproduced by simply providing extracellular HA. Furthermore this dependence was lost once the cells had differentiated to myofibroblasts (Figure 9B).
Figure 9.

Effect of HA on Smad 3 phosphorylation in fibroblasts and myofibroblasts. Fibroblasts (A) or myofibroblasts that had been differentiated by 72 hours incubation with 10 ng/ml TGFβ1 (B), were washed with PBS (×3), growth-arrested in serum-free medium for 48 hours and then stimulated for 60 minutes with 10 ng/ml TGFβ1 in the presence or absence of 25 μg/ml HA. Cell protein was extracted and SDS-PAGE and Western blotting for phosphorylated Smad 3 performed as described. Results are representative of six independent experiments.
Treatment with exogenous HA also inhibited the expression of HC3 and TSG-6, induced by incubation with TGFβ1 (Figure 10, A–B), suggesting that there is a mechanism through which exogenously added HA antagonizes the formation of the pericellular HA coat. Thus both the synthesis of HA and its organization into a coat are linked to the induction of the myofibroblast phenotype.
Figure 10.

Effect of HA on HC3 and TSG6 induction. Fibroblasts were growth-arrested for 48 hours. The medium was then changed and incubations continued in the absence (control) or presence of TGFβ1 for 72 hours with or without 25 μg/ml HA. Cells were then extracted and QPCR performed for HC3 (A) and TSG-6 (B) as described. Ribosomal RNA expression was used as an endogenous control and gene expression was assayed relative to control samples. The comparative CT method was used for relative quantification of gene expression and the results are expressed as the mean ± SEM of four independent experiments. Statistical analysis was performed using the one-way analysis of variance (P < 0.001) test followed by Tukey’s HSD posthoc test and statistical significance was taken as P < 0.05.
Receptor Relocalization during Fibroblast Differentiation
Previous work has shown that during myofibroblastic differentiation there is a relocalization of cell surface HYAL 2.12 Recent reports from others40 and41,42 for review, have suggested that HYAL 2 and CD44 co-localize to lipid rafts in the plasma membrane. The TGF receptor is also reported to shuttle between lipid raft and non-raft pools. To investigate whether there was relocalization of TGF receptors in parallel with CD44 and HYAL 2, we examined TGF receptor 1 (ALK-5) localization by sucrose density centrifugation followed by SDS-PAGE and Western blotting, and cell surface expression of CD44 and HYAL 2 by immunochemistry. HYAL 2 and CD44 co-localized in discrete domains along the plasma membrane in fibroblasts (Figure 11A control). Their pattern of expression was much more diffuse, however, during differentiation to myofibroblasts (Figure 11A TGFβ1), suggesting a re-organization from discrete lipid-rafts to the non-raft pool. Western blotting demonstrated an associated relocalization of TGFR1 to the non-raft pool (Figure 11, B and C), which was reversed by incubation with exogenous HA.
Figure 11.

Receptor relocalization during differentiation. Fibroblasts were growth-arrested for 48 hours. The medium was then changed and incubations continued in the absence (control) or presence of TGFβ1 for 72 hours. A: The cells were fixed and immunostained for HYAL 2 (green) and CD44 (red). B: Cell protein was extracted and subjected to density gradient ultracentrifugation as described. Proteins in each fraction were separated by SDS-PAGE and the ALK-5 receptor identified by Western blotting. C: Results are expressed by densitometric analysis of blots from 4 independent experiments. Statistical analysis was performed using the one-way analysis of variance (P < 0.001) test followed by Tukey’s HSD posthoc test and statistical significance was taken as P < 0.05.
Discussion
The regulation of cell phenotype during tissue repair is an important factor in determining the success of wound healing or the development of scarring and fibrosis. In the present study, the process of TGFβ1-driven fibroblast-to-myofibroblast differentiation was investigated in the light of previous studies indicating that the linear glycosaminoglycan HA may be a modulator of differentiation.29,30 The results demonstrated that HA synthesis was essential for differentiation to occur. However, although the differentiation process was mediated through the Smad signaling pathway, Smad3 phosphorylation was unaffected by blocking HA synthesis, whether at the level of expression of the inducible HAS2 isoform or by chemical inhibition of the synthase, indicating that HA was having its affect either at the level of the TGFβ1 receptor or by a separate mechanism downstream of Smad signaling. The increase in expression of both TSG-6 and HC3 was also independent of Smad phosphorylation, suggesting that although cytoskeletal remodeling and HA coat assembly are both consequences of TGFβ1-triggered differentiation they are mediated by independent pathways. Furthermore, differentiation does not occur if either is antagonized.
TGFβ1 is a member of a family of pleiotropic growth factors that mediate a variety of cell responses and functions. Its effects are transduced through receptor binding and initiation of a complex consisting of receptor types I and II [for review43,44] each of which is a serine/threonine kinase. Individual family members use different type I (ALK) and type II receptors to initiate signaling and each type I receptor phosphorylates a different R-Smad to propagate the signal. For TGFβ1, ALK-4, ALK-5, and ALK-7 are the type I receptors most commonly activated, with their targets being Smad2 and Smad3. In this study selective ALK5 inhibition antagonized all of the responses to TGFβ1, indicating classical TGFβ1 receptor activation. Both ERK and Smad pathways, however, were activated by TGFβ1 binding to its receptor. ERK activation has been described previously35 and been shown to be linked to epithelial/mesenchymal transformation.36,45 Its involvement and importance, however, are cell-type dependent36 and in the current study it appears that ERK activity is important for triggering the mechanisms to do with HA coat formation while Smad activity links directly to the phenotypic change of the cells.
HA is a ubiquitous component of most extracellular matrices and is important for maintaining matrix stability and tissue hydration. It plays a major role in regulating cell functions through interaction with cell-surface receptors (principally CD44 and RHAMM) and with binding proteins (hyaladherins) with which it complexes.24,46,47,48,49,50,51,52 It is therefore an important regulator of tissue re-modeling and has been implicated in a number of biological and pathological processes, including wound healing, embryonic development, tumor growth, and inflammation.53,54,55,56 Previous studies demonstrated the importance of HA in facilitating TGFβ1-mediated phenotypic activation of fibroblasts,29 and that differentiation to the myofibroblast phenotype was associated with the assembly of a HA peri-cellular coat.12 We now show that the assembly of this coat cannot be simply mimicked by adding exogenous HA (even though that HA does have an effect on the cells). Exogenously supplied HA antagonized the re-arrangement of the cytoskeleton and the de novo expression of α-SMA, which are markers of differentiation. The different effect of exogenous HA is most likely a reflection of the different way it interacts with hyaladherins and HA receptors expressed by the cells. Previous studies have made similar observations and shown that the biological actions of HA may be influenced by both its mode of presentation to the cells and by the way in which it is assembled into different pericellular complexes.26,49,50,51,57
CD44 is the principal receptor associated with HA coat assembly51 and in the present study, we observed that CD44 relocalized on the cell surface as the fibroblasts underwent phenotypic change. In fibroblasts, it was localized with a discrete punctate distribution that became diffuse as the cells differentiated. This was coincident with a redistribution of ALK5 (TGFRI) from lipid rafts as shown by Western blotting. HYAL2 relocalization was described in a previous report,12 and other studies describe TGFRI, CD44, and HYAL2 associating within lipid rafts.26,57 In the present work, the relocalization of TGFR1 was antagonized by exogenous HA, while even though there was HA present in the endogenous pericellular coat, this was obviously permissive of the changes. This again highlights the differences in cell response to the two types of HA and re-emphasizes the potential importance of HA macromolecular assembly in the pericellular environment. Whether CD44 is directly involved in mediating the effects of either exogenous or endogenous HA in the present study, or whether other HA receptors are involved is currently being investigated.
In summary, the results presented highlight the intimate involvement of HA in the modulation of myofibroblastic differentiation. While the synthesis and organization of HA are essential and necessary for the differentiation to occur, providing HA exogenously antagonizes differentiation. The mechanisms by which these observations are mediated are currently under investigation. The data do, however, underline the important role that HA plays in pathology and suggest that modulations to affect the way in which cells recognize and respond to HA may have important potential as a way of controlling cell fate.
Footnotes
Address reprint requests to Dr. R. Steadman, Institute of Nephrology, Cardiff University, School of Medicine, Heath Park, Cardiff CF14 4XN, UK. E-mail: steadmanr@cf.ac.uk.
Supported by Kidney Wales Foundation.
A.P. and R.S. made equal contributions to this work.
References
- Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Desmouliere A, Darby IA, Gabbiani G. Normal and pathologic soft tissue remodeling: role of the myofibroblast, with special emphasis on liver and kidney fibrosis. Lab Invest. 2003;83:1689–1707. doi: 10.1097/01.lab.0000101911.53973.90. [DOI] [PubMed] [Google Scholar]
- Hinz B, Gabbiani G. Cell-matrix and cell-cell contacts of myofibroblasts: role in connective tissue remodeling. Thromb Haemost. 2003;90:993–1002. doi: 10.1160/TH03-05-0328. [DOI] [PubMed] [Google Scholar]
- Horowitz JC, Thannickal VJ. Idiopathic pulmonary fibrosis: new concepts in pathogenesis and implications for drug therapy. Treat Respir Med. 2006;5:325–342. doi: 10.2165/00151829-200605050-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy AA. Molecular insights into renal interstitial fibrosis. J Am Soc Nephrol. 1996;7:2495–2508. doi: 10.1681/ASN.V7122495. [DOI] [PubMed] [Google Scholar]
- Eddy AA. Progression in chronic kidney disease. Adv Chronic Kidney Dis. 2005;12:353–365. doi: 10.1053/j.ackd.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Toews GB, White ES, Lynch JP, 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. 2004;55:395–417. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000;15:290–301. doi: 10.1007/s004670000461. [DOI] [PubMed] [Google Scholar]
- Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res. 2000;257:180–189. doi: 10.1006/excr.2000.4869. [DOI] [PubMed] [Google Scholar]
- Evans RA, Tian YC, Steadman R, Phillips AO. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp Cell Res. 2003;282:90–100. doi: 10.1016/s0014-4827(02)00015-0. [DOI] [PubMed] [Google Scholar]
- Jenkins RH, Thomas GJ, Williams JD, Steadman R. Myofibroblastic differentiation leads to hyaluronan accumulation through reduced hyaluronan turnover. J Biol Chem. 2004;279:41453–41460. doi: 10.1074/jbc.M401678200. [DOI] [PubMed] [Google Scholar]
- Spicer AP, Kaback LA, Smith TJ, Seldin MF. Molecular cloning and characterization of the human and mouse UDP-glucose dehydrogenase genes. J Biol Chem. 1998;273:25117–25124. doi: 10.1074/jbc.273.39.25117. [DOI] [PubMed] [Google Scholar]
- Spicer AP, McDonald JA. Characterization and molecular evolution of a vertebrate hyaluronan synthase gene family. J Biol Chem. 1998;273:1923–1932. doi: 10.1074/jbc.273.4.1923. [DOI] [PubMed] [Google Scholar]
- Svee K, White J, Vaillant P, Jessurun J, Roongta U, Krumwiede M, Johnson D, Henke C. Acute lung injury fibroblast migration and invasion of a fibrin matrix is mediated by CD44. J Clin Invest. 1996;98:1713–1727. doi: 10.1172/JCI118970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent TC, Laurent UB, Fraser JR. Serum hyaluronan as a disease marker. Ann Med. 1996;28:241–253. doi: 10.3109/07853899609033126. [DOI] [PubMed] [Google Scholar]
- Laurent UB, Laurent TC, Hellsing LK, Persson L, Hartman M, Lilja K. Hyaluronan in human cerebrospinal fluid. Acta Neurol Scand. 1996;94:194–206. doi: 10.1111/j.1600-0404.1996.tb07052.x. [DOI] [PubMed] [Google Scholar]
- Lewington AJ, Padanilam BJ, Martin DR, Hammerman MR. Expression of CD44 in kidney after acute ischemic injury in rats. Am J Physiol Regul Integr Comp Physiol. 2000;278:R247–R254. doi: 10.1152/ajpregu.2000.278.1.R247. [DOI] [PubMed] [Google Scholar]
- Lewis A, Steadman R, Manley P, Craig K, de la Motte C, Hascall V, Phillips AO. Diabetic nephropathy, inflammation, hyaluronan and interstitial fibrosis. Histol Histopathol. 2008;23:731–739. doi: 10.14670/HH-23.731. [DOI] [PubMed] [Google Scholar]
- Evanko SP, Wight TN. Intracellular localization of hyaluronan in proliferating cells. J Histochem Cytochem. 1999;47:1331–1342. doi: 10.1177/002215549904701013. [DOI] [PubMed] [Google Scholar]
- Evanko SP, Angello JC, Wight TN. Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:1004–1013. doi: 10.1161/01.atv.19.4.1004. [DOI] [PubMed] [Google Scholar]
- Kosaki R, Watanabe K, Yamaguchi Y. Overproduction of hyaluronan by expression of the hyaluronan synthase Has2 enhances anchorage-independent growth and tumorigenicity. Cancer Res. 1999;59:1141–1145. [PubMed] [Google Scholar]
- Legg JW, Lewis CA, Parsons M, Ng T, Isacke CM. A novel PKC-regulated mechanism controls CD44 ezrin association and directional cell motility. Nat Cell Biol. 2002;4:399–407. doi: 10.1038/ncb797. [DOI] [PubMed] [Google Scholar]
- Itano N, Atsumi F, Sawai T, Yamada Y, Miyaishi O, Senga T, Hamaguchi M, Kimata K. Abnormal accumulation of hyaluronan matrix diminishes contact inhibition of cell growth and promotes cell migration. Proc Natl Acad Sci USA. 2002;99:3609–3614. doi: 10.1073/pnas.052026799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Williams JD, Al-Assaf S, Phillips GO, Phillips AO. Hyaluronan and proximal tubular cell migration. Kidney Int. 2004;65:823–833. doi: 10.1111/j.1523-1755.2004.00457.x. [DOI] [PubMed] [Google Scholar]
- Ito T, Williams JD, Fraser DJ, Phillips AO. Hyaluronan regulates transforming growth factor-beta1 receptor compartmentalization. J Biol Chem. 2004;279:25326–25332. doi: 10.1074/jbc.M403135200. [DOI] [PubMed] [Google Scholar]
- Zoltan-Jones A, Huang L, Ghatak S, Toole BP. Elevated hyaluronan production induces mesenchymal and transformed properties in epithelial cells. J Biol Chem. 2003;278:45801–45810. doi: 10.1074/jbc.M308168200. [DOI] [PubMed] [Google Scholar]
- Misra S, Ghatak S, Zoltan-Jones A, Toole BP. Regulation of multidrug resistance in cancer cells by hyaluronan. J Biol Chem. 2003;278:25285–25288. doi: 10.1074/jbc.C300173200. [DOI] [PubMed] [Google Scholar]
- Meran S, Thomas D, Stephens P, Martin J, Bowen T, Phillips A, Steadman R. Involvement of hyaluronan in regulation of fibroblast phenotype. J Biol Chem. 2007;282:25687–25697. doi: 10.1074/jbc.M700773200. [DOI] [PubMed] [Google Scholar]
- Meran S, Thomas DW, Stephens P, Enoch S, Martin J, Steadman R, Phillips AO. Hyaluronan facilitates transforming growth factor-beta1-mediated fibroblast proliferation. J Biol Chem. 2008;283:6530–6545. doi: 10.1074/jbc.M704819200. [DOI] [PubMed] [Google Scholar]
- Kakizaki I, Kojima K, Takagaki K, Endo M, Kannagi R, Ito M, Maruo Y, Sato H, Yasuda T, Mita S, Kimata K, Itano N. A novel mechanism for the inhibition of hyaluronan biosynthesis by 4-methylumbelliferone. J Biol Chem. 2004;279:33281–33289. doi: 10.1074/jbc.M405918200. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- Mucsi I, Skorecki KL, Goldberg HJ. Extracellular signal-regulated kinase and the small GTP-binding protein, Rac, contribute to the effects of transforming growth factor-beta1 on gene expression. J Biol Chem. 1996;271:16567–16572. doi: 10.1074/jbc.271.28.16567. [DOI] [PubMed] [Google Scholar]
- Reimann T, Hempel U, Krautwald S, Axmann A, Scheibe R, Seidel D, Wenzel KW. Transforming growth factor-beta1 induces activation of Ras, Raf-1, MEK, and MAPK in rat hepatic stellate cells. FEBS Lett. 1997;403:57–60. doi: 10.1016/s0014-5793(97)00024-0. [DOI] [PubMed] [Google Scholar]
- Hayashida T, Poncelet AC, Hubchak SC, Schnaper HW. TGF-beta1 activates MAP kinase in human mesangial cells: a possible role in collagen expression. Kidney Int. 1999;56:1710–1720. doi: 10.1046/j.1523-1755.1999.00733.x. [DOI] [PubMed] [Google Scholar]
- Hayashida T, Decaestecker M, Schnaper HW. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-beta-dependent responses in human mesangial cells. FASEB J. 2003;17:1576–1578. doi: 10.1096/fj.03-0037fje. [DOI] [PubMed] [Google Scholar]
- Hayashida T, Schnaper HW. High ambient glucose enhances sensitivity to TGF-beta1 via extracellular signal-regulated kinase and protein kinase Cdelta activities in human mesangial cells. J Am Soc Nephrol. 2004;15:2032–2041. doi: 10.1097/01.ASN.0000133198.74973.60. [DOI] [PubMed] [Google Scholar]
- Hayashida T, Wu MH, Pierce A, Poncelet AC, Varga J, Schnaper HW. MAP-kinase activity necessary for TGFbeta1-stimulated mesangial cell type I collagen expression requires adhesion-dependent phosphorylation of FAK tyrosine 397. J Cell Sci. 2007;120:4230–4240. doi: 10.1242/jcs.03492. [DOI] [PubMed] [Google Scholar]
- Caraci F, Gili E, Calafiore M, Failla M, La Rosa C, Crimi N, Sortino MA, Nicoletti F, Copani A, Vancheri C. TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol Res. 2008;57:274–282. doi: 10.1016/j.phrs.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Bourguignon LY, Singleton PA, Diedrich F, Stern R, Gilad E. CD44 interaction with Na+-H+ exchanger (NHE1) creates acidic microenvironments leading to hyaluronidase-2 and cathepsin B activation and breast tumor cell invasion. J Biol Chem. 2004;279:26991–27007. doi: 10.1074/jbc.M311838200. [DOI] [PubMed] [Google Scholar]
- Stern R, Kogan G, Jedrzejas MJ, Soltes L. The many ways to cleave hyaluronan. Biotechnol Adv. 2007;25:537–557. doi: 10.1016/j.biotechadv.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Stern R. Hyaluronan catabolism: a new metabolic pathway. Eur J Cell Biol. 2004;83:317–325. doi: 10.1078/0171-9335-00392. [DOI] [PubMed] [Google Scholar]
- Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Schnaper HW, Hayashida T, Hubchak SC, Poncelet AC. TGF-beta signal transduction and mesangial cell fibrogenesis. Am J Physiol Renal Physiol. 2003;284:F243–F252. doi: 10.1152/ajprenal.00300.2002. [DOI] [PubMed] [Google Scholar]
- Rochman M, Moll J, Herrlich P, Wallach SB, Nedvetzki S, Sionov RV, Golan I, Ish-Shalom D, Naor D. The CD44 receptor of lymphoma cells: structure-function relationships and mechanism of activation. Cell Adhes Commun. 2000;7:331–347. doi: 10.3109/15419060009015004. [DOI] [PubMed] [Google Scholar]
- Naor D, Nedvetzki S. CD44 in rheumatoid arthritis. Arthritis Res Ther. 2003;5:105–115. doi: 10.1186/ar746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedvetzki S, Gonen E, Assayag N, Reich R, Williams RO, Thurmond RL, Huang JF, Neudecker BA, Wang FS, Turley EA, Naor D. RHAMM, a receptor for hyaluronan-mediated motility, compensates for CD44 in inflamed CD44-knockout mice: a different interpretation of redundancy. Proc Natl Acad Sci USA. 2004;101:18081–18086. doi: 10.1073/pnas.0407378102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbi W, Day AJ, Rugg MS, Fulop C, de la Motte CA, Bowen T, Hascall VC, Phillips AO. Overexpression of hyaluronan synthase 2 alters hyaluronan distribution and function in proximal tubular epithelial cells. J Am Soc Nephrol. 2006;17:1553–1567. doi: 10.1681/ASN.2005080879. [DOI] [PubMed] [Google Scholar]
- Selbi W, de la Motte CA, Hascall VC, Day AJ, Bowen T, Phillips AO. Characterization of hyaluronan cable structure and function in renal proximal tubular epithelial cells. Kidney Int. 2006;70:1287–1295. doi: 10.1038/sj.ki.5001760. [DOI] [PubMed] [Google Scholar]
- Knudson W, Aguiar DJ, Hua Q, Knudson CB. CD44-anchored hyaluronan-rich pericellular matrices: an ultrastructural and biochemical analysis. Exp Cell Res. 1996;228:216–228. doi: 10.1006/excr.1996.0320. [DOI] [PubMed] [Google Scholar]
- Knudson W, Chow G, Knudson CB. CD44-mediated uptake and degradation of hyaluronan. Matrix Biol. 2002;21:15–23. doi: 10.1016/s0945-053x(01)00186-x. [DOI] [PubMed] [Google Scholar]
- Auvinen PK, Parkkinen JJ, Johansson RT, Agren UM, Tammi RH, Eskelinen MJ, Kosma VM. Expression of hyaluronan in benign and malignant breast lesions. Int J Cancer. 1997;74:477–481. doi: 10.1002/(sici)1097-0215(19971021)74:5<477::aid-ijc1>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Hess KA, Chen L, Larsen WJ. Inter-alpha-inhibitor binding to hyaluronan in the cumulus extracellular matrix is required for optimal ovulation and development of mouse oocytes. Biol Reprod. 1999;61:436–443. doi: 10.1095/biolreprod61.2.436. [DOI] [PubMed] [Google Scholar]
- Chen WY, Abatangelo G. Functions of hyaluronan in wound repair. Wound Repair Regen. 1999;7:79–89. doi: 10.1046/j.1524-475x.1999.00079.x. [DOI] [PubMed] [Google Scholar]
- Anttila MA, Tammi RH, Tammi MI, Syrjanen KJ, Saarikoski SV, Kosma VM. High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res. 2000;60:150–155. [PubMed] [Google Scholar]
- Bourguignon LY, Singleton PA, Zhu H, Zhou B. Hyaluronan promotes signaling interaction between CD44 and the transforming growth factor beta receptor I in metastatic breast tumor cells. J Biol Chem. 2002;277:39703–39712. doi: 10.1074/jbc.M204320200. [DOI] [PubMed] [Google Scholar]