

Abstract
The binding affinity of four palm and thumb site representative non-nucleoside inhibitors (NNIs) of HCV polymerase NS5B to wild-type and resistant NS5B polymerase proteins was determined, and the influence of RNA binding on NNI binding affinity was investigated. NNIs with high binding affinity potently inhibited HCV RNA polymerase activity and replicon replication. Among the compounds tested, HCV-796 showed slow binding kinetics to NS5B. The binding affinity of HCV-796 to NS5B increased 27-fold over a 3-h incubation period with an equilibrium Kd of 71 ± 2 nm. Slow binding kinetics of HCV-796 was driven by slow dissociation from NS5B with a koff of 4.9 ± 0.5 × 10−4 s−1. NS5B bound a long, 378-nucleotide HCV RNA oligonucleotide with high affinity (Kd = 6.9 ± 0.3 nm), whereas the binding affinity was significantly lower for a short, 21-nucleotide RNA (Kd = 155.1 ± 16.2 nm). The formation of the NS5B-HCV RNA complex did not affect the slow binding kinetics profile and only slightly reduced NS5B binding affinity of HCV-796. The magnitude of reduction of NNI binding affinity for the NS5B proteins with various resistance mutations in the palm and thumb binding sites correlated well with resistance -fold shifts in NS5B polymerase activity and replicon assays. Co-crystal structures of NS5B-Con1 and NS5B-BK with HCV-796 revealed a deep hydrophobic binding pocket at the palm region of NS5B. HCV-796 interaction with the induced binding pocket on NS5B is consistent with slow binding kinetics and loss of binding affinity with mutations at amino acid position 316.
Hepatitis C virus (HCV)4 constitutes a global health problem. Current therapies are unable to effectively eliminate viral infection in a significant number of patients. The RNA-dependent RNA polymerase (RdRp) of HCV NS5B is an attractive target for the development of orally bioavailable small molecule inhibitors (1, 2). The structure of the NS5B apoenzyme and the NS5B-RNA complex reveals the characteristic right hand architecture of polymerase enzymes, comprising three distinct domains (palm, thumb, and finger) encircling the enzyme active site located in the palm domain (3–6). The structural and biochemical characterization of HCV NS5B polymerase can provide a basis for drug design efforts, and the elucidation of the mechanism of inhibition can guide the optimization of inhibitor efficiency against wild-type and resistant mutants.
Among the extensively investigated non-nucleosides documented to inhibit the RdRp activity of HCV NS5B, derivatives of various benzofuran and benzothiadiazine have been reported to bind to allosteric binding sites in the palm domain of NS5B (7, 8). The palm domain, whose geometry is conserved in virtually all DNA and RNA polymerases, contains catalytic aspartic acids responsible for the nucleotidyl transfer reaction. The benzofuran compound HCV-796 has been shown to have significant antiviral effects in patients chronically infected with HCV (9, 10). In addition, two series of compounds based on the thiophene and benzimidazole scaffolds have been reported to inhibit NS5B by binding to two different binding pockets in the thumb domain of NS5B (11, 12). The thumb domain is connected to the palm domain by a β-hairpin termed the primer grip motif. The C-terminal region of the thumb protrudes toward the active site (3). The thumb binding inhibitors have been proposed to inhibit the RdRp activity of NS5B, perhaps by interfering with template/primer interaction and conformational dynamics of the protein (13, 14).
Despite the elucidation of a number of NNIs that bind to the thumb and palm binding sites, the mechanism by which NNIs cause inhibition of RNA synthesis is unclear. Also, our understanding of the kinetics of NNI interaction with NS5B, the role of NNI binding and kinetics for inhibition, and the inhibitor efficacy on NS5B-resistant mutations remains incomplete. The four representative palm- and thumb-binding NNIs selected in this study have been reported to effectively inhibit replication of subgenomic replicons with low toxicity. Noncompetitive inhibition of NS5B polymerase activity with respect to NTPs has been reported (2, 15, 16). Based on co-crystallization studies with NS5B, it has been proposed that allosteric inhibitors may lock the NS5B protein in an inactive formation by binding tightly to the protein (16, 17). It is important to understand how the binding affinity relates to inhibition potency and resistance to HCV inhibition. Because the intrinsic potency of slowly binding compounds can be underestimated in the short time scale of biochemical studies, insights into slowly binding compounds may help to identify potent inhibitors. Moreover, the effect of the HCV RNA template on binding of NNIs to the enzyme-RNA complex remains to be addressed.
Due to the error-prone nature of HCV polymerase in HCV replication, drug resistance can occur in patients who are treated with antiviral therapy directed at HCV-specific enzymes, and this resistance can limit their efficacy (16). Various in vitro studies using an HCV subgenomic replicon system have identified mutations that can confer resistance to inhibition by NNIs (2, 8, 16). Many of the mutations produce cross-resistance to the same family of inhibitors, which will affect the design of optimal combination therapies. Achieving optimal and sustained binding of these antiviral agents to the NS5B polymerase is crucial to ensure a high probability of clinical success.
In this work, we have used biochemical and biophysical approaches to investigate binding affinities and binding kinetics of structurally diverse palm- and thumb-binding allosteric NS5B inhibitors. The binding of NNIs to wild-type and NNI-resistant NS5B proteins was studied and compared with inhibition and resistance. First, the NNI binding affinity for the NS5B protein was determined in the presence and absence of HCV RNA template, using a newly developed assay measuring the quenching of NS5B intrinsic fluorescence (FQ) in 96-well plates. The time-dependent NNI binding affinities and NNI binding equilibrium were used to identify slowly binding NNIs. Second, various palm and thumb site-specific mutant proteins were used to determine the mechanism of HCV resistance, and the binding affinities of NNIs were compared with the inhibition potencies determined in the HCV RdRp polymerase assay and HCV replicon assay. Finally, co-crystallization of HCV-796 with NS5B proteins from the Con1 and BK strains was performed to address the role of critical residues involved in HCV-796 resistance and NS5B polymorphism.
EXPERIMENTAL PROCEDURES
Compounds
The compounds benzofuran (HCV-796), benzothiadiazine (NNI-1), thiophene-2-carboxylic acid (NNI-3), and benzimidazole (NNI-4) were synthesized at Roche Palo Alto, LLC (Fig. 1). Stocks of 10 mm (2 mm for NNI-1) were prepared in 100% DMSO and stored at −20 °C.
FIGURE 1.

Chemical structure of non-nucleoside inhibitors of HCV NS5B polymerase used in this study.
Plasmid Constructs
Vectors expressing recombinant proteins NS5B570-Con1 (genotype 1b; GenBankTM accession number AJ242654), NS5B570-BK (genotype 1b; GenBankTM accession number P26663), and NS5B570-H77 (genotype 1a; GenBankTM accession number AF011751) contained an N-terminal hexahistidine tag and a 21-amino acid deletion at the C terminus of NS5B (18). The Con1 HCV subgenomic replicon used in this study was based on an adapted dicistronic HCV subgenomic replicon construct previously described (19). Resistance mutations were introduced into this construct by PCR-based site-directed mutagenesis using the QuikChange kit (Stratagene, La Jolla, CA). All constructs were confirmed by DNA sequencing of both strands.
Expression and Purification of HCV NS5B Proteins
The expression and purification of wild-type and mutant NS5B570 proteins were as described (18, 20). Briefly, protein expression was induced in Escherichia coli strain M15 harboring an inducible NS5B expression vector by the addition of 1 mm isopropyl-β-d-thiogalactopyranoside when optical density at 600 nm had reached 1.5–3.5, after which the induced cultures were incubated for 16–18 h at 22 °C. The wild-type and mutant NS5B proteins were purified to homogeneity using a three-step protocol involving sequential column chromatography on nickel-nitrilotriacetic acid (Qiagen), SP-Sepharose HP (GE Healthcare), and Superdex 75 resins (GE Healthcare).
FQ Binding Assay
A fluorescence quenching-based binding assay was developed for the 96-well plate format, and the protein intrinsic fluorescence was read on a Safire 2 microplate reader (Tecan, Research Triangle Park, NC). Förster energy transfer from tryptophan residues of the NS5B protein to NNI molecules bound to NS5B is expected to quench tryptophan fluorescence, providing a basis for the quantification of compound binding to NS5B protein (21). Compound binding to NS5B was measured in a 100-μl reaction volume containing 40 mm Tris (pH 7.5), 4 mm MgCl2, 4 mm dithiothreitol, 40 mm NaCl, 0.0125% maltoside, 10% glycerol, 10% DMSO, and 100 nm wild-type or mutant NS5B protein. NS5B protein (25 μl of 400 nm stock) was added, and the fluorescence quenching was monitored in real time as binding equilibrium was reached at room temperature. Unless described otherwise in the figure legends, the NS5B protein fluorescence was recorded at 10 min after the binding equilibrium was reached, and data were used to calculate Kd values.
The fluorescence emission spectrum of NS5B was scanned from 310 to 400 nm using an excitation wavelength of 280 nm in a Safire 2 microplate reader (Tecan). The maximum emission wavelength of HCV NS5B enzyme was found to be blue-shifted to ∼335–340 nm in fluorescence spectroscopy. Upon excitation at 280 nm, the compounds NNI-1 and NNI-3 did not show apparent fluorescence emission at 330–340 nm, whereas HCV-796 and NNI-4 showed fluorescence emission with peaks at 340 nm. The fluorescence of HCV-796 and NNI-4 was weak at the emission wavelength of 330 nm. Therefore, excitation at 280 nm with emission at 330 nm was used to determine the dissociation constant (Kd) for the four compounds and NS5B in this work. The excitation and emission wavelengths were set at monochromator bandwidths of 10 and 20 nm, respectively. Blank titrations were performed in parallel by titrating solutions of compounds into buffer lacking protein. The data obtained were corrected in order to exclude the signals from background (blank), compound fluorescence, and inner filter effects according to Equation 1,
where Fc represents the corrected fluorescence, F is the experimentally measured fluorescence intensity, Fi is the background compound fluorescence in buffer lacking protein, b is the length (in cm) of the optical path in the 96-well plate, and Aex and Aem are the absorbance of the sample at the excitation and emission wavelengths, respectively.
The binding of compounds to the NS5B protein led to a concentration-dependent decrease in protein fluorescence. The fractional fluorescence (ΔF/Fo) was calculated by dividing the fluorescence change at each compound concentration (ΔF) by the enzyme fluorescence in the absence of compound (Fo). When the Kd value was less than 1 μm (46), the data obtained using a dilution series of each compound were fitted to Equation 2,
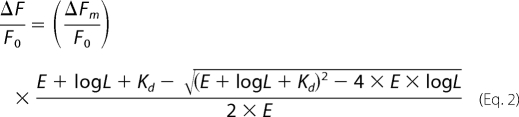 |
in which ΔFm is the maximum fluorescence quenching due to ligand binding to the enzyme at saturating ligand concentration, E is the enzyme concentration, and L is the ligand concentration. Kd is the dissociation constant of E for a ligand L.
When the predicted Kd value was larger than 1 μm (46), fractional fluorescence quenching caused by compound binding was fitted to Equation 3,
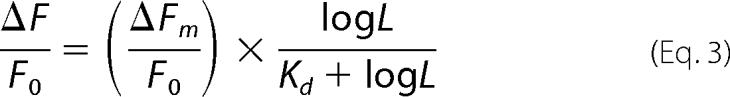 |
To measure NS5B binding to the HCV RNA, two RNA fragments (cIRES-378nt and cIRES-21nt) carrying the complementary sequence of the HCV internal ribosome binding site (cIRES) were used. The cIRES-378nt has 378 nucleotides, which include the full-length cIRES sequence, and the cIRES-21nt (5′-CGCCCCCAAUCGGGGGCUGGC-3′) has 21 nucleotides derived from the first 21 nucleotides of the 3′-end of the cIRES. The two RNAs used in the study showed no fluorescence under the assay conditions. Excitation and emission bandwidths were set to 10 and 20 nm, respectively. The same buffer conditions as above were used in this experiment. The maximum fractional quenching for either cIRES RNA binding to NS5B570-Con1 was ∼25%. The inner filter effect of RNA was also corrected according to Equation 1. The quadratic Equation 2 was used in linear scale to derive dissociation constants based on the fractional fluorescence quenching due to RNA binding. The dissociation constants were found to be the same when the fluorescence quenching results were measured at emission wavelengths of 330 or 340 nm. Microsoft Office ExcelTM 2003 and Graphic PrismTM 4 (GraphicPad Software, Inc., San Diego) were used for data analysis.
To measure NNI binding to the NS5B-cIRES complex, 60 nm cIRES was preincubated with NS5B protein. Prior to the addition of compounds, NS5B-Con1 and cIRES were allowed to equilibrate for 0, 1, or 2 h. The fluorescence signal of the NS5B protein in the presence of 60 nm cIRES was defined as the maximum signal and used to calculate fractional quenching by the compounds. The inner filter effect of RNA was corrected according to Equation 1. The quadratic Equation 2 was used to derive dissociation constants from the fractional quenching of the fluorescence of the NS5B-cIRES complex due to the binding of each compound.
Binding Equilibrium of Compound with NS5B
The equilibrium of compound binding to NS5B was monitored by measuring the fluorescence quenching every 40 s immediately after the addition of NS5B to reaction buffer containing compounds. Blank samples in the presence and absence of enzyme were measured in parallel on the same plate in real time. The observed rate of the binding reaction reaching equilibrium following mixing was analyzed according to Equations 4 and 5,
in which kobs is the rate of the compound binding equilibrium, kon is the rate constant of compound association, and koff is the rate constant of compound dissociation. ΔFobs is the observed fluorescence change, calculated by subtracting the fluorescence in the presence of compound from that in the absence of compound. ΔF0 is the fluorescence change at 0 min, and ΔFm is the maximum fluorescence change at each inhibitor concentration.
The half-life, t½, which represents 50% compound dissociation, was calculated from Equation 6,
IC50 Determination in the HCV NS5B Polymerase Assay
The inhibition potency of NNIs against the RNA-dependent RNA polymerase activity of recombinant NS5B570-Con1 and NS5B570-BK proteins was measured as the incorporation of radiolabeled nucleotide monophosphate into acid-insoluble RNA products, as described (18). The 50% inhibitory concentration (IC50) value refers to the concentration of compound required to reduce the enzymatic activity by 50%. Briefly, IC50 values were measured in 50-μl reaction mixtures containing 20 nm cIRES-378nt RNA template, 1 μCi of tritiated UTP (42 Ci/mmol), a 1 μm concentration of each nucleotide (ATP, CTP, and GTP), 40 mm NaCl, 4 mm dithiothreitol, 4 mm MgCl2, 5 μl of inhibitor (serially diluted in DMSO), and 20 nm enzyme. Kinetic parameters of each enzyme were measured as previously described (22).
EC50 Determinations in the HCV Replicon Assay
The concentration of compound required to inhibit the expression of the HCV replicon reporter by 50% compared with untreated controls (50% effective concentration [EC50]) was determined as described previously using 2209–2223 stably transfected replicon cells (23), except that Huh7-Lunet cells were used for transient transfections (24). The replication capacity of the transient replicons was determined as previously described (20).
Protein Crystallography
NS5B 1–570 Con1 or BK protein (∼7 mg/ml) was mixed in vapor diffusion crystallization drops with an equal volume of well solution (50 mm sodium citrate (pH 4.9), 26% polyethylene glycol 4000, and 7.5% glycerol). The Con1 sample also included 5 mm HCV-796 for co-crystallization. BK apoprotein crystals were soaked with 1 mm HCV-796 for ∼24 h. Crystals were transferred into cryoprotectant (well solution adjusted to 20–25% glycerol) and flash frozen in liquid nitrogen. Data were collected by reciprocal space consulting at beamline 9-2 of the Stanford Synchrotron Radiation Laboratory and reduced with HKL2000 software (25). The molecular replacement solution calculated using Phaser (26) was refined by cycles of Refmac (27, 28), with manual inspection and rebuilding using PyMOL (29) and Coot (30). Molecular images were generated using PyMOL (29). Coordinates are deposited in the Protein Data Bank under Codes 3FQL (Con1) and 3FQK (BK). Compound binding sites were defined as active site, palm 1, palm 2, thumb 1, and thumb 2, based on the location of the binding sites within the NS5B protein structure (Fig. 7).
FIGURE 7.

Locations of NNIs at the palm and thumb binding sites of NS5B polymerase. The overall structure diagram of HCV polymerase shows the finger domain (blue), the palm domain (green), and the thumb domain (red). Inhibitor binding positions are shown for representative non-nucleosides at the allosteric binding sites of NS5B for palm 1 (NNI-1, Protein Data Bank code 2GIQ; bright green), palm 2 (HCV-796; bright blue), thumb 2 (NNI-3, Protein Data Bank code 2GIR; magenta), and thumb 1 (NNI-4, Protein Data Bank code 2BRK; yellow). The locations of resistance mutations are indicated by orange spheres. C316N is a polymorphism found in the NS5B-1b subgenotypes.
RESULTS
Correlation of Binding Affinity and Inhibition Potency of Nonnucleoside Inhibitors of HCV Polymerase
The principle of the tryptophan fluorescence quenching assay has been described to determine compound binding to HCV NS5B and NS3 proteins (31–34). In the present study, a new plate-based fluorescence quenching binding assay was developed to assess whether NNI binding affinity correlates with the potency of inhibition of NS5B polymerase. HCV NS5B genotype 1 proteins possess nine tryptophans located in the palm, thumb, and finger domains of NS5B. Most of the tryptophan residues in NS5B proteins are highly conserved across six genotypes. During assay development, the buffer conditions were adjusted to minimize drift in the tryptophan fluorescence over the course of the binding reaction. A buffer of pH 8.0 was found to reduce the tryptophan fluorescence up to 30% within 1 h, whereas NS5B fluorescence was more stable at lower pH. The addition of detergents such as maltoside also helped to stabilize the NS5B fluorescence signal. NS5B fluorescence was stable for at least 3 h in an optimized assay buffer at pH 7.5 with 0.025% maltoside (Fig. 2A) (see “Experimental Procedures”). Linear intrinsic fluorescence response was observed at NS5B protein concentrations between 6.25 and 200 nm. As an improvement over a cuvette-based approach, the 96-well plate format enabled simultaneous binding tests of multiple compounds to wild-type and mutant NS5B proteins and high throughput analysis of compound binding to NS5B. The negative control of buffer background and the positive control of NS5B protein fluorescence were measured in parallel in real time so that the quenching results could be adjusted accurately, thus reducing interassay variability.
FIGURE 2.

Protein fluorescence of NS5B570-Con1 and binding isotherm of NNI-1 with NS5B570-Con1. Enzyme was tested alone or incubated with increasing concentrations of the compound. The changes of intrinsic protein fluorescence were monitored at the emission spectrum of 310–390 nm, with excitation at 280 nm. The fractional fluorescence quenching of NS5B-Con1 as a function of NNI-1 concentrations was fitted to a quadratic equation (Equation 2) to determine the dissociation constant (Kd). A, the stability of NS5B570-Con1 fluorescence. The excitation/emission wavelength was 280/330 nm. The fluorescence of NS5B titration from 6.25 to 200 nm was monitored up to 3 h 10 min in the binding assay buffer (see “Experimental Procedures”). The values of the NS5B fluorescence in the figure were the values that subtracted the values of 0 nm NS5B blank controls at the corresponding time points. B, fluorescence emission spectrum of 100 nm NS5B570-Con1 in the presence of NNI-1, whose concentration ranged from 0 to 1563 nm in a 2-fold dilution series after a 10-min incubation. The negative control is the signal of the assay buffer in the absence of NNI-1 and NS5B-Con1 (solid square) relative to blanks. The positive control is the signal of NS5B-Con1 fluorescence intensity in the absence of NNI-1 (solid triangles). C, the plot of fractional fluorescence quenching of NS5B-Con1 in the presence of NNI-1 (0–1526 nm) was derived based on the signals at an emission of 330 nm. The Kd value of NNI-1 binding to NS5B-Con1 is 19 ± 10 nm (n = 17).
The spectrum scan of NS5B fluorescence in Fig. 2B shows the typical effect of increasing concentrations of a NNI on NS5B fluorescence. Increasing concentrations of compound decrease NS5B fluorescence intensity without affecting the emission maximum. Compound fluorescence of NNI-1 was the same as the background signal of the buffer only. Interaction of NNI-1, HCV-796, and NNI-4 with HCV NS5B genotype 1 proteins resulted in ∼70% of maximum fluorescence quenching, whereas thumb 2-binding NNI-3 showed ∼50% of maximum quenching. Fig. 2C shows a typical dose-response curve fit using Equation 2 based on the fractional quenching of tryptophan fluorescence by NNI-1. Kd values for representative NNIs binding to different binding sites on NS5B are summarized in Table 1. The Kd values of NNI binding to NS5B were compared with their IC50 values for the inhibition of the NS5B polymerase activity (Table 1). The Kd values of the palm-binding NNI-1 and HCV-796 were comparable with the IC50 values. The Kd value of the thumb 2-binding compound NNI-3 was 6.5-fold lower than the IC50 value, and the thumb 1-binding compound NNI-4 showed similar Kd and IC50 values.
TABLE 1.
The binding affinities and po ly mer ase inhibition potencies for nonnucleoside inhibitors on the NS5B570-Con1 recombinant proteins
| Compound structural class | Binding sites | NS5B570-Con1 (FQ)Kda,b | NS5B570 (Pol) IC50a |
|---|---|---|---|
| μm | μm | ||
| NNI-1 benzothiadiazine | Palm 1 | 0.019 ± 0.010 | 0.009 ± 0.003 |
| HCV-796 benzofuran | Palm 2 | 0.071 ± 0.028 | 0.081 ± 0.019 |
| NNI-3 thiophene | Thumb 2 | 0.026 ± 0.011 | 0.17 ± 0.028 |
| NNI-4 benzimidazole | Thumb 1 | 25 ± 2.4 | 28 ± 4.0 |
a Mean and S.D. values for kinetic constants were determined from at least three experiments.
b The dissociation constant (Kd) values were determined at the binding equilibrium for each compound. TheKd values for NNI-1, -3, and -4 were measured after 10 min of incubation with NS5B, except the value for HCV-796, which was measured after an incubation of 3 h 10 min.
To further assess the relationship between binding affinity and inhibition potency for palm binding compounds, Kd and IC50 values were determined for a larger number of NNI-1 and HCV-796 derivatives using NS5B proteins derived from HCV Con1 and BK strains. The data show a diagonal relationship between Kd and IC50 in linear regression, with R2 values of 0.85 and 0.87 for NS5B-Con1 and NS5B-BK proteins, respectively (Fig. 3, A and B). These results show a good correlation between binding affinity and inhibition of NS5B by palm binding NNIs and suggest that NNI binding affinity to the palm binding sites is predictive of HCV polymerase inhibition potency.
FIGURE 3.

Correlation of Kd values of NS5B binding and IC50 values of NS5B RdRp inhibition for palm-binding compounds. A, Kd versus IC50 for NS5B570-Con1. The R2 of the linear trend line is 0.85. B, Kd versus IC50 for NS5B570-BK. The R2 of the linear trend line is 0.87.
HCV-796 Displays Slow Kinetics of Binding to NS5B Polymerase
The majority of NNIs reached rapid binding equilibrium with NS5B. When binding of NNIs was assessed after a 10-min incubation with NS5B in the fluorescence quenching binding assay, the Kd values of the majority of compounds, including NNI-1, -3, and -4, with NS5B were found to be within 10-fold of their respective IC50 values (Table 1). In contrast, the Kd value of HCV-796 at 10 min was 1.9 ± 0.2 μm (Table 2), which was 23-fold higher than the IC50 value of 0.081 ± 0.019 μm (Table 1). The binding kinetics of HCV-796 with NS5B was therefore established for incubation times up to 6 h. The time-dependent increase in HCV binding affinity was also determined. The Kd value decreased 27-fold from 1.9 ± 0.2 μm at 10 min to 0.07 ± 0.03 μm at 3 h 10 min (Fig. 4A and Table 2). The apparent Kd value became constant after 2 h 10 min. These results indicate that ∼2 h was needed for HCV-796 to reach binding equilibrium. Thus, in contrast to the three other palm- and thumb-binding compounds, HCV-796 shows slow binding kinetics to the NS5B protein.
TABLE 2.
Values of the dissociation constantKd of HCV-796 binding to NS5B570-Con1 in the absence and presence of HCV cIRES-378nt RNA
All of the values shown are mean and S.D. values calculated from three or more independent experiments. The experiments were carried out as described under “Experimental Procedures.”
| HCV cIRES | Preincubation of NS5B and cIRES |
Kd |
|||||
|---|---|---|---|---|---|---|---|
| 10 mina | 40 mina | 1 h 10 mina | 2 h 10 mina | 3 h 10 mina | 4 h 10 mina | ||
| μm | |||||||
| − | − | 1.9 ± 0.2 | 0.33 ± 0.12 | 0.17 ± 0.06 | 0.10 ± 0.04 | 0.07 ± 0.03 | 0.06 ± 0.02 |
| + | 0 h | 3.2 ± 0.3 | 0.73 ± 0.13 | 0.40 ± 0.06 | 0.24 ± 0.05 | 0.17 ± 0.03 | 0.14 ± 0.02 |
| + | 1 h | 2.7 ± 0.5 | 0.66 ± 0.09 | 0.39 ± 0.08 | 0.22 ± 0.03 | 0.19 ± 0.05 | 0.13 ± 0.03 |
| + | 2 h | 3.6 ± 1.7 | 0.75 ± 0.17 | 0.42 ± 0.09 | 0.21 ± 0.06 | 0.17 ± 0.03 | 0.13 ± 0.03 |
a Time for HCV-796 to reach binding equilibrium with NS5B or NS5B-cIRES.
FIGURE 4.

The binding of HCV-796 to NS5B570-Con1 in the fluorescence quenching binding assay. The binding experiments were performed in the absence of HCV cIRES RNA. A, the binding of HCV-796 with NS5B570-Con1 at 10 min, 40 min, 1 h 10 min, 2 h 10 min, 3 h 10 min, and 4 h 10 min. The Kd values are shown in Table 2. B, equilibrium of HCV-796 binding to NS5B570-Con1. Progress toward the binding equilibrium was assessed every 40 s, and concentrations of HCV-796 were 0.5, 1, 2, 4, and 8 μm. C, the rate of NS5B binding as a function of HCV-796 concentrations. The dissociation rate constant (koff) was derived from nonlinear regression curve fitting to Equations 4 and 5, and the t½ was calculated from Equation 6 (see “Experimental Procedures”). The koff value of HCV-796 was 4.9 ± 0.5 × 10−4 s−1, and the t½ was 25 ± 6 min (n = 3). D, the binding of NNI-1 with NS5B570-Con1 at the binding equilibrium of 10 min, 40 min, 1 h 10 min, 2 h 10 min, 3 h 10 min, and 4 h 10 min. E, the equilibrium of NNI-1 binding to NS5B570-Con1. Fluorescence quenching, which indicated progress toward binding equilibrium, was measured every 40 s. Concentrations of NNI-1 were 5, 10, 20, 40, and 80 nm.
To characterize the slow binding properties of HCV-796, the binding of HCV-796 to NS5B570-Con1 protein was measured every 40 s from the beginning of the binding reaction. HCV-796 slowly reached a binding equilibrium with NS5B-Con1 (Fig. 4B), whereas NNI-1 rapidly attained binding equilibrium (Fig. 4E). No time-dependent Kd changes were observed for NNI-1 (Fig. 4D). Similarly, NNI-3 and NNI-4 reached rapid binding equilibrium with NS5B (data not shown). The consistent Kd values suggest that the binding of NNI-1, -3, and -4 to the NS5B protein reaches equilibrium within 10 min. Based on the observed rate of fluorescence quenching in the titration of HCV-796 (Fig. 4C), the rate constants of compound association and dissociation (kon and koff) were calculated as kon = 4.95 ± 0.21 × 103 m−1 s−1 and a koff of 4.85 ± 0.99 × 10−4 s−1 (Equations 4 and 5). The calculated mean Kd from the kinetic binding analysis was 0.098 μm, consistent with the Kd determined from equilibrium binding analysis. The half-life (t½) for 50% dissociation of HCV-796 from the NS5B570-Con1 genotype 1b protein was 24.7 ± 5.7 min. The koff values of HCV-796 binding to other genotype 1 NS5B proteins were 1.39 ± 0.54 × 10−3 s−1 for NS5B570-BK (1b) and 4.85 × 10−3 ± 0.42 × 10−3 s−1 for NS5B570-H77 (1a). The t½ values of HCV-796 dissociation were 9.4 ± 3.6 min from NS5B570-BK and 2.4 ± 0.2 min from NS5B570-H77. Therefore, HCV-796 slowly dissociates from NS5B and thus shows slow binding kinetics to NS5B proteins of genotype 1a and 1b, although dissociation is significantly faster from BK and H77 proteins as compared with Con1.
Surface plasmon resonance was also used to analyze HCV-796 binding to the pocket in palm 2 relative to the binding of other NNIs at other allosteric sites. Surface plasmon resonance gave kon and koff values of HCV-796 binding to NS5B570-Con1 of 5.5 × 104 m−1 s−1 and 9.1 × 10−4 s−1, respectively, and thus a mean Kd value within two S.D. values from the solution-based binding analyses (see Table S1). The results were therefore consistent with those determined using fluorescence quenching (Fig. 4, B and C). The binding of the other three NNIs (NNI-1, -3, and-4) to NS5B570-Con1 showed faster (10- to 30-fold) koff values in the range of 10−2 s−1 determined in surface plasmon resonance (Table S1), which was consistent with the characteristic rapid binding equilibrium observed in the fluorescence quenching experiments.
Comparison of NNI Binding to Free NS5B and NS5B-RNA Complex
The NS5B polymerase interacts with HCV RNA template during RNA polymerization. Previous data suggested that the genomic RNA-bound form of NS5B was resistant to inhibition by NNIs (18). The effect of HCV-RNA on HCV-796 binding to the NS5B-RNA complex was investigated. HCV-cIRES, a complementary sequence of internal ribosomal binding site that directs de novo synthesis of the positive strand RNA, was used in this study. The fluorescence quenching assay system allowed us to determine nucleic acid binding to a protein. First, the binding affinity of NS5B and RNA was determined for a 378-nucleotide, full-length cIRES RNA (cIRES-378nt), and it was compared with the binding of a short, 21-nucleotide RNA oligonucleotide (cIRES-21nt), which is a GC-rich stem-loop region at the 3′-end of the cIRES (Fig. 5A). At binding equilibrium RNA binding to NS5B resulted in ∼25% quenching of NS5B fluorescence. Maximal fluorescence quenching was achieved after 10 min of incubation with RNA, and it remained constant thereafter. The quenching in fluorescence observed following the addition of RNA was not due to the destabilization or photobleaching of the NS5B protein, because NS5B fluorescence in the presence of different RNA concentrations remained constant over the course of the incubation. The binding affinity of NS5B to the cIRES-378nt RNA was 22.5-fold higher than to the cIRES-21nt RNA, and the respective Kd values were 6.9 ± 0.3 and 155.1 ± 16.2 nm.
FIGURE 5.

The binding of HCV-796 to NS5B570-Con1 in the nucleoprotein complex of NS5B570-Con1·HCV-cIRES RNA. A, the binding of NS5B570-Con1 to HCV-cIRES RNA. The experiments were carried out with 100 nm NS5B570-Con1, as described under “Experimental Procedures.” The binding curve shows the fractional quenching of NS5B570-Con1 as a result of binding to cIRES-378nt (white diamonds) or cIRES-21nt (white squares). The Kd values are 6.9 ± 0.3 nm for cIRES-378nt and 155.1 ± 16.2 nm for cIRES-21nt (n = 3). B, the binding of HCV-796 to NS5B570-Con1 complexed with HCV-cIRES RNA at the binding equilibrium after 10 min, 40 min, 1 h 10 min, 2 h 10 min, 3 h 10 min, and 4 h 10 min. The NS5B protein was preincubated with RNA for 1 h before adding the compound to initiate the binding equilibrium as described under “Experimental Procedures.” The level of fractional quenching was calculated based on the maximum fluorescence of NS5B-RNA in the absence of the compound.
To assess NNI binding to the NS5B-RNA complex, 60 nm cIRES-378nt RNA was used to preincubate with NS5B. Fig. 5A showed that 60 nm RNA (>5-fold Kd) had saturated the binding of 100 nm NS5B. The binding of HCV-796 to the NS5B-cIRES complex was determined without preincubation of NS5B with cIRES or following preincubation for 1 or 2 h. The Kd value of HCV-796 was determined at 3 h 10 min, when binding equilibrium had been reached. cIRES RNA binding to NS5B was associated with only a small reduction of 2–3-fold in the affinity of HCV-796 binding to NS5B compared with that in the absence of RNA at equilibrium (Table 2). The kinetics of HCV-796 binding to NS5B was also similar with either free NS5B or NS5B-cIRES RNA complex (Table 2 and Fig. 5B). NNI-1 at the palm 1 binding site bound to NS5B-RNA complex with similar affinity as compared with free NS5B (Table 3). NNI-3 and NNI-4 at thumb binding sites showed 3–6-fold lower binding affinity to NS5B-RNA complex than free NS5B (Table 3). These results suggest that NNIs used in this study can bind to binding sites on palm and thumb of NS5B in the presence of RNA and that RNA binding to NS5B is not likely to cause major changes in the structure of the NNI binding sites on NS5B.
TABLE 3.
Values of the dissociation constantKd of the binding of NNI-1, NNI-3, and NNI-4 with NS5B570-Con1 in the absence and presence of HCV cIRES-378nt RNA
All of the values shown are mean and S.D. values calculated from three or more independent experiments. The experiments were carried out as described under “Experimental Procedures.”
| Compounds | HCV cIRES | Preincubation of NS5B and cIRES |
Kd |
|
|---|---|---|---|---|
| 10 mina | 1 h 10 mina | |||
| μm | ||||
| NNI-1 | − | − | 0.019 ± 0.010 | 0.029 ± 0.013 |
| + | 0 h | 0.019 ± 0.008 | 0.027 ± 0.012 | |
| + | 1 h | 0.018 ± 0.006 | 0.022 ± 0.009 | |
| NNI-3 | − | − | 0.026 ± 0.011 | 0.025 ± 0.005 |
| + | 0 h | 0.086 ± 0.012 | 0.099 ± 0.036 | |
| + | 1 h | 0.082 ± 0.020 | 0.105 ± 0.018 | |
| NNI-4 | − | − | 25 ± 2.4 | 26 ± 12 |
| + | 0 h | 123 ± 9.0 | 72 ± 1.8 | |
| + | 1 h | 162 ± 24 | 89 ± 0.2 | |
a Time of NNI incubation with NS5B or NS5B-cIRES.
Resistance Mutations Reduce NNI Binding Affinity to NS5B in a Site-specific Manner
Observations from clinical studies and in vitro studies using the HCV replicon system have identified mutations on NS5B that can confer resistance to NNIs in an apparent site-specific manner. The benzothiadiazine series, including NNI-1, was reported to select for major resistance mutations at amino acid positions Met414 and Tyr448 in the palm domain, which lies near the enzyme active site (35, 36). Mapping of replicon cells resistant to HCV-796 identified specific mutations in NS5B involving changes at residues Cys316 and Ser365 at the palm binding site (8, 24). Viral resistance selection against thiophene-based carboxylic acid derivatives NNI-3 identified major substitutions at Leu419 and Met423 at the base of one of the thumb domain binding pockets (20), whereas the selection of resistance mutations against the benzimidazole 5′-carboxylic acid series localized at the top of the thumb domain with Pro495 as a key residue (12, 37). For the present study, resistance mutations in binding sites in palm 1 (M414T), thumb 2 (L419M), and thumb 1 (P495L) were selected in order to determine NNI binding specificity (Table 3 and Fig. 7). C316N is a natural polymorphism of NS5B at the palm 2 binding site in the consensus sequences of HCV 1b subtypes. The ability of C316N to reduce inhibitory potency of HCV-796 has been reported (8, 9).
In the panel of NS5B mutant proteins shown in Table 4, the reduced binding affinities of NNIs specifically correlated with the mutations in their respective palm or thumb binding sites. The binding of NNI-1 was decreased 70-fold by the M414T mutation at the palm 1 binding site but not by the single mutations C316N, L419M, or P495L that characterize other binding sites on NS5B. Similarly, the binding affinity of HCV-796 was specifically reduced 14-fold by C316N at the palm 2 site but was not affected by mutations in the thumb binding sites. The binding of NNI-3 was reduced 12-fold by L419M at the thumb 2 binding site. NNI-4 binding was reduced by the thumb 1 mutation P495L. No cross-resistance effect on NNI binding affinity at the palm or thumb binding sites was observed by mutations located at the other allosteric binding sites in this panel of selected mutations. The panel of mutant NS5B proteins could therefore be used in high-throughput binding screening mode to rapidly determine the binding sites of new inhibitors of NS5B.
TABLE 4.
Effects of characteristic mutations at the palm and thumb binding sites of NS5B po ly mer ase on the binding affinities of NNIs
All of theKd values shown are mean and S.D. values calculated from at least three independent experiments. TheKd values are in boldface type when the values of compound binding to mutant proteins are more than 3-fold larger than the respective compound binding to the wild-type NS5B protein.
| Compounds | Binding sites |
Kd |
||||
|---|---|---|---|---|---|---|
| Wild type | M414T | C316Na | L419M | P495L | ||
| μm | ||||||
| NNI-1b Benzothiadiazine | Palm 1 | 0.016 ± 0.01 | 1.3 ± 0.04 | 0.032 ± 0.005 | 0.023 ± 0.004 | 0.018 ± 0.005 |
| HCV-796b Benzofuran | Palm 2 | 0.071 ± 0.02 | 0.043 ± 0.016 | 0.96 ± 0.23 | 0.064 ± 0.016 | 0.045 ± 0.009 |
| NNI-3b Thiophene | Thumb 2 | 0.024 ± 0.015 | 0.048 ± 0.014 | 0.062 ± 0.016 | 0.32 ± 0.06 | 0.067 ± 0.013 |
| NNI-4c Benzimidazole | Thumb 1 | 13 ± 2.3 | 14 ± 2.2 | NAd | 15 ± 2.9 | 40 ± 8.4 |
a C316N is a natural polymorphism in HCV genotype 1b; the residue is C316 in Con1 strain and N316 in the BK strain.
b TheKd values of NNI-1, HCV-796, and NNI-3 were tested on NS5B570_Con1 (GT1b). TheKd values for NNI-1, -3, and -4 were measured after 10 min of incubation with NS5B, except for HCV-796, which was measured after an incubation of 3 h 10 min.
c TheKd values of NNI-4 were tested on NS5B570-BK (GT 1b).
d NA, not available.
Reduced NNI Binding Affinity Is the Key Mechanism of HCV Resistance
In light of site-specific binding determined for the four NNIs on the palm and thumb binding sites of NS5B (Table 4), the reductions in binding affinity were compared with reductions in the sensitivity of HCV polymerase activity and replicon replication to the different inhibitors. An expanded panel of single resistance mutations at NNI binding sites was selected: M414T and Y448H in the palm 1 site, C316N/Y and S365T in the palm 2 site, L419M in the thumb 2 site, and P495L in the thumb 1 site (Fig. 7). The S282T mutation conferring resistance to nucleoside inhibitors (e.g. 2′-C-methyl-cytidine) was also selected as a representative mutation in the active site of HCV RdRp (38). The -fold shifts in Kd values due to the resistance mutations were compared with the -fold shifts in IC50 values. In Fig. 6, A and B, the -fold reduction in Kd values was generally consistent with the -fold reduction in NS5B and replicon inhibitory potency for NNI-1 and HCV-796 and consistent with palm-binding interaction of these compounds. As shown in Fig. 6C, NNI-3 binding to NS5B was significantly affected by mutation L419M, characterizing it as a thumb binding compound. Mutations in the palm domain (M414T, Y448H, and C316N/Y) or the active site (S282T) did not substantially affect binding. The polymerase IC50 values were slightly (3.3-fold) reduced by palm site mutations C316N and C316Y, and NS5B binding (Kd values) and replicon inhibition were similar to wild-type within 3-fold. We did not attempt to determine the statistical significance of these small changes in NS5B enzyme inhibition values relative to wild-type enzyme. The binding affinity and inhibitory potency of the four allosteric inhibitors were not affected by S282T, the active site mutation that confers resistance to 2′-C-methyl-cytidine (NM283) (38). These results indicate that the specific loss of NNI binding affinity to resistant polymerase mutants correlates with the loss of NNI inhibitory potency.
FIGURE 6.

Effects of palm and thumb resistance mutations on binding affinity of inhibitors to NS5B and the potency of the inhibitors to suppress polymerase activity and HCV replicon replication. A, NNI-1; B, HCV-796; C, NNI-3. Compound binding affinity and polymerase-inhibitory activity were tested on NS5B570-Con1 proteins carrying single mutations (M414T, Y448H, C316N, C316Y, S365T, L419M, P495L, or S282T) in the fluorescence quenching binding assay and HCV RNA polymerase assay. Inhibition on the HCV replicon assay was tested as described under “Experimental Procedures” in an HCV bicistronic system carrying the single mutations indicated above. -Fold changes in Kd of mutant proteins were calculated relative to the Kd value measured for wild-type NS5B570-Con1. -Fold changes in IC50 for NS5B and EC50 for HCV replication were calculated based on the respective IC50 values measured for wild-type NS5B protein or the EC50 values for HCV replication. EC50 values of HCV replication with a polymerase bearing the P495 mutation could not be measured because the compounds were toxic to the replicon. NA, not available. White bars, -fold changes in Kd; gray bars, -fold changes in IC50 for NS5B polymerase activity; black bars, -fold changes in EC50 for HCV replication.
Although the binding of NNI-1 at the palm 1 binding site was not affected by C316N, the binding affinity of NNI-1 was reduced 202-fold by C316Y (Fig. 6B), a clinically selected resistance mutation against HCV-796 at the palm 2 binding site (8). In addition, the binding of NNI-1 and HCV-796 were decreased 15- and 226-fold, respectively, by the S365T mutation. The sensitivity of inhibition of the compounds for HCV RdRp and replicon replication was significantly reduced by both the C316Y and S365T single mutations (Fig. 6, A and B) (24). Conversely, the effects of M414T were specific to the palm 1 binding compound NNI-1. The decreased binding affinity and inhibition potency of the two palm-binding compounds to the C316Y and S365T mutant proteins suggests that the palm 1 and palm 2 binding sites partially overlap, as previously suggested (8).
Co-crystal Structures Reveal an Induced Hydrophobic Binding Pocket for HCV-796
We determined the crystal structures of HCV-796 bound to both Con1 and BK strains of NS5B (Table 5). The inhibitor binds near the polymerase active site in a very enclosed, elongated, predominantly hydrophobic pocket between the primer grip motif (residues 364–369) and the central β sheet (strands 214–219, 319–325, and 310–316) in the core of the palm domain. In this structure, Arg200 adopts a different rotamer from that observed in other NS5B structures, which makes this pocket accessible. Residue 316, cysteine in Con1 and asparagine in BK, contacts the inhibitor. The cysteine sulfur of the Con1 sequence fills the concave space between the inhibitor methyl amide and cyclopropyl moieties (Fig. 8A). The BK Asn side chain at this position approaches the inhibitor carbonyl at a reasonable distance for polar interaction. Although this geometry of N316 accommodates the inhibitor surface, it is not ideal for hydrogen bonding (Fig. 8B). It deprives the Asn316 side chain oxygen of its favorable interactions with Arg200 in the apo form of the enzyme. The loss of optimal interaction with Arg200 and Cys316 interaction is consistent with reduced binding affinity of HCV-796 to C316N protein on the Con1 backbone. The change of Asn to Cys at residue 316 on the BK backbone recovered the binding affinity of HCV-796. Modeling a tyrosine at this position to reflect the clinically selected resistance mutation with reduced sensitivity to HCV-796 shows significant steric clash with the inhibitor or the protein itself, regardless of the tyrosine rotamer tested (Fig. 8C). The conformational adaptations necessary to relieve these extensive clashes are consistent with the >200-fold reduction in binding affinity and inhibitory potency for HCV-796 with NS5B-C316Y.
TABLE 5.
Crystallography data for NS5B po ly mer ase bound to HCV-796
| Data set | NS5B570-Con1 | NS5B570-BK |
|---|---|---|
| Copies | 1 | 2 |
| Space group | P212121 | P212121 |
| Cell dimensionsa,b,c (Å) | 60.04, 60.90, 155.23 | 86.29, 106.20, 125.94 |
| Resolution range (Å)a | 40–1.80 (1.86–1.80) | 50–2.2 (2.28–2.20) |
| % Completenessa | 94.2 (66.7) | 93.3 (91.8) |
| I/σ(I)a | 27.51 (1.91) | 10.73 (1.68) |
| Rsym(%)a | 6.5 (50.7) | 13.4 (68.9) |
| Observationsa | 542,754 | 1,393,833 |
| Unique reflectionsa | 50,278 (3510) | 55,845 (5438) |
| Rcrystb | 18.96 | 20.93 |
| Rfreec | 22.36 | 26.42 |
| AverageBfactor (Å2) | ||
| Protein | 20.0 | 27.1 |
| Water molecules | 32.1 | 29.3 |
| Compound | 14.9 | 30.0 |
| Root mean square deviation bond length (Å) | 0.008 | 0.010 |
| Root mean square deviation bond angle (degrees) | 1.157 | 1.211 |
| Ramachandran plot (percentage of amino acids in region) | ||
| Favored | 93.2 | 92.2 |
| Allowed | 6.8 | 7.8 |
| Generous | 0.0 | 0.0 |
| Disallowed | 0.0 | 0.0 |
a Numbers in parentheses are for the highest resolution shell.
bRcryst= Σhkl‖Fobs| − |〈Fc〉‖/Σhkl|Fobs|.
c Rfree,Rcrystwith 5% ofFobsexcluded from refinement.
FIGURE 8.

The co-crystal structures of HCV-796 binding to the palm domain of HCV NS5B polymerase. A, crystal structure details of HCV-796 binding to NS5B570-Con1 (genotype 1b) with cysteine at position 316 (pink highlight). B, crystal structure details of HCV-796 binding to NS5B570-BK (1b) carrying asparagine at position 316 (pink highlight). C, modeling of HCV-796 binding to NS5B protein with tyrosine at position 316 (orange highlight).
The Con1 crystal structure reported in this study is the first of this strain represented in the Protein Data Bank, which also establishes a novel crystal form. Of note, a register shift near the C terminus of the protein causes significant rearrangement in the interior polymerase cavity. The C-α of Tyr555 is shifted by ∼5.5 Å, whereas the hydroxyl at the tip of the side chain is moved over 13 Å. In the new position, this tyrosine side chain occludes a portion of the palm 1 binding pocket and is only 3.7 Å from the cyclopropyl moiety of HCV-796, further constricting the only obvious route of egress.
DISCUSSION
The NS5B polymerase represents an attractive target for the identification of antiviral agents to treat chronic HCV infection. The non-nucleoside inhibitors selected for this study have been shown to bind the palm or thumb binding sites and effectively inhibit replication of HCV subgenomic replicons (1, 2). The analysis of NNI binding affinity to NS5B suggests a correlation of binding affinity with inhibitory potency of NNIs, in particular within a large set of palm binding compounds. Notably, this study identified HCV-796 as an unusual slow binding inhibitor of NS5B as compared with a large set of NNIs tested. In addition, the co-crystallization of NS5B and HCV-796 revealed a compound-induced binding pocket, consistent with slow binding kinetics of HCV-796.
Several lines of evidence in this study establish that HCV-796 is a slow dissociation compound to NS5B. The binding kinetics of HCV-796 is different from those of the other three NNIs (Fig. 1). First, HCV-796 exhibited a time-dependent increase in NS5B binding affinity and slow binding equilibrium in the fluorescence quenching binding assay. Second, a slow dissociation rate constant on the order of 10−4/s was determined for the HCV-796/NS5B interaction in both fluorescence quenching and surface plasmon resonance experiments. In addition, HCV-796 showed slow binding kinetics of interaction with the NS5B-RNA complex. Although a clinical trial of HCV-796 was recently stopped due to elevation in liver enzymes, HCV-796 remains the first allosteric inhibitor shown to have significant antiviral potency in HCV-infected patients. Previously, a number of HCV protease inhibitors, including SCH-503034 and ITMN-191, were reported to be slow binding inhibitors (39–41). Slow binding with slow dissociation is also a notable feature of non-nucleoside inhibitors of the reverse transcriptase of the human immunodeficiency virus (42). Tight binding and a long dissociation half-life are hallmarks of slow binding inhibitors and are expected to provide pharmacological benefit with regard to sustained antiviral effects at the target site. A rapid screening system for the identification of slow binding inhibitors is therefore useful for early characterization of antiviral lead compounds.
The co-crystal structure of HCV-796 with NS5B protein that was solved as part of this study supports the findings of slow binding kinetics and effects of resistance mutations on binding affinity of HCV-796. Relative to the apo crystal structure conformation of NS5B, the binding of HCV-796 causes multiple structural rearrangements, notably a rotamer shift of Arg200 and a setback of the primer grip loop backbone. These conformational changes induce the HCV-796 binding pocket formation. In contrast to the broad open surface exposed in the palm 1 binding pocket, this new palm 2 binding pocket is deep, narrow, and enclosed descending from the palm 1 binding site. HCV-796 and its structural variants, including HCV-086, display not only slow dissociation rate constants but also slow association constants (data not shown). Although some potent palm 1-binding compounds have been observed to have slow binding kinetics, the ubiquity of this characteristic for palm 2-binding compounds studied to date suggests that the NNI binding site in palm 2 has unique structural features. The deep enclosure within the core of the conserved and highly structured palm domain, featuring narrow entry and exit portals and further gating provided by Arg200 motions, may explain this slow binding. However, the slow binding of many compounds cannot be predicted based on the structures of binding pockets. In addition, compounds binding in the same pocket display different binding kinetics. A subtle movement or a minor conformational change in a side chain in a binding pocket may reverse the slow binding properties of a compound. Nevertheless, the slow binding of the HCV-796 series to the palm 2 binding site is unaffected by the binding of the HCV RNA template to NS5B. Moreover, the resistance mutations in this study, including C316N and C316Y, do not eliminate the slow binding properties of HCV-796. Therefore, the deep binding pocket in palm 2 is likely to contribute to the characteristically slow binding of HCV-796. The slow binding kinetics of HCV-796 were most pronounced in the Con1 strain. It is tempting to rationalize these data in light of the unique structural rearrangement of the C-terminal tail of that protein observed in the crystal structure. This rearrangement further narrows the only channel into or out of the binding site. However, further Con1 structures are needed to determine whether this structural shift represents an inherent predisposition of the Con1 sequence with accompanying physiological consequences, is simply an artifact of the novel crystal form, or is a coincidental arrangement of a putatively labile portion of the protein.
The fluorescence quenching assay system could also be used for the assessment of RNA binding to NS5B and its impact on NNI binding. The NS5B protein has a substantially higher binding affinity to the 378-nt, full-length cIRES RNA than to the short, 21-nt 3′-end RNA of cIRES (Fig. 5A). This finding is consistent with the hypothesis of oligomerization of NS5B on longer RNA molecules to contribute to overall RNA binding affinity. An apparent binding affinity of ∼10 nm has previously been reported for the binding of NS5B to a 1442-nt RNA in a gel mobility shift assay (32). Conversely, high dissociation constants have been reported for NS5B binding to short RNAs. A Kd of ∼250 nm for a 16-mer single-stranded RNA and a 13/16-mer double-stranded RNA substrate was measured by equilibrium fluorescence titration (43). In addition, a Kd of 420 nm was determined for a 14-mer RNA using fluorescence polarization (14, 44). These previous data are in agreement with our results. The long cIRES RNA therefore is likely to provide a substrate for the cooperative binding of NS5B protein. Note that the short 3′-end stem-loop structure of cIRES-21nt is sufficient to initiate the de novo synthesis of viral RNA, but the initiation is inefficient. It is conceivable that oligomerization-dependent, strong binding affinity of the NS5B enzyme for the full-length cIRES RNA also increases the efficiency of RNA replication initiation.
The binding of RNA to NS5B did not appear to alter the structure of the palm 2 NNI binding pocket. cIRES RNA binding to NS5B shows minimal interference with the binding affinity of HCV-796 (Table 2). The slow binding kinetics of HCV-796 remains unchanged in the presence of RNA. The fact that HCV-796 maintained its high binding affinity to NS5B in the presence of HCV RNA (Table 2) indicates that RNA binding does not affect the initial binding of HCV-796 to NS5B. Although all compounds included in this study were either not affected or only moderately affected by RNA binding (Tables 2 and 3), we also found that certain structural classes of thumb-binding NNIs for NS5B were dramatically affected by RNA (data not shown). RNA contacts at the two thumb binding sites of NS5B have been demonstrated by co-crystallization and cross-linking studies (4, 6, 44). Contacts between HCV RdRp and the RNA template have been reported to change during different steps in RNA-dependent RNA synthesis. The binding of RNA may therefore have a different effect on the thumb and palm subdomains. A significant movement of the thumb subdomain during the formation of the NS5B-RNA complex in HCV replication may alter NNI binding affinity. We are currently comparing the effects of HCV RNA on NNI binding to NS5B at thumb binding pockets with the effects at palm binding pockets.
The NNI binding profile of wild-type and mutant NS5B proteins provides direct evidence of the site-specific binding by the palm and thumb binding compounds (Table 4). Because the resistance mutations cause consistent changes in Kd values of NS5B binding and in IC50 values of RdRp inhibition and replicon replication (Fig. 6), the binding profile from the high throughput FQ binding assay can be used to rapidly predict the binding site of novel leads and their resistance profile. The loss of binding affinity to mutant enzymes observed here was consistent with the emergence of site-specific resistance. We used a panel of NS5B recombinant proteins with characteristic mutations at palm binding sites (M414T and C316N) and thumb binding sites (L419M and P495L) in order to identify binding sites for novel allosteric inhibitors of NS5B (Table 4 and Fig. 7). Recently, changes in the IC50 value as a function of site mutation in NS5B were proposed to be useful for the early determination of NNI binding sites on NS5B (45). Thus, the use of a panel of site-specific NS5B mutant proteins in the binding assay like the one described here can be helpful for identifying binding pockets of allosteric inhibitors before co-crystal structures are available.
Residue 316 of the NS5B enzyme is critical to resistance development against palm-binding compounds, such as NNI-1 and HCV-796, and there is also natural sequence polymorphism at this position, which may be clinically significant (8). The introduction of the single mutation C316N in the Con1 protein reduces HCV-796 binding affinity to NS5B by 14-fold, which is comparable with the reduction in potency of polymerase inhibition and replication caused by the mutation. These results, coupled with the interaction between HCV-796 and the Asn316 residue of NS5B-BK in the co-crystal structure, which appear less energetically favorable than the interaction between the compound and C316 of NS5B-Con1, suggest that a single change at residue 316 of NS5B can cause profound changes in the efficacy of a compound (Fig. 8, A and B). Clinical selection of C316Y as the major HCV-796 resistance mutation was also consistent with a key role of Cys316 in HCV-796 binding affinity. C316Y caused a loss of ∼100-fold in binding affinity of the compound for the mutant NS5B as well as in the ability of the compound to inhibit the enzyme. The modeling of the large side chain of C316Y indicates a steric clash with HCV-796 (Fig. 8C). In addition, the single mutations C316Y and S365T were found to confer high resistance and reduced binding affinity of NNI-1 to palm 1 and of HCV-796 to palm 2. The fact that many single mutations identified in HCV replicon significantly affect compound binding and polymerase inhibition suggests a clinically significant low barrier to resistance for allosteric NNIs. The understanding of the mode of NNI binding and cross-resistance profiles between NNIs will help guide optimal combination therapies and the design of new compounds that retain binding affinity and activity against mutant NS5B variants.
Although the reduced NNI binding affinity to NS5B results in major resistance effects and NNI polymorphisms, a few resistance mutations work by different mechanisms. For instance, the single mutation H95R has been reported as a resistance mutation of benzothiadiazine (14), but this mutation did not affect compound binding to the NS5B-Con1 protein. H95R has been implicated in binding to the RNA template (6), and it has been proposed to affect the ability of the polymerase to recognize the viral genome. Further study of NNI binding to NS5B protein with RNA template is likely to address this hypothesis.
HCV polymerase is a unique antiviral target that possesses at least four allosteric binding sites suitable for accommodating structurally diverse classes of antiviral compounds. Information regarding the mode of NNI binding has made important contributions to the development of potent inhibitors through compound screening and clinical development. With the focus of HCV drug discovery shifting toward the optimization of sustained binding affinity and higher resistance barriers in the development of potent inhibitors, detailed study of compound binding will facilitate selection of clinical leads. Evolution of NS5B allosteric inhibitors has shown promise for developing these compounds into clinical candidates. Combination treatment that includes HCV polymerase inhibitors will be required to counter the emergence of resistance and provide the best treatment coverage of the various genotypes and their respective subtypes of HCV.
Supplementary Material
Acknowledgments
We thank Dr. Jim Barnett and members of the Molecular Protein Science Group for providing the wild-type and mutant recombinant proteins of HCV NS5B. We thank Todd Elworthy, Robert Than Hendricks, Lina Setti, and Jason Harris for synthesizing the compounds used in this work.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1.
The atomic coordinates and structure factors (codes 3FQK and 3FQL) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- HCV
- hepatitis C virus
- cIRES
- complementary internal ribosome entry site
- FQ
- fluorescence quenching
- NNI
- non-nucleoside inhibitors
- nt
- nucleotide(s)
- RdRp
- RNA-dependent RNA polymerase.
REFERENCES
- 1.Beaulieu P. L. ( 2006) Curr. Opin. Drug Discov. Devel. 9, 618– 626 [PubMed] [Google Scholar]
- 2.Beaulieu P. L. ( 2007) Curr. Opin. Investig. Drugs 8, 614– 634 [PubMed] [Google Scholar]
- 3.Bressanelli S., Tomei L., Rey F. A., De Francesco R. ( 2002) J. Virol. 76, 3482– 3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bressanelli S., Tomei L., Roussel A., Incitti I., Vitale R. L., Mathieu M., De Francesco R., Rey F. A. ( 1999) Proc. Natl. Acad. Sci. U. S. A. 96, 13034– 13039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lesburg C. A., Cable M. B., Ferrari E., Hong Z., Mannarino A. F., Weber P. C. ( 1999) Nat. Struct. Biol. 6, 937– 943 [DOI] [PubMed] [Google Scholar]
- 6.O'Farrell D., Trowbridge R., Rowlands D., Jager J. ( 2003) J. Mol. Biol. 326, 1025– 1035 [DOI] [PubMed] [Google Scholar]
- 7.Dhanak D., Duffy K. J., Johnston V. K., Lin-Goerke J., Darcy M., Shaw A. N., Gu B., Silverman C., Gates A. T., Nonnemacher M. R., Earnshaw D. L., Casper D. J., Kaura A., Baker A., Greenwood C., Gutshall L. L., Maley D., DelVecchio A., Macarron R., Hofmann G. A., Alnoah Z., Cheng H. Y., Chan G., Khandekar S., Keenan R. M., Sarisky R. T. ( 2002) J. Biol. Chem. 277, 38322– 38327 [DOI] [PubMed] [Google Scholar]
- 8.Howe A. Y., Cheng H., Johann S., Mullen S., Chunduru S. K., Young D. C., Bard J., Chopra R., Krishnamurthy G., Mansour T., O'Connell J. ( 2008) Antimicrob. Agents Chemother. 52, 3327– 3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Villano S., Howe A. Y., Raible D., Harper D., Speth J., Bichier G. ( 2006) Hepatology 44, 607A [Google Scholar]
- 10.Villano S., Raible D., Harper D., Speth J., Chandra P., Shaw P., Bichier G. ( 2007) Hepatology 46, S24 [Google Scholar]
- 11.Chan L., Pereira O., Reddy T. J., Das S. K., Poisson C., Courchesne M., Proulx M., Siddiqui A., Yannopoulos C. G., Nguyen-Ba N., Roy C., Nasturica D., Moinet C., Bethell R., Hamel M., L'Heureux L., David M., Nicolas O., Courtemanche-Asselin P., Brunette S., Bilimoria D., Bedard J. ( 2004) Bioorg. Med. Chem. Lett. 14, 797– 800 [DOI] [PubMed] [Google Scholar]
- 12.Kukolj G., McGibbon G. A., McKercher G., Marquis M., Lefebvre S., Thauvette L., Gauthier J., Goulet S., Poupart M. A., Beaulieu P. L. ( 2005) J. Biol. Chem. 280, 39260– 39267 [DOI] [PubMed] [Google Scholar]
- 13.Di Marco S., Volpari C., Tomei L., Altamura S., Harper S., Narjes F., Koch U., Rowley M., De Francesco R., Migliaccio G., Carfi A. ( 2005) J. Biol. Chem. 280, 29765– 29770 [DOI] [PubMed] [Google Scholar]
- 14.Tomei L., Altamura S., Bartholomew L., Bisbocci M., Bailey C., Bosserman M., Cellucci A., Forte E., Incitti I., Orsatti L., Koch U., De Francesco R., Olsen D. B., Carroll S. S., Migliaccio G. ( 2004) J. Virol. 78, 938– 946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howe A. Y., Cheng H., Thompson I., Chunduru S. K., Herrmann S., O'Connell J., Agarwal A., Chopra R., Del Vecchio A. M. ( 2006) Antimicrob. Agents Chemother. 50, 4103– 4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomei L., Altamura S., Paonessa G., De Francesco R., Migliaccio G. ( 2005) Antiviral Chem. Chemother. 16, 225– 245 [DOI] [PubMed] [Google Scholar]
- 17.Liu Y., Jiang W. W., Pratt J., Rockway T., Harris K., Vasavanonda S., Tripathi R., Pithawalla R., Kati W. M. ( 2006) Biochemistry 45, 11312– 11323 [DOI] [PubMed] [Google Scholar]
- 18.Ma H., Leveque V., De Witte A., Li W., Hendricks T., Clausen S. M., Cammack N., Klumpp K. ( 2005) Virology 332, 8– 15 [DOI] [PubMed] [Google Scholar]
- 19.Krieger N., Lohmann V., Bartenschlager R. ( 2001) J. Virol. 75, 4614– 4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Pogam S., Kang H., Harris S. F., Leveque V., Giannetti A. M., Ali S., Jiang W. R., Rajyaguru S., Tavares G., Oshiro C., Hendricks T., Klumpp K., Symons J., Browner M. F., Cammack N., Najera I. ( 2006) J. Virol. 80, 6146– 6154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lackowicz J. R. ( 1999) Principles of Fluorescence Spectroscopy, 2nd Ed., pp. 446– 514, Kluwer Academic/Plenum Publishers, New York [Google Scholar]
- 22.Klumpp K., Leveque V., Le Pogam S., Ma H., Jiang W. R., Kang H., Granycome C., Singer M., Laxton C., Hang J. Q., Sarma K., Smith D. B., Heindl D., Hobbs C. J., Merrett J. H., Symons J., Cammack N., Martin J. A., Devos R., Najera I. ( 2006) J. Biol. Chem. 281, 3793– 3799 [DOI] [PubMed] [Google Scholar]
- 23.Le Pogam S., Jiang W. R., Leveque V., Rajyaguru S., Ma H., Kang H., Jiang S., Singer M., Ali S., Klumpp K., Smith D., Symons J., Cammack N., Najera I. ( 2006) Virology 351, 349– 359 [DOI] [PubMed] [Google Scholar]
- 24.McCown M. F., Rajyaguru S., Le Pogam S., Ali S., Jiang W. R., Kang H., Symons J., Cammack N., Najera I. ( 2008) Antimicrobial Agents Chemother. 52, 1604– 1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otwinowski Z., Minor W. ( 1997) Methods Enzymol. 276, 307– 326 [DOI] [PubMed] [Google Scholar]
- 26.McCoy A. J., W., G.-K. R., Storoni L. C., Read R. J. ( 2005) Acta Crystallogr. Sect. D 61, 458– 464 [DOI] [PubMed] [Google Scholar]
- 27.Collaborative Computational Project N., ( 1994) Acta Crystallogr. Sect. D 50, 760– 76315299374 [Google Scholar]
- 28.Murshudov G. N., Vagin A. A., Dodson E. J. ( 1997) Acta Crystallogr. Sect. D 53, 240– 255 [DOI] [PubMed] [Google Scholar]
- 29.Delano W. L. ( 2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA [Google Scholar]
- 30.Emsley P., Cowtan K. ( 2004) Acta Crystallogr. Sect. D 60, 2126– 2132 [DOI] [PubMed] [Google Scholar]
- 31.Bougie I., Bisaillon M. ( 2003) J. Biol. Chem. 278, 52471– 52478 [DOI] [PubMed] [Google Scholar]
- 32.Bougie I., Charpentier S., Bisaillon M. ( 2003) J. Biol. Chem. 278, 3868– 3875 [DOI] [PubMed] [Google Scholar]
- 33.Frick D. N., Rypma R. S., Lam A. M., Gu B. ( 2004) J. Biol. Chem. 279, 1269– 1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howe A. Y., Bloom J., Baldick C. J., Benetatos C. A., Cheng H., Christensen J. S., Chunduru S. K., Coburn G. A., Feld B., Gopalsamy A., Gorczyca W. P., Herrmann S., Johann S., Jiang X., Kimberland M. L., Krisnamurthy G., Olson M., Orlowski M., Swanberg S., Thompson I., Thorn M., Del Vecchio A., Young D. C., van Zeijl M., Ellingboe J. W., Upeslacis J., Collett M., Mansour T. S., O'Connell J. F. ( 2004) Antimicrob. Agents Chemother. 48, 4813– 4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen T. T., Gates A. T., Gutshall L. L., Johnston V. K., Gu B., Duffy K. J., Sarisky R. T. ( 2003) Antimicrob. Agents Chemother. 47, 3525– 3530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mo H., Lu L., Pilot-Matias T., Pithawalla R., Mondal R., Masse S., Dekhtyar T., Ng T., Koev G., Stoll V., Stewart K. D., Pratt J., Donner P., Rockway T., Maring C., Molla A. ( 2005) Antimicrob. Agents Chemother. 49, 4305– 4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomei L., Altamura S., Bartholomew L., Biroccio A., Ceccacci A., Pacini L., Narjes F., Gennari N., Bisbocci M., Incitti I., Orsatti L., Harper S., Stansfield I., Rowley M., De Francesco R., Migliaccio G. ( 2003) J. Virol. 77, 13225– 13231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Migliaccio G., Tomassini J. E., Carroll S. S., Tomei L., Altamura S., Bhat B., Bartholomew L., Bosserman M. R., Ceccacci A., Colwell L. F., Cortese R., De Francesco R., Eldrup A. B., Getty K. L., Hou X. S., LaFemina R. L., Ludmerer S. W., MacCoss M., McMasters D. R., Stahlhut M. W., Olsen D. B., Hazuda D. J., Flores O. A. ( 2003) J. Biol. Chem. 278, 49164– 49170 [DOI] [PubMed] [Google Scholar]
- 39.Malcolm B. A., Liu R., Lahser F., Agrawal S., Belanger B., Butkiewicz N., Chase R., Gheyas F., Hart A., Hesk D., Ingravallo P., Jiang C., Kong R., Lu J., Pichardo J., Prongay A., Skelton A., Tong X., Venkatraman S., Xia E., Girijavallabhan V., Njoroge F. G. ( 2006) Antimicrob. Agents Chemother. 50, 1013– 1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rajagopalan P. T. R., Stevens S., Lam D., Stoycheva A., Brandhuber B., Zhang H., Serebryany V., Gale M., Jr., Blatt L. M., Beigelman L., Seiwert S. D., Kossen K. ( 2007) in 58th Annual Meeting of the American Association for the Study of Liver Diseases, Boston, MA, November 2–6, 2007, Hepatology, Abstr. A 1386, John Wiley & Sons, Inc., Hoboken, NJ [Google Scholar]
- 41.Liu Y., Saldivar A., Bess J., Solomon L., Chen C. M., Tripathi R., Barrett L., Richardson P. L., Molla A., Kohlbrenner W., Kati W. ( 2003) Biochemistry 42, 8862– 8869 [DOI] [PubMed] [Google Scholar]
- 42.Spence R. A., Kati W. M., Anderson K. S., Johnson K. A. ( 1995) Science 267, 988– 993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cramer J., Jaeger J., Restle T. ( 2006) Biochemistry 45, 3610– 3619 [DOI] [PubMed] [Google Scholar]
- 44.Kim Y. C., Russell W. K., Ranjith-Kumar C. T., Thomson M., Russell D. H., Kao C. C. ( 2005) J. Biol. Chem. 280, 38011– 38019 [DOI] [PubMed] [Google Scholar]
- 45.Pauwels F., Mostmans W., Quirynen L. M., van der Helm L., Boutton C. W., Rueff A. S., Cleiren E., Raboisson P., Surleraux D., Nyanguile O., Simmen K. A. ( 2007) J. Virol. 81, 6909– 6919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Copeland R. A. ( 2005) Evaluation of Enzyme Inhibitors in Drug Discovery, pp. 1– 271, John Wiley & Sons, Inc., Hoboken, NJ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.