

Abstract
Atypical hemolytic uremic syndrome (aHUS) is a thrombotic microangiopathy associated with mutations in complement proteins, most frequently in the main plasma alternative pathway regulator factor H (FH). The hotspot for the FH mutations is in domains 19–20 (FH19–20) that are indispensable for FH activity on C3b bound covalently to host cells. In aHUS, down-regulation of cell-bound C3b by FH is impaired, but it is not clear whether this is due to an altered FH binding to surface-bound C3b or to cell surface structures. To explore the molecular pathogenesis of aHUS we tested binding of 14 FH19–20 point mutants to C3b and its C3d fragment, mouse glomerular endothelial cells (mGEnC-1), and heparin. The cell binding correlated well, but not fully, with heparin binding and the cell binding site was overlapping but distinct from the C3b/C3d binding site that was shown to extend to domain 19. Our results show that aHUS-associated FH19–20 mutants have different combinations of three primary defects: impaired binding to C3b/C3d, impaired binding to the mGEnC-1 cells/heparin, and, as a novel observation, an enhanced mGEnC-1 cell or heparin binding. We propose a model of the molecular pathogenesis of aHUS where all three mechanisms lead eventually to impaired control of C3b on the endothelial cell surfaces. Based on the results with the aHUS patient mutants and the overlap in FH19–20 binding sites for mGEnC-1/heparin and C3b/C3d we conclude that binding of FH19–20 to C3b/C3d is essential for target discrimination by the alternative pathway.
Atypical hemolytic uremic syndrome (aHUS)2 is a familial disease characterized by erythrocyte fragmentation and hematuria, damaged renal endothelium, vascular microthrombi, and thrombocytopenia (1). The syndrome leads ultimately to end-stage renal disease with a high mortality rate (2). In aHUS cases point mutations have been found in complement components C3, factor B, CD46, factor I, and factor H (FH), all of which play a role in the activation or control of the alternative pathway (3–8). More than half of the mutations have been found to originate in the HF1 gene that encodes FH and FH-like protein 1.
The alternative pathway is initiated spontaneously by hydrolysis of C3 to C3H2O that forms the C3-convertase C3H2OBb (9, 10). This enzyme complex converts numerous C3 molecules to C3b that are covalently bound onto practically any nearby surface (11). On a so-called activator surface, such as a microbe, the surface-bound C3b molecules are not efficiently eliminated and therefore new C3bBb complexes are formed leading to more C3b depositions and eventually effective opsonization or damage of the target cell. On non-activator surfaces, such as viable self (host) cells, factor I cleaves C3b to inactive C3b (iC3b) in the presence of one of the cofactors (CD46, CD35, FH, and FHL-1) (12–16). FH is the only one of these cofactors that mediates recognition of self-surfaces making the alternative pathway capable of discriminating between activating and non-activating surfaces (17–19).
The two main functions of FH are to prevent the alternative pathway activation in plasma and on self-surfaces. This 150-kDa glycoprotein consists of 20 tandemly arranged short consensus repeat domains that are composed of ∼60 amino acids. Domains 1–4 are essential for the cofactor and decay accelerating activity (20). In the middle region of FH (domains 5–15) there are two binding sites for C-reactive protein (21), one or two sites for glycosaminoglycans (GAGs) (22–25), and one site for C3c part of C3b (C3b/C3c) (25, 26). The C-terminal domains 19–20 (FH19–20) possess binding sites for the thiol ester domain of C3b (C3d or C3dg, TED domain) and GAGs (26, 27).
The most common types of mutations found in aHUS are FH missense mutations located within FH19–20 that was recently solved as crystal and NMR structures (2, 28, 29). The C terminus of FH is crucial in self-cell protection as demonstrated by the severity of the aHUS cases and also in a recent mouse model of aHUS where domains 16–20 had been deleted (30, 31). Histopathology of aHUS in these mice had all the characteristics of human aHUS being concordant with the similarity of binding sites for C3b, heparin, and human umbilical vein endothelial cells between human and mouse FH domains 18–20 (32). Binding of mouse or human FH to glomerular endothelial cells has not been characterized despite the fact that in aHUS damage occurs mainly in the small vessels, especially in the glomeruli.
The molecular pathogenesis leading to the clinical aHUS in patients with FH mutations remains elusive. The suggested molecular mechanisms for some aHUS-associated mutations include defective binding of the mutated FH to GAGs, endothelial cells, or C3b/C3d (28, 29, 33, 34). The aim of this study was to define the effects of nine aHUS-associated FH mutations and five other structurally closely located mutations on binding of FH19–20 to C3b, C3d, mouse glomerular endothelial cells, and heparin. We identified three primary defects of the mutants: impaired C3b/C3d binding, enhanced mGEnC-1/heparin binding, and impaired mGEnC-1/heparin binding that could lead via three mechanisms to incapability of FH to eliminate C3b on plasma-exposed self-cells. The results clarify the mechanism of target discrimination of the alternative pathway by the C terminus of FH.
EXPERIMENTAL PROCEDURES
Mutagenesis, Expression, and Purification of the Mutant Proteins
Fourteen point mutations were introduced to the FH19–20 encoding DNA sequence in the pPICZαB vector as described earlier (29). Five of the mutations have been published earlier (29). Of the nine new mutants six had been associated with aHUS, whereas three were non-aHUS associated. The residue Trp-1157 was mutated to leucine instead of arginine found in aHUS to avoid global changes in the structure of FH19–20. The residue Arg-1210 was substituted with alanine instead of cysteine found in aHUS to prevent a surface-exposed cysteine to make covalent bonds (35). The previously published mutation of residue Arg-1182 to alanine was done before the mutation R1182S was described. The residues Asp-1119 and Gln-1139 were mutated because they are located at the interface between the four protomers found in the crystal structure of FH19–20 where they may contribute to oligomerization of FH19–20. For the non-aHUS-associated residues alanine substitution was chosen to preserve the fold of the protein and the backbone dihedral angles.
For mutagenesis the following primers were used (mutated codon in italic font): CCT ATT GAC AAT GGG GGC ATT ACT TCA TTC CCG (for mutation D1119G), TT GAG TAC CAA TGC GCG AAC TTG TAT CAA CTT GAG GG (Q1139A), GT AGA AAT GGA CAA TTG TCA GAA CCA CCA AAA TGC (W1157L), CA TTA AGG TGG AGA GCC AAA CAG AAG CTT TAT TCG (T1184R), ACA GCC AAA CAG AAG CGT TAT TCG AGA ACA GG (L1189R), AA CGG GGA TAT GCT CTT TCA TCA CGT TCT CAC ACA TTG C (R1206A), A CGG GGA TAT CGT CTT TCA TCA GCT TCT CAC ACA TTG C (R1210A), and GT TCT CAC ACA TTG CAA ACA ACA TGT TGG GAT GG (R1215Q). The plasmids harboring mutated FH19–20 were sequenced and used in the Pichia pastoris expression system using 1% methanol induction as described previously (29). Purification of the above mentioned mutant proteins, the wild-type FH19–20, and the previously described mutant constructs (R1182A, W1183L, K1186A, K1188A, and E1198A) were performed using heparin affinity chromatography after filtration, 2-fold dilution with H2O, and pH adjustment of the culture supernatant to 6.0–6.5. Thereafter the mutants were further purified with a Sephadex 75 10/300 GL gel filtration column using the ÄKTA HPLC system (GE Healthcare). The recombinant proteins were resolved by 15% SDS-PAGE gels in reducing and non-reducing conditions and purity was assessed after visualization by Coomassie Brilliant Blue staining. Antigenicity of the proteins was verified using Western blotting with polyclonal goat anti-human factor H antibody (Calbiochem), horseradish peroxidase-conjugated donkey anti-goat antibody (Jackson ImmunoResearch, West Grove, PA), and Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life Sciences).
The recombinant C3d was expressed and purified as described earlier (36), except that in place of DEAE-Sephacel, the first chromatographic step employed a column of CM-Sepharose Fast Flow (GE Healthcare) equilibrated with 10 mm sodium acetate, 20 mm NaCl, and 1 mm EDTA, pH 5.5. After washing, the column was step eluted with the same buffer containing 0.4 m NaCl. The used C3b protein was prepared from plasma-purified C3 using trypsin as described previously (37). Concentrations of all the proteins were measured using the BCA assay (Thermo Scientific).
Radioligand Assay
The wild-type FH19–20 was labeled with 125I using IODO-GEN (specific activity of 2.4 × 106 cpm/μg). Nunc Polysorp Break Apart plates (Nunc) were coated with 5 μg/ml of purified C3b or recombinant C3d in 50% phosphate-buffered saline (PBS) (a mixture of PBS with distilled water in a 1:1 ratio) for 17 h at +4 °C. The wells were blocked with 1% bovine serum albumin in 50% PBS for 2 h at +22 °C and washed once with 50% PBS. Serial dilutions of the non-labeled constructs in 50% PBS were mixed with 125I-FH19–20 (final concentration 1 μg/ml) in separate polypropylene 96-well microplates (Greiner Bio One, Frickenhausen, Germany) followed by transfer onto the C3b- or C3d-coated wells and incubation for 2 h at +22 °C. After two washes with 50% PBS, radioactivity of the separated wells was measured with a γ-counter (Wallac, Turku, Finland). The inhibition curves were fitted using non-linear regression and “log(inhibitor) versus response” model. The mean 50% inhibitory concentrations (IC50) were calculated from the fitted curves using GraphPad Prism version 5.01 for Windows (GraphPad Software, CA). The assay was performed three times in duplicate wells.
Binding of the FH19–20 Proteins to Mouse Glomerular Endothelial Cells as Determined in Enzyme Immunoassay
Conditionally immortalized mouse glomerular endothelial cells (mGEnC-1) with all features of primary glomerular endothelial cells were cultured in 96-well plates as described in Rops et al. (38). The cells were activated with 10 ng/ml of tumor necrosis factor-α (Peprotech, Rocky Hill, NJ) for 18 h to mimic the inflammatory conditions of aHUS (39). The cells were washed with PBS and the FH19–20 proteins, diluted serially in PBS with 2% bovine serum albumin, were incubated in the wells for 2 h at 37 °C in 5.0% CO2 atmosphere. Binding of the proteins was detected using the polyclonal rabbit anti-FH19–20 antibody (a kind gift from Dr. Jens Hellwage, Jena, Germany) followed by horseradish peroxidase-conjugated F(ab′)2 donkey anti-rabbit IgG (Jackson ImmunoResearch) in PBS with 2% bovine serum albumin. Cells were washed twice with 0.05% Tween in PBS after incubation with the first antibody, and 3 times after incubation with the second antibody. Finally, cells were incubated with tetramethylbenzidine substrate solution (SFRI Laboratories, Berganton, France) for 15 min at 22 °C. The reaction was stopped with 2 m H2SO4 and the absorbance was measured at 450 nm. The response curves were fitted using non-linear regression and log(agonist) versus response model. The mean 50% effective concentrations (EC50) were calculated from the fitted curves using GraphPad Prism 5.01 for Windows. The experiment was performed three times in duplicate.
Heparin Affinity Chromatography
Binding of the FH19–20 mutants to heparin was measured with a 1-ml HiTrap Heparin column (GE Healthcare) connected to the ÄKTA® purifier HPLC system (GE Healthcare). The mutants (45 μg) were injected into the column in 900 μl of 50% PBS and eluted using linear salt gradient from 50% PBS to 0.5 m NaCl in 50% PBS. The elution was monitored in real time by absorbance of UV light at 280 nm. The ionic strength of the elution buffer was monitored in real time with a conductivity meter. The experiment was repeated using Hitachi LaChrom HPLC (Merck) and a gradient from PBS to 1 m NaCl in PBS.
Statistical Analyses
Values are expressed as mean ± S.D. and all the statistical analyses were performed using Microsoft Excel. The F-test was used to compare whether variances between means are equal or unequal. Depending on the result of the F-test, t test assuming equal or unequal variances was used to compare means of IC50 and EC50.
RESULTS
Generation of FH19–20 Mutant Constructs
Six residues mutated in aHUS patients and three structurally closely located residues were used as targets for site-directed mutagenesis of the FH19–20 construct. The generated mutants were D1119GaHUS, W1157LaHUS, T1184RaHUS, L1189RaHUS, R1210AaHUS, R1215QaHUS, D1119G/Q1139A, Q1139A, and R1206A. Previously published mutants R1182AaHUS, W1183LaHUS, K1186A, K1188A, and E1198AaHUS were also used in binding experiments (29).
The mutant constructs were expressed in P. pastoris and purified using heparin affinity chromatography and gel filtration to reach >95% purity. The proteins were analyzed in non-reducing conditions (Fig. 1) to look for disulfide-linked oligomers that would be indicative of incorrect disulfide pairing in the folding process. The proteins migrated similarly under reducing conditions (data not shown). All the mutants were detectable by polyclonal anti-FH antibody (Fig. 1). The mutant FH19–20 proteins were approximately the same yield and size as the wild-type protein, suggesting that glycosylation sites are not introduced and stability is not affected.
FIGURE 1.

SDS-PAGE and Western blot analysis of the recombinant FH19–20 mutant constructs. Samples of the proteins (0.3 μg) were resolved by SDS-PAGE gels in non-reducing Laemmli sample buffer. The proteins were visualized by Coomassie Blue staining (upper panel) and Western blotting (lower panel), where the proteins were detected with polyclonal goat anti-human FH antibody and horseradish peroxidase-conjugated donkey anti-goat antibody. The aHUS-associated mutations are in bold. Dashed lines indicate composition of separate lanes from the gels.
Several aHUS-associated Mutations Cause Impaired C3b and C3d Binding
Binding of wild-type and mutant FH19–20 proteins to C3b and C3d was analyzed using radioligand assays where purified C3b or C3d was coated onto the plastic wells and inhibition of 125I-FH19–20 binding by the mutant proteins was analyzed. Nine of 14 mutant proteins inhibited binding of 125I-FH19–20 to C3b less efficiently than the wild-type FH19–20 indicating decreased binding to C3b (IC50 difference 3.7–46-fold, p < 0.05). These were namely D1119G/Q1139A, Q1139A, W1157LaHUS, R1182AaHUS, W1183LaHUS, T1184RaHUS, K1188A, R1206A, and R1210AaHUS (Fig. 2). Five of the mutant constructs, D1119GaHUS, K1186A, L1189RaHUS, E1198AaHUS, and R1215QaHUS, had an essentially wild-type-like affinity to C3b (Fig. 2). Nine of the mutants were used also in an experiment where C3d was used instead of C3b. The results were similar except that the mutant T1184RaHUS inhibited the 125I-FH19–20-C3d interaction more weakly than the 125I-FH19–20/C3b interaction (Fig. 3). Taken together several, but not all, aHUS-associated mutations cause impaired binding to C3b/C3d.
FIGURE 2.

Inhibition of 125I-FH19–20 binding to C3b with mutant proteins. Binding of the fluid-phase 125I-FH19–20 to plastic surface-coated C3b was inhibited using serial dilutions of the unlabeled FH19–20 mutants. After washing, radioactivity was detected with a γ-counter and the results were fitted to inhibition curves. The results are shown in two panels. A, the mutants that bound to C3b similarly to the wild-type FH19–20; B, the mutants that showed decreased binding. Results of a representative experiment are shown. C, for each FH19–20 construct the IC50 value was calculated from three independent experiments performed in duplicate. Standard deviations for IC50 are presented as error bars. Labels for the aHUS-associated mutations are in bold; cpm, counts per minute; *, p < 0.05; ●, FH19–20 (wild-type); ○, D1119G; ■, D1119G/Q1139A; □, Q1139A; ▴, W1157L; ▾, T1184R; +, L1189R; ♦, R1206A; Δ, R1210A; ▿, R1215Q; ×, R1182A; ⊗, W1183L; ⊕, K1186A; ∞, K1188A; and 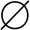 , E1198A.
, E1198A.
FIGURE 3.

Inhibition of 125I-FH19–20 binding to C3d with mutant constructs. Binding of the fluid-phase 125I-FH19–20 to plastic surface-coated C3d was inhibited using serial dilutions of the unlabeled FH19–20 mutants. After washing, radioactivity was detected with a γ-counter and the results were fitted to inhibition curves. The results are shown in two panels. A, the mutants that bound to C3b similarly to the wild-type FH19–20; B, the mutants that showed decreased binding. Results of a representative experiment are shown. C, for each FH19–20 construct IC50 value was calculated from three independent experiments performed in duplicate. Standard deviations for IC50 are presented as error bars. Labels for the aHUS-associated mutations are in bold; cpm, counts per minute; *, p < 0.05; ●, FH19–20 (wild-type); ○, D1119G; ■, D1119G/Q1139A; □, Q1139A; ▴, W1157L; ▾, T1184R; +, L1189R; ♦, R1206A; Δ, R1210A; and ▿, R1215Q.
Mutations Associated with aHUS Have Variable Effects on FH19–20 Binding to Mouse Glomerular Endothelial Cells
The wild-type FH19–20 and 14 mutants were tested for binding to differentiated mGEnC-1 cells (38) using enzyme immunoassay (Fig. 4). Based on differences in binding (EC50; Fig. 4D), four of the constructs, T1184RaHUS, K1186A, L1189RaHUS, and E1198AaHUS, showed increased (Fig. 4B) and three, R1182AaHUS, R1206A, and R1210AaHUS, showed reduced binding to mGEnC-1 (Fig. 4C). The rest of the mutants, D1119GaHUS, D1119G/Q1139A, Q1139A, W1157LaHUS, W1183LaHUS, K1188A, and R1215QaHUS, bound similarly to wild-type FH19–20 (Fig. 4A). Taken together, it is evident that different aHUS-associated mutations in FH19–20 have variable effects on binding to mouse glomerular endothelial cells.
FIGURE 4.

Binding of the wild-type and mutant FH19–20 proteins to tumor necrosis factor-α-activated mouse glomerular endothelial cells. Serial dilutions of the wild-type and mutant FH19–20 constructs were applied onto the mGEnC-1 cells and binding was detected using enzyme immunoassay. Data are shown in three panels. A, mutants that bound similarly to the wild-type FH19–20; B, mutants that bound stronger than the wild-type.
Binding of FH19–20 Mutants to Heparin and Mouse Glomerular Endothelial Cells Correlate Well
The wild-type and 13 mutant FH19–20 proteins were analyzed for their affinity to heparin by applying them to a heparin column and monitoring the elution with increasing ionic strength (Fig. 5). Five mutants had normal binding to heparin, i.e. the same as the wild-type FH19–20 (Fig. 5A). Two of the mutant constructs had increased affinity to heparin as judged by the higher ionic strength needed for elution (Fig. 5B), whereas six mutants were eluted at lower ionic strength, and therefore bound heparin less efficiently than wild-type FH19–20 (Fig. 5C). Eight of the 13 mutants have been associated with aHUS and two of those, T1184RaHUS and L1189RaHUS, had increased affinity to heparin, whereas three, R1182AaHUS, R1210AaHUS, and R1215QaHUS, had decreased affinity, and three, D1119GaHUS, W1157LaHUS, and W1183LaHUS, had affinity similar to FH19–20 (Fig. 5D). The results of the heparin affinity chromatography and the mGEnC-1 binding assays correlated well, although there was one exception, because the K1186A protein showed a reduced binding to heparin, but an increased binding to the mouse glomerular endothelial cells (Figs. 4D and 5D). In summary, different mutations in FH19–20 lead to similar heparin and glomerular endothelial cell binding characteristics.
FIGURE 5.

Heparin affinity chromatography of the wild-type and mutant FH19–20 proteins. The FH19–20 wild-type and mutant proteins were applied to the heparin column in 50% PBS and eluted using linear salt gradient from 0 to 0.5 m NaCl in 50% PBS. Elution of the proteins from the column was monitored by measuring the absorbance of UV light at 280 nm. Data are shown in three panels. A, mutants that were eluted at the same ionic strength as the wild-type FH19–20; B, mutants that were eluted at a higher ionic strength than the wild-type FH19–20; and C, mutants that were eluted at the lower ionic strength than the wild-type FH19–20. Ionic strength of the buffer was followed with a conductivity meter (dashed line). D, ionic strength of the buffer at which the apex of the A280 signal peak for each protein was detected. Labels of the aHUS-associated mutations are in bold.
The C3b/C3d Binding Site, but Not the Glomerular Endothelial Cell Binding Site, Extends to Domain 19
We highlighted the mutated amino acid residues on the crystal structure of FH19–20 (29) according to the results of the mutant protein binding data. The mutations that interfered with binding of FH19–20 to C3b and C3d were located in both domains 19 and 20 (Fig. 6A). The longest distance between the mutations that impaired C3b/C3d binding is 52 Å (distance between Cα of Trp-1157 and Arg-1206), whereas the diameter of the lentil-shaped C3d is ∼50 Å (Protein Data Bank code 1C3D (36)). The mutations that impaired binding of FH19–20 to mGEnC-1 cells and/or heparin were all located as a cluster within domain 20 (Fig. 6B). The diameter of this cluster is ∼30 Å and heparin octasaccharide contains seven sulfate groups within this distance on one face (1HPN (40)). The mutations T1184RaHUS, K1186A, L1189RaHUS, and E1198AaHUS with an increased binding affinity to mGEnC-1 were located next to this cluster indicating that the mutations extend the mGEnC-1/heparin cell binding site on FH19–20 (Fig. 6B).
FIGURE 6.

Binding sites for C3b and mouse glomerular endothelial cells on the FH19–20 structure. A, map of the C3b binding site based on the results presented in Table 1. The indicated mutation of the light blue colored residues decreased binding to C3b, whereas the mutations of the black residues had no effect on C3b binding. B, map of the mGEnC-1 cell binding site based on the results presented in Fig. 5. The indicated mutations of the purple colored residues decreased affinity to mGEnC-1 cells, whereas the red colored increased affinity to mGEnC-1 cells. Mutations of the black residues had no effect on mGEnC-1 cell binding. Amino acids indicated in bold are associated with aHUS.
Function of FH in aHUS Is Impaired by Three Primary Defects
When data were combined from the assays that measured binding of the FH19–20 mutant constructs to C3b/C3d, mGEnC-1, and heparin it became evident that the effects of all the studied aHUS-associated mutations are not similar (Table 1). The primary defects of the mutants when compared with the wild-type FH19–20 fell into three groups, (i) impaired C3b/C3d binding, (ii) enhanced mGEnC-1/heparin binding, and (iii) impaired mGEnC-1/heparin binding. Five of the mutants showed only one of these primary defects, whereas three showed a combination of two defects (Table 1). One mutant (D1119GaHUS) did not show any defects in the performed analyses.
TABLE 1.
Summary of the binding experiments with the FH19–20 mutants
Arrows pointing up indicate increase in binding and arrows pointing down decrease in binding; horizontal arrows indicate no change in the binding when compared to the wild-type FH19–20.
The asterisks indicate: * According to aHUS database (www.fh-hus.org). ** Published in Jokirantaet al.2006. *** Reduced binding to C3b, wild-type-like binding to C3d.

DISCUSSION
Atypical hemolytic uremic syndrome is a rare but severe disease that typically affects children and adolescents. Mutations in several complement proteins have been reported from the patients but more than half of these are accounted for by mutations in the HF1 gene that codes FH. Molecular mechanisms underlying the association of mutations in FH with aHUS have, however, largely remained elusive. In this study we have characterized the effects of nine aHUS-associated and five non-associated mutations on binding of FH19–20 to C3b, C3d, mouse glomerular endothelial cells, and heparin. The results show that the aHUS-associated FH mutations cause different combinations of three primary defects. We also show, for the first time, the binding site for mouse glomerular endothelial cells in FH domain 20 and demonstrate that the binding site for C3b/C3d is likely to extend from domain 20 to domain 19. Finally, the results indicate that FH19–20 needs to bind to C3b/C3d to enable full activity of FH.
Three primary defects were identified in our set of nine aHUS mutations: impaired binding to C3b/C3d, enhanced binding to mouse glomerular endothelial cells, and impaired binding to glomerular endothelial cells (Table 1). Five of the aHUS-associated mutants had impaired binding to C3b (W1157LaHUS, R1182AaHUS, W1183LaHUS, T1184RaHUS, and R1210AaHUS) and two of these did not have either of the other primary defects. This indicates that defects in C3b binding of FH19–20 alone can lead to aHUS and that binding of FH19–20 to C3b/C3d is essential for full activity of FH (Fig. 7, aHUS mechanism i). A role of impaired C3b/C3d binding in aHUS has also been previously suggested and so far a total of 10 mutations have been described with this defect (W1157RaHUS, R1182AaHUS, W1183LaHUS, W1183RaHUS, V1197AaHUS, E1198AaHUS, R1210CaHUS, R1215GaHUS, P1226SaHUS, and S1191L/V1197A) (29, 33–35)). Therefore all the available data indicates that the loss of C3b/C3d binding is typical in aHUS and may lead to the disease in the absence of other functional defects.
FIGURE 7.

Proposed mechanism of FH function on surface-bound C3b and the pathogenic mechanisms of aHUS associated with FH mutations. Under the physiological conditions FH may be attached to the cell surface GAGs such as heparan sulfate. When C3b is deposited onto the surface the C terminus of FH is released from the cell surface GAGs to bind to the C3d part of C3b. Thereafter the N terminus of FH acts efficiently as a cofactor for factor I (FI) in cleavage of C3b to inactive C3b (iC3b) (upper panel). The three primary binding defects of mutated FH in aHUS can lead to restricted C3b elimination via three basic mechanisms. (i) FH mutations that decrease C3b/C3d binding lead to impaired C3b elimination on a cell surface (right lower panel). (ii) If FH binding to GAGs on a cell surface is clearly increased, the ability of the mutated FH molecules to bind to C3b is decreased due to the overlapping binding sites. Thereby inactivation of C3b can be ineffective (right lower panel). (iii) If affinity of FH to the cell surface GAGs is impaired the deposited C3b may not be efficiently inactivated due to lack of FH in close proximity (left lower panel). Both mechanisms i and ii are associated with a relative decrease of FH affinity for C3b/C3d compared with heparin.
In this study we describe a novel defect of FH in aHUS, where three aHUS-associated mutants (T1184RaHUS, L1189RaHUS, and E1198AaHUS) have enhanced binding to mouse glomerular endothelial cells and heparin with normal C3b or C3d binding (Table 1). This gain-of-function does not lead to sequestration of FH to inappropriate compartments of the body because the patients with these mutations have at least normal plasma levels of FH (41–43). Therefore the findings with these mutants suggest novel concepts for both the target discrimination between the activator/nonactivator surfaces by FH and the pathogenesis of aHUS in these patients. In target discrimination the typical model is that on nonactivator surfaces the C terminus of FH binds to the cell surface molecules such as heparan sulfate leading to enhanced elimination of the surface-bound C3b molecules by the N-terminal effector functions of FH. The model clearly contains the right players but it fails to consider that the C terminus of FH might eventually need to bind to the C3d part of C3b for effective elimination of the surface-bound C3b molecules. On the basis of our data it can be suggested that in a physiological situation heparin forms a temporary encounter complex with FH19–20 and subsequently there is transfer of FH19–20 to the C3d portion of C3b, because of the higher affinity of the protein-protein interaction relative to that of the protein/GAG interaction (Fig. 7).
The suggested model of the alternative pathway discrimination is supported by the results that binding of the C terminus of FH to heparin and C3b is competitive (44) making a ternary complex between FH19–20, heparin (or other GAGs), and C3b unlikely. The key question is if it matters for function of full FH whether its C terminus is bound to heparin or the C3d portion of C3b on a surface. Our results with the mutant W1183L, found from an aHUS patient, indicate that it matters. This mutant shows normal heparin binding and decreased C3b/C3d binding indicating that normal heparin binding cannot compensate for loss of the C3b/C3d binding because the patient suffered from clinical aHUS. Therefore it is clear that a heparin-bound FH is not itself functionally fully active if it is unable to bind to the C3d part of C3b. This means that C3b/C3d binding is essentially needed for the physiological function of FH19–20 on cell-bound C3b. With this clarified model the explanation of the molecular pathogenesis of mutants T1184R, L1189R, and E1198A is logical. Because the heparin and C3b binding sites on FH19–20 are overlapping the enhanced heparin binding can lead to decreased C3b/C3d binding. Because normal C3b/C3d binding is needed for the fully functional molecule the elimination of cell-bound C3b is impaired leading to clinical aHUS. In this way the gain-of-function in heparin or cell binding eventually leads to loss-of-function in elimination of C3b (Fig. 7, mechanism ii).
The third primary defect was described only by one mutant, R1215QaHUS, that had wild-type-like binding to C3b/C3d and impaired binding to heparin and possibly also to mGEnC-1 (Figs. 4D and 5D). Although the result for the mGEnC-1 binding was not statistically significant in our experiments, the role of Arg-1215 in cell binding is likely because a different mutation of the same residue, R1215GaHUS, has previously been shown to impair binding of FH8–20 to C3b, heparin, and human umbilical vein endothelial cells (34). Thus it may be that impaired binding of heparin/cells alone without loss of C3b/C3d binding can also lead to aHUS, as has been suggested before (Fig. 7, mechanism iii) (34, 45). This mutant, R1215QaHUS, however, is the only mutant published to date where a heparin binding defect has been observed in the absence of defect in C3b/C3d binding.
We detected at least one functional defect with eight of the nine aHUS-associated mutants (Table 1). The only mutant that appeared functionally normal in our analyses was the mutant D1119GaHUS. The mutation has been described from one individual whose sibling was also affected but his/her sequence analysis has not been published (42). At the time of the publication it was not known that in aHUS there are mutations also in other complement proteins and therefore, it is possible that the disease was not caused by the D1119GaHUS mutation because other known aHUS-associated mutations have not been excluded.
The results of this study indicate that the C3b/C3d binding site is located in FH domains 19 and 20, and glomerular endothelial cell, and heparin binding sites in domain 20 (Fig. 6). Due to the similarity of the used mouse glomerular endothelial cells to human glomerular endothelial cells (39) it is likely that the observed FH binding site is the same for human cells. The binding site for C3b/C3d in domain 20 has been analyzed previously, but on domain 19 the only amino acid studied so far has been Trp-1157 (33). In that study, mutation W1157RaHUS decreased C3b binding but introduction of a charged arginine in place of the partially buried hydrophobic tryptophan may cause more distant changes in the overall structure without being part of the interacting surface. We now show that in addition to the W1157LaHUS mutation, the Q1139A mutation causes impairment in C3b/C3d binding (Figs. 2C and 3C). The Gln-1139 residue is surface exposed and therefore it is not likely to cause any global changes to the structure. Thereby the current results, showing that mutations D1119G/Q1139A, Q1139A, and W1157LaHUS in domain 19 lead to impaired C3b/C3d binding, suggests that the C3b/C3d binding site extends from domain 20 to domain 19. This observation would also explain the six different missense mutations in domain 19 found in aHUS patients.
A heparin binding site on FH19–20 has previously been characterized in a nuclear magnetic resonance study of FH19–20, where heparin tetrasaccharide perturbed NMR spectral peaks of eight residues in domain 20 (Arg-1182, Lys-1186, Lys-1188, Lys-1202, Arg-1203, Arg-1215, Lys-1230, and Arg-1231), whereas no perturbations were observed in domain 19 (28). Our study corroborates these results as we show that mutations of positively charged residues R1182AaHUS, K1186A, K1188A, R1206A, R1210AaHUS, and R1215QaHUS in domain 20 decrease heparin binding, whereas mutations in domain 19 do not have an effect on heparin binding. Interestingly, aHUS-associated mutations that enhanced heparin binding (Fig. 5D) introduce positive (T1184RaHUS, L1189RaHUS) or remove negative charge (E1198AaHUS) in the proximity of the heparin binding cluster in the structure of FH19–20. Thus T1184RaHUS, and L1189RaHUS seem to extend the binding site for heparin in domain 20 and E1198AaHUS removes repulsive charge from the edge of the cluster.
Based on the identified binding sites we conclude that the glomerular endothelial cell binding site on FH19–20 is on domain 20 where it overlaps with the C3b/C3d binding site, and that the cell binding site is nearly identical with the heparin binding site (Fig. 6 and Table 1). This data fits well with the previous suggestion that the C3d and heparin binding sites on the FH15–20 background were found to be distinct but overlapping (44). Our results on the glomerular endothelial cell binding site on domain 20 (Fig. 6B) are concordant with the previously published studies on human umbilical vein endothelial cells, where mutations in domain 20 on FH8–20 and FH15–20 backgrounds decreased binding to cells (33, 34, 45). The binding of the mutants to cells thus correlates well with their binding to heparin indicating that the binding site for cells and heparin is located in domain 20 only. The only exception was mutant K1186A, which had increased binding to mGEnC-1 and impaired binding to heparin (Table 1). This indicates two things. First, FH19–20 binding to heparin that has been used as a model for cell surface heparan sulfate proteoglycans seems to be a relatively good model for endothelial cell binding. Second, the use of porcine heparin preparations may not reflect in all cases the actual binding of FH to sulfated GAGs on cells. Currently, we are further exploring the exact role of GAGs at the glomerular endothelial surface in binding FH.
The data in this study shows that all aHUS-associated FH19–20 mutations do not cause the same primary functional defect but one or two of the three observed abnormalities in C3b/C3d or glomerular endothelial cell/heparin binding. It is likely that all three primary functional defects caused by the FH mutations in aHUS lead eventually to the same process of inefficient clearance of surface-bound C3b via three mechanisms (Fig. 7). In this study we have demonstrated a novel defect of enhanced binding of aHUS-associated mutants to glomerular endothelial cells or heparin that adds to the previously suggested two defects of impaired C3b/C3d binding and impaired cell/heparin binding. In addition, we have also mapped the binding site of FH19–20 for the glomerular endothelial cells to domain 20 and show that cell binding correlates well, but not fully, with heparin binding. This site is distinct from the C3b/C3d binding site that, for the first time, is shown to extend to domain 19. Finally, we have proposed a model to explain how the target discrimination by the alternative pathway essentially needs binding of FH C terminus to the C3d part of C3b.
Acknowledgments
We thank Marjatta Ahonen, Kirsti Widing, and Marinka Bakker for kind help and excellent technical assistance, M.Sc. Karita Haapasalo for providing the purified C3, and Prof. Seppo Meri for fruitful discussions.
This work was supported in part by the Academy of Finland Projects 201506 and 202529, The Sigrid Jusélius Foundation, Dutch Kidney Foundation Grant C05.2152, and The Helsinki University Central Hospital Funds.
- aHUS
- atypical hemolytic uremic syndrome
- FH
- factor H
- GAG
- glycosaminoglycan
- FH19–20
- domains 19 to 20 of FH
- PBS
- phosphate-buffered saline
- mGEnC-1
- mouse glomerular endothelial cell line 1.
REFERENCES
- 1.Ruggenenti P., Noris M., Remuzzi G. ( 2001) Kidney Int. 60, 831– 846 [DOI] [PubMed] [Google Scholar]
- 2.Kavanagh D., Goodship T. H., Richards A. ( 2006) Br. Med. Bull. 5, 5. [DOI] [PubMed] [Google Scholar]
- 3.Fremeaux-Bacchi V., Dragon-Durey M. A., Blouin J., Vigneau C., Kuypers D., Boudailliez B., Loirat C., Rondeau E., Fridman W. H. ( 2004) J. Med. Genet. 41, e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frémeaux-Bacchi V., Miller E. C., Liszewski M. K., Strain L, Blouin J., Brown A. L., Moghal N., Kaplan B. S., Weiss R. A., Lhotta K., Kapur G., Mattoo T., Nivet H., Wong W., Gie S., Hurault de Ligny B., Fischbach M., Gupta R., Hauhart R., Meunier V., Loirat C., Dragon-Durey M. A., Fridman W. H., Janssen B. J., Goodship T. H., Atkinson J. P. ( 2008) Blood 112, 4948– 4952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goicoechea de Jorge E., Harris C. L., Esparza-Gordillo J., Carreras L., Arranz E. A., Garrido C. A., López-Trascasa M., Sánchez-Corral P., Morgan B. P., Rodríguez de Córdoba S. ( 2007) Proc. Natl. Acad. Sci. U. S. A. 104, 240– 245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noris M., Brioschi S., Caprioli J., Todeschini M., Bresin E., Porrati F., Gamba S., Remuzzi G.International Registry of Recurrent and Familial HUS/TTP ( 2003) Lancet 362, 1542– 1547 [DOI] [PubMed] [Google Scholar]
- 7.Richards A., Kemp E. J., Liszewski M. K., Goodship J. A., Lampe A. K., Decorte R., Müslümanoğlu M. H., Kavukcu S., Filler G., Pirson Y., Wen L. S., Atkinson J. P., Goodship T. H. ( 2003) Proc. Natl. Acad. Sci. U. S. A. 100, 12966– 12971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warwicker P., Goodship T. H., Donne R. L., Pirson Y., Nicholls A., Ward R. M., Turnpenny P., Goodship J. A. ( 1998) Kidney Int. 53, 836– 844 [DOI] [PubMed] [Google Scholar]
- 9.Lesavre P. H., Müller-Eberhard H. J. ( 1978) J. Exp. Med. 148, 1498– 1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pangburn M. K., Schreiber R. D., Müller-Eberhard H. J. ( 1981) J. Exp. Med. 154, 856– 867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Law S. K., Levine R. P. ( 1977) Proc. Natl. Acad. Sci. U. S. A. 74, 2701– 2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison R. A., Lachmann P. J. ( 1980) Mol. Immunol. 17, 219– 228 [DOI] [PubMed] [Google Scholar]
- 13.Harrison R. A., Lachmann P. J. ( 1980) Mol. Immunol. 17, 9– 20 [DOI] [PubMed] [Google Scholar]
- 14.Ruddy S., Austen K. F. ( 1969) J. Immunol. 102, 533– 543 [PubMed] [Google Scholar]
- 15.Pangburn M. K., Schreiber R. D., Müller-Eberhard H. J. ( 1977) J. Exp. Med. 146, 257– 270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seya T., Turner J. R., Atkinson J. P. ( 1986) J. Exp. Med. 163, 837– 855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pangburn M. K., Müller-Eberhard H. J. ( 1978) Proc. Natl. Acad. Sci. U. S. A. 75, 2416– 2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weiler J. M., Daha M. R., Austen K. F., Fearon D. T. ( 1976) Proc. Natl. Acad. Sci. U. S. A. 73, 3268– 3272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whaley K., Ruddy S. ( 1976) J. Exp. Med. 144, 1147– 1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gordon D. L., Kaufman R. M., Blackmore T. K., Kwong J., Lublin D. M. ( 1995) J. Immunol. 155, 348– 356 [PubMed] [Google Scholar]
- 21.Jarva H., Jokiranta T. S., Hellwage J., Zipfel P. F., Meri S. ( 1999) J. Immunol. 163, 3957– 3962 [PubMed] [Google Scholar]
- 22.Blackmore T. K., Sadlon T. A., Ward H. M., Lublin D. M., Gordon D. L. ( 1996) J. Immunol. 157, 5422– 5427 [PubMed] [Google Scholar]
- 23.Ormsby R. J., Jokiranta T. S., Duthy T. G., Griggs K. M., Sadlon T. A., Giannakis E., Gordon D. L. ( 2006) Mol. Immunol. 43, 1624– 1632 [DOI] [PubMed] [Google Scholar]
- 24.Pangburn M. K., Atkinson M. A., Meri S. ( 1991) J. Biol. Chem. 266, 16847– 16853 [PubMed] [Google Scholar]
- 25.Schmidt C. Q., Herbert A. P., Kavanagh D., Gandy C., Fenton C. J., Blaum B. S., Lyon M., Uhrín D., Barlow P. N. ( 2008) J. Immunol. 181, 2610– 2619 [DOI] [PubMed] [Google Scholar]
- 26.Jokiranta T. S., Hellwage J., Koistinen V., Zipfel P. F., Meri S. ( 2000) J. Biol. Chem. 275, 27657– 27662 [DOI] [PubMed] [Google Scholar]
- 27.Blackmore T. K., Hellwage J., Sadlon T. A., Higgs N., Zipfel P. F., Ward H. M., Gordon D. L. ( 1998) J. Immunol. 160, 3342– 3348 [PubMed] [Google Scholar]
- 28.Herbert A. P., Uhrín D., Lyon M., Pangburn M. K., Barlow P. N. ( 2006) J. Biol. Chem. 281, 16512– 16520 [DOI] [PubMed] [Google Scholar]
- 29.Jokiranta T. S., Jaakola V. P., Lehtinen M. J., Pärepalo M., Meri S., Goldman A. ( 2006) EMBO J. 25, 1784– 1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferreira V. P., Herbert A. P., Hocking H. G., Barlow P. N., Pangburn M. K. ( 2006) J. Immunol. 177, 6308– 6316 [DOI] [PubMed] [Google Scholar]
- 31.Pickering M. C., de Jorge E. G., Martinez-Barricarte R., Recalde S., Garcia-Layana A., Rose K. L., Moss J., Walport M. J., Cook H. T., de Córdoba S. R., Botto M. ( 2007) J. Exp. Med. 204, 1249– 1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng Z. Z., Hellwage J., Seeberger H., Zipfel P. F., Meri S., Jokiranta T. S. ( 2006) Mol. Immunol. 43, 972– 979 [DOI] [PubMed] [Google Scholar]
- 33.Józsi M., Heinen S., Hartmann A., Ostrowicz C. W., Hälbich S., Richter H., Kunert A., Licht C., Saunders R. E., Perkins S. J., Zipfel P. F., Skerka C. ( 2006) J. Am. Soc. Nephrol. 17, 170– 177 [DOI] [PubMed] [Google Scholar]
- 34.Manuelian T., Hellwage J., Meri S., Caprioli J., Noris M., Heinen S., Jozsi M., Neumann H. P., Remuzzi G., Zipfel P. F. ( 2003) J. Clin. Investig. 111, 1181– 1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sánchez-Corral P., Pérez-Caballero D., Huarte O., Simckes A. M., Goicoechea E., López-Trascasa M., de Córdoba S. R. ( 2002) Am. J. Hum. Genet. 71, 1285– 1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagar B., Jones R. G., Diefenbach R. J., Isenman D. E., Rini J. M. ( 1998) Science 280, 1277– 1281 [DOI] [PubMed] [Google Scholar]
- 37.Koistinen V., Wessberg S., Leikola J. ( 1989) Complement Inflamm. 6, 270– 280 [DOI] [PubMed] [Google Scholar]
- 38.Rops A. L., van der Vlag J., Jacobs C. W., Dijkman H. B., Lensen J. F., Wijnhoven T. J., van den Heuvel L. P., van Kuppevelt T. H., Berden J. H. ( 2004) Kidney Int. 66, 2193– 2201 [DOI] [PubMed] [Google Scholar]
- 39.Rops A. L., van den Hoven M. J., Baselmans M. M., Lensen J. F., Wijnhoven T. J., van den Heuvel L. P., van Kuppevelt T. H., Berden J. H., van der Vlag J. ( 2008) Kidney Int. 73, 52– 62 [DOI] [PubMed] [Google Scholar]
- 40.Mulloy B., Forster M. J., Jones C., Davies D. B. ( 1993) Biochem. J. 293, 849– 858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pérez-Caballero D., González-Rubio C., Gallardo M. E., Vera M., López-Trascasa M., Rodríguez de Córdoba S., Sánchez-Corral P. ( 2001) Am. J. Hum. Genet. 68, 478– 484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richards A., Buddles M. R., Donne R. L., Kaplan B. S., Kirk E., Venning M. C., Tielemans C. L., Goodship J. A., Goodship T. H. ( 2001) Am. J. Hum. Genet. 68, 485– 490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaziri-Sani F., Holmberg L., Sjöholm A. G., Kristoffersson A. C., Manea M., Frémeaux-Bacchi V., Fehrman-Ekholm I., Raafat R., Karpman D. ( 2006) Kidney Int. 69, 981– 988 [DOI] [PubMed] [Google Scholar]
- 44.Hellwage J., Jokiranta T. S., Friese M. A., Wolk T. U., Kampen E., Zipfel P. F., Meri S. ( 2002) J. Immunol. 169, 6935– 6944 [DOI] [PubMed] [Google Scholar]
- 45.Jokiranta T. S., Cheng Z. Z., Seeberger H., Jòzsi M., Heinen S., Noris M., Remuzzi G., Ormsby R., Gordon D. L., Meri S., Hellwage J., Zipfel P. F. ( 2005) Am. J. Path. 167, 1173– 1181 [DOI] [PMC free article] [PubMed] [Google Scholar]