

Abstract
Damage of presynaptic mitochondria could result in release of proapoptotic factors that threaten the integrity of the entire neuron. We discovered that α-synuclein (Syn) forms a triple complex with anionic lipids (such as cardiolipin) and cytochrome c, which exerts a peroxidase activity. The latter catalyzes covalent hetero-oligomerization of Syn with cytochrome c into high molecular weight aggregates. Syn is a preferred substrate of this reaction and is oxidized more readily than cardiolipin, dopamine, and other phenolic substrates. Co-localization of Syn with cytochrome c was detected in aggregates formed upon proapoptotic stimulation of SH-SY5Y and HeLa cells and in dopaminergic substantia nigra neurons of rotenone-treated rats. Syn-cardiolipin exerted protection against cytochrome c-induced caspase-3 activation in a cell-free system, particularly in the presence of H2O2. Direct delivery of Syn into mouse embryonic cells conferred resistance to proapoptotic caspase-3 activation. Conversely, small interfering RNA depletion of Syn in HeLa cells made them more sensitive to dopamine-induced apoptosis. In human Parkinson disease substantia nigra neurons, two-thirds of co-localized Syn-cytochrome c complexes occurred in Lewy neurites. Taken together, these results indicate that Syn may prevent execution of apoptosis in neurons through covalent hetero-oligomerization of cytochrome c. This immediate protective function of Syn is associated with the formation of the peroxidase complex representing a source of oxidative stress and postponed damage.
Lewy bodies (LBs),3 mitochondrial impairment, and oxidative stress are cardinal features of Parkinson disease (PD) and several related neurodegenerative disorders (1, 2). Aggregation of α-synuclein (Syn), an abundant protein in synaptic terminals, plays a major role in the formation of LBs (3, 4). Neither the mechanisms of LB production nor their pathogenic or protective roles in neurodegeneration are well understood.
In nigrostriatal dopaminergic synaptic terminals, mitochondria, harboring a host of death-initiating factors, are in peril of damage by reactive oxygen species generated by disrupted electron transport and/or oxidative metabolism of dopamine (DA). Because cytochrome c (cyt c)-dependent formation of apoptosomes and activation of caspases designates a point of no return in the apoptotic program, release of proapoptotic factors from synaptic mitochondria could threaten the integrity of the entire neuron. How neurons protect themselves against inadvertent release of death signals from damaged synaptic mitochondria is not known.
The N-terminal fragment of Syn contains six variants of an 11-amino acid consensus motif that include an apolipoprotein-like class A2 helix participating in binding of different lipids, particularly anionic phospholipids (5). This domain is believed to be important for Syn functions in regulation of neuronal lipid metabolism, particularly turnover of a mitochondria-specific phospholipid, cardiolipin (CL) (6). However, the relevance of the Syn lipid binding capacity in regulating neuronal injury (apoptotic) responses has not been established.
It is believed that oxidative stress participates in the accumulation of LB and Lewy neurites (LN) through yet to be identified pathways (7). Reportedly, Syn is co-localized with cyt c in LBs (8), indicating a potential interaction between the two proteins. Because cyt c is a redox-active hemeprotein (9, 10), its presence in the LBs in conjunction with Syn may also provide a mechanistic link of LBs with oxidative stress. We have recently reported that cyt c interacts with CL in mitochondria early in apoptosis and with phosphatidylserine (PS) in the plasma membrane after its release into the cytosol (11, 12). In both cases, this results in redox activation of cyt c and the production of complexes with high peroxidase activity that effectively catalyze peroxidation of the respective phospholipids (13).
Based on these facts, we hypothesize and provide experimental evidence that Syn acts as a sacrificial scavenger of cytosolic cyt c inadvertently released from synaptic mitochondria to prevent its migration into the soma, i.e. spread of the proapoptotic signal and cell death. This vital function is realized through the emergence of a peroxidase activity of the cyt c-Syn-phospholipid complex that cross-links its components and yields covalently conjugated protein-lipid hetero-oligomers. The latter maintain lingering peroxidase activity. Thus protection against acute apoptotic cell death comes with a penalty of Syn-cyt c aggregation into a peroxidase complex capable of inducing protracted oxidative stress. Our results present a novel biochemical mechanism likely involved in Lewy body formation and explain a known paradox of a dual protective and deleterious role that Syn plays in neuronal cells.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatment
HeLa, HL-60, and SH-SY5Y cells were purchased from the American Type Culture Collection and cultured in 1:1 mixture of Eagle's minimum essential medium and Ham's F-12 medium supplemented with 10% of fetal bovine serum (FBS), 1.5 g/liter sodium bicarbonate, 2 mm l-glutamine, 0.5 mm sodium pyruvate, and 0.05 mm nonessential amino acids. For apoptosis induction, HeLa cells were incubated with tert-butyl hydroperoxide (t-BuOOH) (400 μm) or ActD (200 ng/ml) for 16 h; SH-SY5Y cells were incubated with t-BuOOH (10 μm for 16 h) or ActD (10 μg/ml for 18 h). At the end of incubation, the attached cells were harvested by trypsinization and pooled with the detached cells from supernatant for PS externalization analysis and immunostaining. Cyt c-deficient HeLa cells were generated using siRNA-expressing plasmid (pSEC hygro vector, Ambion) as described previously (12) and cultured in Dulbecco's modified Eagle's medium supplemented with 15% FBS, 25 mm HEPES, 50 mg/liter uridine, 110 mg/liter pyruvate, 2 mm glutamine, 1× nonessential amino acids, 0.05 mm 2′-mercaptoethanol. Syn knockdown HeLa cells were generated using siRNA-expressing plasmid (pSEC hygro vector, Ambion). Target sequence (AAGAGGGTGTTCTCTATGTAG) was cloned into pSEC hygro vector, and the resultant plasmid was transfected into HeLa cells. Positive clones were selected by hygromycin.
Delivery of Syn into MECs
Mouse embryonic cells (MECs) (courtesy of X. Wang, University of Texas, Dallas) were derived from 8- to 9-day-old mouse embryos by Li et al. (14). MECs were cultured in Dulbecco's modified Eagle's medium supplemented with 15% FBS, 25 mm HEPES, 50 mg/liter uridine, 110 mg /liter pyruvate, 2 mm glutamine, 1× nonessential amino acids, 0.05 mm 2′-mercaptoethanol, 0.5 × 106 units/liter mouse leukemia inhibitory factor. Syn protein was delivered into cells using Chariot (Active Motif, Carlsbad, CA) according to the manufacturer's instructions. Briefly, cells were seeded at a density of 0.03 × 106/well in a 24-well plate and allowed to attach overnight. Chariot-Syn complex (2 μl, 0.5 μg) was incubated with cells for 3 h for integration. After that, cells were treated with 50 ng/ml ActD for 18 h. At the end of incubation, attached cells were harvested by trypsinization and pooled with detached cells from supernatant. Caspase-3/7 activity was determined using a caspase-3/7 Glo kit (Promega, San Luis Obispo, CA).
Preparation of Liposomes
Liposomes containing dioleoyl-phosphatidylcholine (DOPC) and tetraoleoyl-CL (TOCL) (or other anionic lipids) (lipid/DOPC ratio 1:1), were prepared in 20 mm HEPES, pH 7.4, by sonication under N2 and used immediately after preparation. To prevent redox cycling with free metals, diethylenetriaminepentaacetic acid (DTPA) (100 μm) was added to all solutions used.
Preparation of Fibrillated (Aged) Syn
Fibrillated (aged) Syn was prepared by incubation of wild-type Syn and its mutants (200 μm) in 20 mm HEPES, 100 μm DTPA, pH 7.4, with shaking at 200 rpm for 6 days at 37 °C.
Isolation of Mitochondria
Mitochondria were isolated as described previously (12). Briefly, harvested cells were resuspended in isolation buffer containing 300 mm mannitol, 10 mm HEPES-KOH, pH 7.4, 0.2 mm EDTA, 0.1% bovine serum albumin, and protease inhibitor mixture (Roche Applied Science) homogenized on ice with a glass homogenizer, and then centrifuged at 1000 × g for 10 min at 4 °C. The resulting supernatants were centrifuged at 14,000 × g for 15 min at 4 °C. The resulting pellet was collected as the mitochondrial fraction. Protein concentration was determined using Bio-Rad assay.
Conditions for Model Biochemical Experiments
Recombinant Syn was purchased from Chemicon International Inc. (Temecula, CA). Synuclein was diluted in water (to a final concentration of 1 mg/ml), divided into aliquots, and stored at −20 °C until use. In all model experiments in Fig. 1, b–d, and Fig. 2a, the following conditions were utilized: Syn (1.5 μm) was incubated with 0.5 μm cyt c and TOCL/DOPC liposomes (TOCL/Syn ratio 25:1) in 20 mm HEPES, pH 7.4, for 60 min at 37 °C. Incubation volume was 50 μl. 50 μm H2O2 was added to the incubation mixture every 15 min. The reaction was stopped by addition of 5 μl of catalase (0.1 mg/ml).
FIGURE 1.

Electrophoretic evidence for H2O2-induced hetero-oligomerization of Syn with cyt c in the presence of TOCL (a–c) and other anionic phospholipids (d). a, PAGE-based assessment of competitive binding of CL with Syn. A typical polyacrylamide gel was stained for Syn with SilverStain Snap kit. Note that the monomeric form of Syn migrating to the cathode in the absence of TOCL changes its migration behavior upon binding with CL. Titration with different amounts of NAO, which has a known high affinity for CL (16), reconstitutes the Syn migration profile, because of competitive binding of TOCL with NAO. The amount of free Syn was quantified by densitometry using Bio-Rad Multi-Imager and Multi-Analyst software. b, SDS-PAGE of aggregates formed after incubation of Syn with cyt c/CL/H2O2. Staining with anti-Syn (left panel) and anti-cyt c antibodies (right panel) reveals that all for components Syn, cyt c, TOCL, and H2O2 were required for the formation of hetero-oligomers (containing both Syn and cyt c, detectable on both panels, arrows). A weak H2O2-induced aggregation of Syn with cyt c was observed in the absence of TOCL (arrowheads) in line with the known induction of peroxidase activity of cyt c during incubation with H2O2 (8). As expected, significant aggregation of cyt c occurs (asterisk) in the absence of Syn as cyt c-TOCL complexes exert significant peroxidase activity causing H2O2-dependent aggregation (12). However, the aggregates are formed due to homo-oligomerization of cyt c, and they do not contain Syn (compare with the lack of staining for Syn on the left panel). c, native electrophoresis in agarose gel and Western blotting of complexes formed after incubation of Syn with cyt c and TOCL. Staining was with anti-Syn (left panel) and anti-cyt c antibodies (right panel). Note that Syn and cyt c did not interact with each other in the absence of TOCL. In the absence of H2O2, Syn and cyt c migrated mostly as single monomeric proteins. TOCL changed migration profile of Syn and cyt c. TOCL plus H2O2 caused oligomerization of Syn-cyt (asterisks) as evidenced by the appearance of aggregates at the origin of the gel and dense smeared pattern on the bottom of the gel. Thus, all four components, Syn, cyt c, CL, and H2O2, were required for the catalytic formation of cross-linked polymers. Note that no nonspecific reactivity was displayed by anti-Syn or anti-cyt c antibodies to either monomeric or aggregated forms of cyt c and Syn, respectively. d, SDS electrophoresis of aggregates formed after incubation of Syn with cyt c and H2O2 in the presence of different anionic phospholipids as follows: PS, PI, and PA. A nonanionic phospholipid PC was used as a control (shown for Syn). Staining with anti-Syn antibody (left panel) and anti-cyt c antibody (right panel) revealed that all four components, Syn, cyt c, H2O2, and anionic phospholipids, were required (arrows) for the formation of hetero-oligomers (containing both Syn and cyt c and detectable on both panels). Note that all four tested phospholipids were active in inducing H2O2-dependent hetero-oligomerization of Syn with cyt c; PI was significantly less effective than other phospholipids. In the absence of cyt c, none of the phospholipids tested caused oligomerization of Syn (data not shown). Typical gels representative of five experiments are shown.
FIGURE 2.

Formation of hetero-oligomeric complexes and covalent cross-linked aggregates of cyt c/TOCL with wild-type Syn (intact and aged) and its mutants A53T and A30P. a, native PAGE of Syn-cyt c-TOCL hetero-oligomeric complexes (staining with SilverSNAP stain kit, ThermoFisher) (left panel). Syn (8 μm) was incubated with TOCL/DOPC liposomes (TOCL/Syn ratio 20:1) and cyt c (8 μm) for 60 min at 37 °C. Assessment of Syn binding to CL/cyt c using NBD-CL fluorescence (right panel) is shown. Inset, typical fluorescence spectra obtained from the following: CL/NBD-CL liposomes (uppermost curve), CL/NBD-CL-cyt c complexes (lowest curve), and CL/NBD-CL/cyt c after titration with wild-type (Wt) Syn (1) and mutant forms of Syn A53T (2) and A30P (3) (1.75 μm). b, SDS-PAGE (12%) with subsequent Western blot analysis (using anti-Syn antibody) of cross-links formed after incubation of wild-type (wt) Syn, A53T Syn, and A30P Syn with cyt c/TOCL in the presence of H2O2. Wild-type Syn, A30P, and A53T mutants underwent hetero-oligomeric covalent cross-linking with cyt c in a similar way. c, SDS-PAGE (7.5%) with subsequent Western blot analysis (using anti-Syn antibody) of Syn-cyt c-CL covalent cross-links formed by incubation of nonfibrillated and fibrillated (aged) Syn with cyt c/TOCL in the presence of H2O2. Comparable covalent cross-linking of nonfibrillated and fibrillated Syn was observed in the presence of cyt c, TOCL, and H2O2.
Western Blot Analysis
Proteins were separated by SDS-PAGE run in Mini-Protean 3 system (Bio-Rad) in Tris/glycine buffer. 0.8% agarose gel electrophoresis was run in horizontal gel system “Mupid-21” (Cosmo Bio Co., Ltd.) in nondenaturing buffer (43 mm imidazole, 35 mm HEPES, pH 7.4). In experiments with the HeLa cells, proteins were extracted with 1% SDS after cells were washed with PBS. The separated proteins were electrotransferred to nitrocellulose membrane. After blocking with 5% nonfat milk dissolved in phosphate-buffered saline/Tween 20 (PBS-T, 0.05%) or Tris-buffered saline/Tween 20 (TBS-T) for 1 h, membrane was incubated with primary antibodies (anti-synuclein, anti-cyt c, anti-dityrosine, or anti-5,5-dimethyl-1-pyrroline-N-oxide (DMPO) antibodies) overnight at 4 °C. The membranes were washed 3–4 times followed by incubation with horseradish peroxidase- or alkaline phosphatase-conjugated goat anti-rabbit or goat anti-mouse antibodies for 60 min at room temperature. The protein bands were visualized by SuperSignal West Pico Chemiluminescent Substrate (Pierce) for horseradish peroxidase-conjugated secondary antibody or Lumi-Phos Western blot (Pierce) for alkaline phosphatase-conjugated secondary antibody as described by the manufacturer. The density of bands was determined by scanning with Epi Chemi II Darkroom (UVP BioImaging Systems, Upland, CA).
Peroxidase Activity Measurements
Peroxidase activity of cyt c was determined by methods utilizing fluorescence, gel electrophoresis, and EPR spectroscopy as follows. (i) Fluorescence of resorufin, an oxidation product of Amplex Red (λex 570 nm, λem 585 nm) was measured using Shimadzu RF-5301PC spectrofluorophotometer (Tokyo, Japan). For determination of peroxidase activity, different amounts of Syn were incubated with 0.5 μm cyt c, liposomes (TOCL/DOPC ratio 1:1; TOCL/cyt c ratio 25:1), 50 μm Amplex Red, 50 mm H2O2. (ii) Peroxidase activity in gel was determined after native electrophoresis in 0.8% agarose. Gels were incubated in solution containing SuperSignal West Pico Chemiluminescent Substrate (Pierce), and chemiluminescence was determined by scanning with Epi Chemi II Darkroom (UVP BioImaging Systems, Upland, CA). (iii) EPR spectroscopy of etoposide phenoxyl radicals produced by oxidation of etoposide was performed at 25 °C under the following conditions: 3,350 G center field; 50 G sweep width, 0.5 G field modulation; 10 milliwatt microwave power; 0.1-s time constant; 2,000 receiver gain; and 4-min time scan. Different amounts of Syn were incubated with 5 μm cyt c, liposomes (TOCL/DOPC ratio 1:1; TOCL/cyt c ratio 25:1), 100 μm etoposide, and 100 μm H2O2.
Low Temperature EPR Measurements of Protein-derived (Tyrosyl) Radicals
Cyt c (80 μm) was incubated with DOPC/TOCL liposomes (DOPC/TOCL = 1:1; 4 mm total lipid) and Syn for 5 min at room temperature in 25 mm HEPES-Na buffer, pH 7.4, 100 μm DTPA (N2-conditions); then H2O2 (800 μm) was added. The reaction was stopped after 5 s by freezing the samples in liquid nitrogen. The EPR spectra were recorded at 77 K under the following conditions: 3230 G, centered field; 100 G, sweep width; 5 G, field modulation; 5 milliwatts, microwave power; 0.1 s, time constant; and 2-min time scan. Dependence of relative magnitude (percentage of the maximal magnitude) of EPR signals of tyrosyl radicals on square root from microwave power (in milliwatts) was presented as saturation curves. The spin-lattice relaxation time was determined by fitting the experimental curve of radical signal saturation to the theoretical one as described previously (15).
Fluorescence Spectroscopy Analysis of Dityrosine Adducts
Samples were digested with proteinase K (20 μg/ml) at 37 °C overnight. Digested samples were precipitated with cold perchloric acid (final concentration 0.53 n). Samples were allowed to stand 10 min on an ice bath and centrifuged for 15 min at 3,000 × g. After neutralization with 2 n KOH, the precipitate of potassium perchlorate crystals was discarded by centrifugation (15 min at 3,000 × g). Supernatants were diluted with 0.2 m HEPES, pH 9.0, and fluorescence was measured (λex 315, λem 420 nm).
Assessment of Syn Binding to CL/Cyt c Using NBD-CL Fluorescence
CL/cyt c complexes were formed by incubation of DOPC/CL liposomes (5 μm total lipids, including 1 μm CL and 0.05 μm 1,1′,2-trioleoyl-2′-[12-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-cardiolipin (NBD-CL, custom-synthesized by Avanti Polar Lipids, Alabaster, AL) and cyt c (0.05 μm) for 2 min. NBD-CL fluorescence spectra were recorded in the range of 500–650 nm (excitation wavelength of 480 nm, slits 10 and 10 nm) using a Shimadzu F5301-PC spectrofluorometer. NBD fluorescence was monitored 2 min after addition of Syn aliquots (0.25 μm each).
Mass Spectrometry (MS) Analysis of Syn-Cyt c Covalent Cross-links
After SDS-gel electrophoresis, proteins were visualized by Coomassie staining, and bands corresponding to high molecular weight Syn-cyt c complexes were excised. Destaining was achieved by several washes with 25 mm NH4CO3, 50% CH3CN. Gel pieces were reduced in the presence of dithiothreitol (DTT) followed by alkylation with iodoacetamide. Gel pieces were washed, dehydrated, and dried. The complexes were subjected first to overnight in-gel digestion with trypsin (25 ng/μl in 25 mm ammonium bicarbonate buffer, pH 7.8) followed by overnight digestion with endoproteinase-Glu-C (25 ng/μl in the same buffer). Peptides were extracted with 5% formic acid, 50% acetonitrile and evaporated to near dryness. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry was performed on a Bruker Ultraflex mass spectrometer in positive reflector mode (20 kV) with a matrix of α-cyano-4-hydroxycinnamic acid (Sigma). At least 500 laser shots were averaged to get each spectrum. Masses were calibrated to known peptide standards (purchased from Applied Biosystems) on the same day of analysis. 30-μl aliquots of each of the digests were acidified with 1.5 μl of 5% trifluoroacetic acid (Sigma), and then taken up into a C18 ZipTip (Millipore) that had been prepared as per the manufacturer's instructions. The bound peptides were desalted with two 15-μl washes of 0.1% trifluoroacetic acid and then eluted with 2 μl of aqueous, acidic acetonitrile (67% CH3CN, 0.1% trifluoroacetic acid). The eluant was mixed with 1 μl of freshly prepared cyano-4-hydroxycinnamic acid stock solution (20 mg/ml cyano-4-hydroxycinnamic acid in aqueous acetonitrile as above), and 1-μl portions of this mixture were spotted onto a MALDI sample plate for air-drying. Potential cross-linked candidates identified by the MALDI run were subjected to sequence analysis by liquid chromatography-electrospray ionization tandem-mass spectrometry (LC-ESI-MS). LC-ESI-MS was performed on a Micromass triple quadrupole mass spectrometer (Waters). A microcapillary column (10-cm × 75-μm inner diameter) was packed in-house using 5-μm C18 particles (PerSeptive Biosystems). Flow rates were generated with a Rainin high performance liquid chromatography (HPLC) system equipped with an LC-Packings microflow processor and maintained at 180 nl/min. The Syn-cyt c tryptic/Glu-C digest fragments were loaded onto the microcapillary column, washed in 0.1% acetic acid in water (buffer A), and eluted with a linear gradient of acetonitrile containing 0.1% acetic acid (buffer B) over 30 min. Fragmentation of potential cross-linked species identified by LC-ESI-MS was performed using a collision energy ramp and argon as the collision gas.
CL/Syn Binding Constant Assessments
DOPC/CL liposomes (5 nmol of total lipids, 1:1) were incubated with Syn (100 pmol) for 15 min. The mixture was then incubated in the presence of acridine 10-nonyl bromide (NAO, 2–10 nmol) in 20 mm HEPES buffer, pH 7.4, for 30 min at room temperature. Samples (10 μl) were applied to 7.5% PAGE, and electrophoresis was performed (1 h at 120 V). Unbound Syn was stained with SilverSNAP stain kit from Pierce and quantified by optical density. CL/ Syn binding constants were calculated using Equation 1,
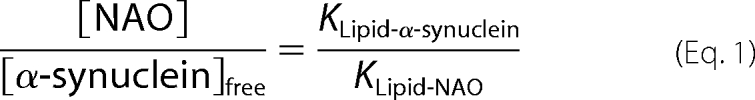 |
where Klipid-NAO = 2 × 106 m−1 for CL (16).
Quantification and Mass Spectrometric Analysis of CL Hydroperoxides
TLCL and its oxidized species were extracted from the incubation medium by Folch procedure (17) and evaporated under N2. DOPC/TLCL liposomes (250 μm at a ratio of 1:1) were incubated with cyt c (5 μm) and H2O2 (100 μm were added every 15 min during incubation) in phosphate-buffered saline (PBS) (pH 7.4 + 100 μm DTPA) in the absence and presence of Syn (15 μm). TLCL hydroperoxides were determined by fluorescence HPLC of products formed in microperoxidase 11-catalyzed reaction with a fluorogenic substrate, Amplex Red, N-acetyl-3,7-dihydroxyphenoxazine (Molecular Probes, Eugene, OR) as described previously (12, 18). For ESI-MS analysis, TLCL and its oxidation products were resuspended in chloroform/methanol 1:2 v/v (20 pmol/μl) and analyzed by direct infusion (flow rate of 5 μl/min) into a quadrupole linear ion trap mass spectrometer LXQ (Thermo Electron, San Jose, CA) The electrospray probe was operated at a voltage differential of 5 kV in the negative ion mode. Source temperature was maintained at 150 °C.
Cell-free Apoptosis System (S-100 System)
Human HL-60 cells were grown in RPMI 1640 medium supplemented with 15% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate. The cytosol extracts (S-100) were obtained as described previously (19) with minor modification. Briefly, the cells were washed twice in cold phosphate-buffered saline, pH 7.4, and the resulting pellet was resuspended in buffer containing 25 mm HEPES-KOH, pH 7.0, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 1 mm DTT, 0.1 mm phenylmethylsulfonyl fluoride, 0.05% digitonin, and 1% protease inhibitor mixture (Sigma) for 2 min at 4 °C. Cells were centrifuged at 4 °C for 10 min at 10,000 × g. The resulting supernatant was further centrifuged at 4 °C for 50 min at 100,000 × g. The supernatant was collected as S-100 and kept at −80 °C until further use. For caspase-3 activation, S-100 (5 μg/μl) was incubated with 1 mm dATP and 1 μm cyt c for 90 min at 37 °C, and caspase-3 activity was normalized as 100%. Syn (13 μm) was added alone or in complex with TOCL (at a ratio of 3:1). The caspase-3 activity was evaluated as described in the manufacturer's manual (Invitrogen, Enzchek caspase-3 assay kit).
Rat Rotenone Model of Parkinson Disease
All animal use was in accordance with National Institutes of Health guidelines and was approved by the Pittsburgh University Institutional Animal Care and Use Committee. Surgeries were performed as described previously (20). Briefly, male Lewis rats (300–350 g) received 3.0 mg/kg/day rotenone for up to 4 weeks through subcutaneous osmotic mini-pumps (Alzet Corp., Palo Alto, CA). Control rats received vehicle (DMSO/polyethylene glycol, 1:1). Rotenone-infused rats were euthanized at the time of severe systemic illness characterized by rigidity and akinesia that prevented adequate feeding and grooming. Control rats were euthanized at similar time intervals. Rats were euthanized using isoflurane (Abbott). The brains were removed without prior perfusion and dissected midsagittally into two hemispheres. One hemisphere was immersion-fixed in 4% paraformaldehyde in phosphate buffer and used for immunochemistry. From the other hemisphere, specific brain regions were dissected and immediately frozen in dry ice and stored at −80 °C.
Fluorescence Resonance Energy Transfer (FRET) Protocol
Co-localization (molecular proximity) of cyt c and Syn in histological preparations was assessed by analysis of FRET between fluorescently labeled specific antibodies. Because FRET efficiency strongly depends on the distance between the fluorophores, increased FRET levels reflect the close proximity of the antibodies and therefore of the proteins of interest (reviewed in Ref. 21). Primary antibodies were directly labeled using the DyLight 549 and DyLight 649 antibody labeling kits (ThermoFisher, Rockford, IL). Lyophilized primary antibodies were resuspended in 0.05 m borate buffer, pH 8.5, to a final concentration of 1 mg/ml; 100 μg were used for labeling. The dyes were reconstituted in 20 μl of dimethylformamide; 8 μl of DyLight 549 or 5 μl of DyLight 649 was added to the antibody solution, and the mixture was incubated for 45 min at room temperature. The labeled antibodies were purified, and the excess dye was removed by processing the solution with a Micro Bio-Spin chromatography column 6 (Bio-Rad) according to the manufacturer's instructions. Thus modified antibody (in 10 mm Tris-HCl buffer, pH 7.4) was used for treatment of tissue sections obtained according to standard histological procedures. Images were acquired using a laser scanning confocal microscope (Fluoview1000, Olympus) equipped with spectral detector technology, which provides accurate wavelength separation of the emitted light. FRET was detected by reading the acceptor emission (DyLight 649-conjugated primary antibody) at 674 nm while exciting the donor (DyLight 549-conjugated primary antibody) with the 543 nm laser.
Immunostaining of Syn and Cyt c in Cells
Paraformaldehyde (4%)-fixed cytospin preparations were permeabilized with 0.2% Triton X-100. Cyt c was stained with mouse anti-cyt c antibody (Pharmingen, 1:100) followed by FITC-conjugated goat anti-mouse IgG (Pharmingen, 1:100), which exhibits a green fluorescence. Syn was stained with rabbit anti-Syn antibody (Chemicon International, 1:200) followed by Texas Red-conjugated sheep anti-rabbit IgG (GeneTex, 1:200), which shows red fluorescence.
Immunostaining of Syn and Cyt c in Human and Rat Brains
Double immunofluorescence analysis was performed as described previously (22). In brief, de-waxed substantia nigra sections from PD/LB disease (LBD) cases were heated in target retrieval solution at 95 °C for 30 min, blocked with Immunon protein blocking agent (Shandon, Pittsburgh, PA), and incubated at 4 °C overnight with rabbit anti-cyt c (1:50, Cell Signaling Technology, Beverly, MA) and mouse anti-α-Syn (1:1500, Zymed Laboratories Inc.). The sections were then incubated with Alexa Fluor 488 donkey anti-mouse and Cy3-conjugated donkey anti-rabbit IgG (1:500, Jackson ImmunoResearch, West Grove, PA). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. To minimize bleed through from the green into the red channel, images were always collected sequentially. For negative controls, primary antibody was replaced with nonimmune rabbit or mouse IgG. Sections were visualized using Olympus IX 71 fluorescent microscope.
Statistical Analysis
The results are presented as either means ± S.D. or means ± S.E. values from at least three experiments, and statistical analyses were performed by paired Student's t test unless specified otherwise. The statistical significance of differences was set at p < 0.05.
RESULTS
Syn Cross-links Cyt c in a Peroxidase Process Requiring Anionic Phospholipids
To explore the possibility of peroxidase-catalyzed cross-linking of Syn with cyt c, we utilized a model system that included Syn, cyt c, and an anionic phospholipid. Given the high affinity of cyt c for CL, we initially used CL as a prototypic anionic phospholipid. Because Syn can also bind lipids, we performed estimates of the binding capacity of Syn toward CL. Migration of Syn as a single band was changed to a smeared profile in the presence of CL (Fig. 1a). Addition of NAO, as a competitive complexer of CL, reinstated the single band of Syn at NAO/CL ratios of >1:2. Based on these estimates, the binding constant for Syn with TOCL is on the order of 8.5 ± 1.0 × 106 m−1 for nonfibrillated Syn. Previous studies demonstrated that, upon binding with acidic phospholipids, Syn undergoes structural rearrangements toward α-helical conformation (22). For PS, the high affinity binding with Syn was stimulated by its homo-oligomerization and the formation of (proto)fibrils (23, 24). Therefore, we assessed the binding constant for CL with fibrillated (aged) Syn that has been prepared according to Smith et al. (23). We found that aged Syn had a significantly higher affinity for CL than nonfibrillated Syn (1.8 ± 0.5 × 107 m−1, p < 0.02, versus nonfibrillated Syn). These results correspond with previous reports showing that protofibrils and fibrils of Syn interact with lipid membranes more effectively than the soluble monomeric protein (23–25). These data indicate that Syn oligomers with bound CL may act as substrates for peroxidase activity of cyt c (see below).
To test whether PD-associated mutants of Syn-A30P and A53T were also able to form hetero-oligomeric complexes with cyt c, we performed native PAGE with subsequent silver staining. We utilized conditions that are compatible with the expression of sufficiently high peroxidase activity of cyt c-TOCL complexes (1:20 mol/mol). We found that at a Syn-cyt c ratio of 1:1, the affinity of wild-type Syn for binding with cyt c/TOCL was markedly higher than for both Syn mutants (A30P and A53T). This was documented by almost complete disappearance of Syn monomer due to its oligomerization with cyt c/TOCL for the wild-type protein and a similar but only a partial decrease of the Syn monomeric forms for both mutants (Fig. 2a, left panel). To quantitatively assess the formation of hetero-oligomeric complexes of cyt c/TOCL/Syn across different Syn concentrations, we used fluorescently labeled CL, NBD-CL. The assay is based on the ability of cyt c to quench fluorescence of NBD-CL (Fig. 2a, right panel, inset), likely due to the proximity of the NBD chromophore to the heme moiety of cyt c (26). Competition between cyt c and Syn for CL binding, upon addition of Syn, diminishes the quenching effect resulting in the increased fluorescence intensity (Fig. 2a, right panel, inset). We found that wild-type Syn was more effective in reconstituting the NBD fluorescence than both of the Syn mutants (A53T and A30P) (Fig. 2, inset, spectra 2 and 3, respectively). When the fluorescence intensity differences were plotted against several concentrations of added Syn (Fig. 2a, right panel), one can see that both mutant forms of Syn were less potent as fluorescence enhancers than the wild-type protein.
We then studied peroxidase reactions and cross-linking of Syn-CL-cyt c complexes. SDS-PAGE analyses and staining with anti-cyt c and anti-Syn antibodies showed that different hetero-oligomers of Syn and cyt c, as well as very high molecular weight aggregates, were produced by the H2O2-mediated cyt c-catalyzed cross-linking of proteins (Fig. 1b). Reducing conditions (treatment with 20 mm DTT) did not affect the accumulation of oligomerized protein aggregates (data not shown). Because SDS can mimic and interfere with the interactions of anionic phospholipids with Syn and cyt c, we performed native agarose gel electrophoresis. This revealed that the covalent hetero-oligomerization of Syn and cyt c was induced by H2O2 in the presence of CL (Fig. 1c). All four components, Syn, cyt c, CL, and H2O2, were required for the formation of cross-linked polymers. Even in the presence of a large excess of Syn (Syn-cyt c ratio of 10:1), oligomerization did not occur without TOCL and H2O2.
In the presence of H2O2 and TOCL, wild-type Syn, A30P, and A53T mutants underwent hetero-oligomeric covalent cross-linking with cyt c (at different Syn-cyt c ratios, from 1:5 to 40:1) in a similar way as assessed by Western blot analysis and staining with anti-Syn antibodies (Fig. 2b). Moreover, we observed that in the presence of cyt c, TOCL, and H2O2, both intact and fibrillated Syn underwent covalent cross-linking in a similar way (Fig. 2c). The lack of significant difference in covalent conjugation with cyt c and CL between fibrillated and nonfibrillated Syn was observed not only with wild-type Syn but also with its mutants, A53T and A30P.
Although the presence of an anionic phospholipid was critical for the hetero-oligomerization of Syn and cyt c, the requirement in CL was not absolute as other negatively charged phospholipids such as phosphatidic acid (PA), phosphatidylinositol (PI), and PS were effective in supporting the hetero-oligomerization as well (Fig. 1d). However, a noncharged PC was insufficient for the stimulation of protein hetero-oligomerization (Fig. 1d).
Syn Is a Preferred Substrate for Reactive Intermediates of the Peroxidase Complex
To test whether reactive intermediates of peroxidase activity of cyt c in its complexes with Syn and CL are essential for protein oxidative hetero-oligomerization, we employed immuno-spin trapping (27) and EPR techniques. A spin trap, DMPO, has been shown to form spin adducts with protein radicals generated during the peroxidase activation of cyt c by H2O2 (28). Utilizing anti-DMPO antibodies, we confirmed the H2O2-dependent production of protein derived radicals by both nonoligomerized as well as by oligomerized complexes of Syn-CL-cyt c (Fig. 3a). Importantly, interaction of DMPO with protein radicals effectively inhibited hetero-oligomerization in a concentration-dependent manner (Fig. 3a) corroborating the involvement of the peroxidase mechanism in hetero-oligomerization.
FIGURE 3.

Evidence for the involvement of peroxidase reactive intermediates in hetero-oligomerization of Syn-cyt c-TOCL in the presence of H2O2. a, immuno-spin trapping of protein-derived radicals during H2O2-dependent hetero-oligomerization of Syn with cyt c in the presence of TOCL. Staining of polyacrylamide gels with anti-DMPO antibody (left panel) demonstrates the production of protein-derived radicals. Staining with anti-Syn (middle panel) and anti-cyt c (right panel) antibodies is shown. Note that the production of hetero-oligomeric forms of Syn-cyt c-TOCL aggregates is progressively inhibited by increasing concentrations of DMPO indicating that the formation of DMPO adducts competitively inhibited oligomerization. No oligomerization of Syn with cyt c and TOCL occurred in the absence of H2O2 (data not shown). Typical gels representative of three experiments are shown. b, low temperature EPR measurements of H2O2-induced protein-derived (tyrosyl) radicals of cyt c-CL complexes are shown in the left panel. Syn quenched the formation of tyrosyl radicals of cyt c-CL complexes in a concentration-dependent manner. The inset shows a typical EPR spectra of protein-derived (tyrosyl) radicals of cyt c-CL complexes in the absence (trace 1) and presence of Syn (trace 2), cyt c/α-synuclein = 1:0.5. Right panel shows power-saturation curves of protein-derived tyrosyl radical EPR signals of cyt c-CL complexes (trace 1) and cyt c-CL-Syn complexes (trace 2) (cyt c/α-synuclein = 1:0.5). c, assessments of dityrosine cross-links formed by incubation of Syn-cyt c-TOCL with H2O2 by fluorescence intensity (λex 315 nm and λem 420 nm). Formation of dityrosine adducts increased by 1.6-fold upon addition of H2O2 to cyt c/TOCL. When Syn was present in this incubation system along with H2O2, the fluorescence intensity was further increased by 3.2-fold (mean ± S.D., *, p < 0.05 versus cyt c/TOCL/H2O2). d, SDS-PAGE with subsequent Western blot analysis of covalent conjugates with dityrosine cross-links formed from syn-cyt c-TOCL hetero-oligomeric complexes in the presence of H2O2. Staining was performed using anti-dityrosine antibody (lanes A), anti-Syn antibody (lanes B), and anti-cyt c antibody (lanes C). A strong positive signal was observed with anti-dityrosine antibody in cross-linked high molecular weight aggregates (nondissociable in SDS). e, MS/MS spectrum of m/z 725.8 (M + 2H)2+ species. This species was obtained from the digestion of high molecular weight aggregates of Syn-cyt c-CL formed in the presence of H2O2. Note the presence of ions at m/z 468, 495, 625, 730, and 896 corresponding to various daughter ions of the Syn-(132–137) and cyt c-(74–79) cross-link.
EPR spectroscopy revealed a characteristic broad signal (half-width 15.5 G) of protein-immobilized radicals (with a g factor of 2.005) that has been tentatively ascribed to Tyr˙ radicals generated by cyt c-CL complexes treated with H2O2 (12, 29). The signal was also detectable in the presence of Syn (Fig. 3b, left panel). As a participant of the peroxidase reaction leading to hetero-oligomerization, Syn is expected to interact with the cyt c protein radicals, likely resulting in quenching of these radicals because of the formation of Tyr-Tyr bonds. Indeed, increasing the ratio of Syn to cyt c/CL caused a significant quenching of the signal such that half-maximal magnitude of the signal was achieved at a ratio of 0.4:1. This is compatible with the expected central role of cyt c in generating the radical intermediates and relatively “passive” substrate participation of Syn leading to Tyr-Tyr hetero-oligomerization of the latter in a triple complex of cyt c-TOCL-Syn. More detailed studies of power saturation dependence of the signal demonstrated its relatively fast saturability in the cyt c/TOCL system (Fig. 3b, right panel). Based on the comparable data in the literature, one can assume that the saturation course is indicative of a spin-lattice relaxation time of tyrosyl radical T1 = (1.3 ± 0.3)·10−5 s (30, 31). The proximity of Tyr˙ radicals to the heme iron moiety was unaffected by Syn based on a very similar saturation pattern of the radical in the triple system cyt c/TOCL/Syn exposed to H2O2.
To further characterize formation of dityrosine cross-links in Syn-cyt c-TOCL hetero-oligomeric complexes in the presence of H2O2, we performed the following: (i) fluorescence spectroscopy; (ii) Western blotting with anti-dityrosine antibody; and (iii) MS analysis. Measurements of characteristic dityrosine fluorescence demonstrated a 1.6-fold increase in dityrosine adducts upon addition of H2O2 to cyt c/TOCL (Fig. 3c). When Syn was present in this incubation system along with H2O2, the fluorescence intensity was further increased by 3.2-fold.
Western blot analysis with anti-dityrosine antibody showed a strong positive signal in cross-linked high molecular weight aggregates (nondissociable in SDS) that were weaker in the absence of Syn or H2O2 (Fig. 3d). Syn enhanced the formation of dityrosine-based covalent cross-links, in line with measurements of di-tyrosine fluorescence (Fig. 3c). These results are also in agreement with the above data showing significant quenching of the characteristic EPR signal of H2O2-induced Tyr˙ radicals of cyt c/CL by Syn.
Finally, MS analysis of covalent aggregates further confirmed the formation of cyt c/Syn cross-links as a result of recombination of protein Tyr˙ radicals. After SDS-gel electrophoresis, bands corresponding to potential high molecular weight Syn-cyt c aggregates were cut from the Coomassie-stained gel and subjected to an in-gel digest with trypsin at 37 °C for 24 h followed by digestion with endoproteinase Glu-C for an additional 24 h. The MALDI-TOF profile of the multienzyme in-gel digest included fragments from both Syn as well as cyt c, which included one species of m/z 1450 whose m/z matched that of a potential Syn-cyt c cross-link (not shown). This species was further analyzed by LC-ESI-MS. Fragmentation of a proteolytic fragment with m/z 725.8 documented the cross-link involving Tyr-133 of Syn (GYQDYE) and Tyr-74 of cyt c (YIPGTK). This cross-link preferred to run as the doubly charged species (m/z 725.8) as opposed to the parent molecular ion ((M + H)+, m/z 1450) under our instrumental conditions (not shown). Fragmentation of the doubly charged species revealed ions with m/z 468, 495, 625, 730, and 896 corresponding to G(Y/Y)I (without CO), G(Y/Y)I, GYQ(D/Y), YQD(Y/Y), and G(Y/Y)IPGTK, respectively (Fig. 3e). Because these fragments are all singly charged species, it is not unusual to detect ions with masses above that of the doubly charged parent ions.
To further prove the involvement of the peroxidase mechanism in protein aggregation, we assessed the effects of three prototypical peroxidase phenolic substrates, DA, Amplex Red, and etoposide. Consistently, all three compounds caused concentration-dependent inhibition of protein hetero-oligomerization (Fig. 4, a and b). Conversely, Syn displayed a concentration-dependent inhibition of the peroxidase activity of the complexes with the phenolic peroxidase substrates (Fig. 4b). Very low micromolar concentrations of Syn were sufficient to inhibit the peroxidase activity of cyt c-CL complexes. With Amplex Red, the IC50 for inhibition by Syn was 0.2 μm in the presence of 2 orders of magnitude higher concentrations of Amplex Red (50 μm). Low micromolar Syn concentrations were also effective in inhibiting the peroxidase activity in the presence of 2 orders of magnitude higher concentrations of DA or etoposide (Fig. 4b). Staining of native agarose gels for the peroxidase activity, using West Pico chemiluminescence reagent as a substrate, demonstrated that the high activity detectable in cyt c-CL complexes, was markedly diminished in the presence of Syn (Fig. 4c). Furthermore, PD-associated Syn mutants, A30P, A53T, and A53T/A30P, were tested for their peroxidase activity. We found that in the presence of Syn mutants (A30P and A53T/A30P), the peroxidase activity of cyt c-TOCL complexes was higher than in the presence of wild-type Syn. Combined, these data suggests that Syn acted as a preferred substrate for the peroxidase activity of cyt c-CL complexes.
FIGURE 4.

Syn is preferred substrate for reactive intermediates of the peroxidase complex. a, effects of two prototypical peroxidase phenolic substrates Amplex Red (left panel) and DA (right panel) on oligomerization of Syn-cyt c-TOCL in the presence of H2O2. Staining with anti-Syn antibody. Note that both compounds caused concentration-dependent inhibition of protein hetero-oligomerization. Typical gels representative of three experiments are shown. b, inhibition of peroxidase activity of Syn-cyt c-TOCL complexes with Amplex Red, DA, and etoposide. Left panel, Syn inhibits the peroxidase activity of Syn-cyt c-TOCL complexes, with Amplex Red as a substrate, in a concentration-dependent manner. Note that the IC50 for Syn was 0.2 μm in the presence of 50 μm Amplex Red. The results of three independent experiments are shown. Middle panel, Syn inhibits the peroxidase activity of Syn-cyt c-TOCL complexes, with etoposide as a substrate, in a concentration-dependent manner. Note that the IC50 for Syn was 0.5 μm in the presence of 100 μm etoposide. A typical ESR spectrum of etoposide phenoxyl radical generated by the H2O2-dependent peroxidase activity is shown (inset). Right panel, Syn inhibits the peroxidase activity of Syn-cyt c-TOCL complexes, with DA as a substrate (200 μm). Data are presented as mean ± S.D. (n = 3). c, native agarose gels of Syn-cyt c-CL and cyt c-CL complexes stained for peroxidase activity with SuperSignal West Pico chemiluminescence substrate (left panel). Note that the peroxidase activity of cyt c-TOCL complexes (single arrow) toward the chemiluminescence substrate is inhibited when Syn is included in the preformed complex (double arrows) and then exposed to H2O2/chemiluminescence substrate. Typical gels representative of three experiments are shown. Quantification of residual peroxidase activity of wild-type (wt) and mutated forms of Syn (A53T, A30P, A53T/A30P)-cyt c-TOCL complexes (right panel). The peroxidase activity of cyt c-TOCL complexes was higher in the presence of Syn mutants (A30P and A53T/A30P) compared with wild-type Syn (mean ± S.D.; *, p < 0.05 versus wild-type Syn). d, Syn protects polyunsaturated TLCL against oxidation induced by cyt c/H2O2. Cyt c-induced accumulation of oxidized TLCL (as assessed by fluorescence HPLC (left panel) and MS analysis of doubly charged ions [M − 2H]2− (right panel)) was markedly suppressed by Syn. This was accompanied by the formation of high molecular weight aggregates of Syn-cyt c-TLCL (see Western blot with Syn antibody (left panel, inset). Data are presented as mean ± S.E.; *, p < 0.05 versus TLCL + cyt c + H2O2. Accumulation of monohydroperoxy/monohydroxy derivatives of TLCL molecular species was observed after incubation of liposomes with cyt c and H2O2 (m/z 724.2 + [O] = 732.2; 724.2 + [OO] = 739.7; 724.2 + [O] + [OO] = 748.2; 724.2 + [2OO] = 755.9; 724.2 + [O] + [2OO] = 763.8; 724.2 + [3OO] = 771.8; 724.2 + [O] + [3OO] = 779.7; 724.2 + [4OO] = 787.9; and 724 + [O] + [4OO] = 795.7). This effect was markedly reduced by Syn. Representative mass spectra from three independent experiments are presented.
To determine whether the important peroxidase role of Syn is realized with another physiologically relevant substrate, we monitored oxidation of polyunsaturated TLCL by cyt c in the presence and absence of Syn. Using MS analysis and fluorescence HPLC, we found that Syn was able to protect TLCL against oxidation (Fig. 4d). As expected, oxidation of Syn by cyt c/TLCL was accompanied by accumulation of characteristic covalent aggregates detectable on SDS-polyacrylamide gels (Fig. 4d, inset).
Syn and Syn-CL Complex Inhibit Apoptotic Caspase Activation
We reasoned that the covalent binding of cyt c through its Syn-CL-mediated peroxidase cross-linking should effectively decrease the amounts of cyt c required for its participation in apoptosome formation and caspase activation. To experimentally address this, we employed a well characterized proapoptotic cell-free system composed of S-100 fraction (obtained from human leukemia HL-60 cells lacking endogenous Syn) and activated apoptosis by cyt c/dATP (19). We demonstrated that, in the presence of CL, exogenously added Syn indeed was able to significantly suppress activation of caspase-3/7. Preincubation of cyt c-Syn-CL complexes with H2O2 completely abolished the proapoptotic effects of cyt c toward caspase-3 activation in S-100 fraction (Fig. 5a). We found that wild-type Syn and two mutants, A53T and A30P, displayed equal inhibitory effects against caspase-3 activation after preincubation with cyt c/TOCL/H2O2. This suggests that covalent hetero-oligomeric cross-links produced by peroxidase activity of cyt c with wild-type Syn as well as A30P and A53T mutants under conditions of oxidative stress result in equally effective inhibition of cyt c interactions with Apaf-1 and caspase activation. These results correspond with our data obtained by PAGE demonstrating similar effectiveness of wild-type Syn and its A30P and A53T mutants in H2O2-dependent formation of covalent cross-links shown in Fig. 2b.
FIGURE 5.

Syn and Syn-CL complex inhibits apoptotic caspase activation. a, Syn-TOCL complexes inhibit caspase-3 activated by cyt c/dATP in a cell-free proapoptotic system (S-100 fraction isolated from HL-60 cells). Note that Syn in combination with TOCL was most effective in preventing cyt c-dependent caspase-3 activation. Wild-type Syn and two mutants, A53T and A30P, displayed equal inhibitory effects against caspase-3 activation after preincubation with cyt c/TOCL/H2O2. S-100 (5 μg of protein/μl) fractions were incubated with 1 μm cyt c for 90 min at 37 °C, in the absence and in the presence of complex of Syn-TOCL. Bar 1, S-100; bar 2, 1 + cyt c; bar 3, 2 + TOCL/Syn; bar 4, complex of wild-type Syn-TOCL-cyt c was preincubated with H2O2 and then added into 1; bar 5, complex of A53T Syn-TOCL-cyt c was preincubated with H2O2 and then added into 1; bar 6, complex of A30P Syn-TOCL-cyt c was preincubated with H2O2 and then added into 1. All the data are presented as % of caspase-3 activation in complete system (bar 2) taken as 100%. Data are presented are means ± S.D. (n = 6). The statistical significance was calculated using analysis of variance, followed by Tukey's procedure with the family-wise error rate of p < 0.05 to perform our pairwise comparisons of selected group means. Asterisk indicates a statistically significant difference resulting from the Tukey comparisons. b, densitometric and Western blot analysis (inset) with anti-Syn antibody in wild-type (WT), nontargeting siRNA (C), two different clones (A5 and A7) of HeLa Syn knockdown. Data are presented as mean ± S.D. (n = 3/condition). *, p < 0.05 versus wild type; #, p < 0.05, A7 versus A5. c, biomarkers of apoptosis-PS externalization (left panel) and caspase-3/7 activation (middle panel) increase and cell viability (right panel) decrease in Syn-deficient HeLa A5 and A7 cells after exposure to DA. Data are presented as mean ± S.D. (n = 3/condition). *, p < 0.05, 400 μm versus 0 μm; #, p < 0.05, A7 versus A5. d, delivery of Syn into MECs confers resistance to proapoptotic caspase-3/7 activation. Note that robust activation of caspase-3/7 MECs stimulated with ActD was markedly attenuated in Syn-containing MECs (delivered by the direct introduction of Syn into cells). Delivery of a nonspecific protein, green fluorescent protein (GFP), exerted no effect on ActD-induced caspase-3/7 activation in MECs. Data are presented as mean ± S.D. (n = 4/condition). *, p < 0.05, ActD versus ActD + Syn.
To further test whether Syn is able to protect cells against apoptosis, we used HeLa cells containing Syn and knocked down its expression to less than 10% of its content in wild-type cells by siRNA protocol to generate clones HeLa A5 and A7 (Fig. 5b). We then compared cell viability and apoptotic responses of HeLa cells and HeLa A5 and A7 cells to DA (Fig. 5c). Cell viability was decreased, and two biomarkers of apoptosis, PS externalization and caspase-3/7 activation, were increased in Syn-deficient HeLa cells incubated for 24 h in the presence of DA (400 μm). Furthermore, the resistance of cells to apoptosis induced by DA was proportional to the levels of Syn expression. These results indicate that Syn acts as a protector against apoptosis, and its deficiency is associated with increased sensitivity to DA-induced apoptosis.
In a separate series of experiments, we used a reverse approach and delivered Syn into MECs, naturally expressing very low levels of Syn. The efficiency of Syn delivery was confirmed by immunohistochemistry. Western blot analysis demonstrated a 3-fold increase in Syn content compared with controls. In MEC, standard challenge with a proapoptotic agent, ActD, caused a robust activation of caspases 3/7 (Fig. 5d). Significantly lower caspase activation was found in MECs with delivered Syn. Delivery of a nonspecific protein, green fluorescent protein, exerted no effect on ActD-induced caspase-3/7 activation in MECs. Thus both in a cell-free system and in two types of cells, Syn afforded protection against cell death induced by different proapoptotic stimuli. Notably, Machida et al. (32) have demonstrated that increasing expression levels of Syn enhanced resistance to death-promoting stimuli in SH-SY5Y cells.
Syn and Cyt c Co-localize after Proapoptotic Stimulation in Cells
In LBs, Syn has been reported to be co-localized with cyt c (8). If cyt c co-aggregation with Syn is essential for its anti-apoptotic action, the production of aggregates with co-localized Syn and cyt c is expected to occur in cells triggered to apoptosis. We utilized two different proapoptotic stimuli, a nonoxidant ActD and a pro-oxidant, t-BuOOH, to initiate apoptosis in Syn-containing HeLa cells. Proapoptotic stimulation with ActD resulted in the appearance of cells containing inclusions with co-localized cyt c and Syn (Fig. 6a). These inclusions were not detectable in cyt c-deficient HeLa cells (HeLa 1.2) engineered (using siRNA protocol) to express 14% of their constitutive levels of cyt c (Fig. 6a). Importantly, we were able to detect t-BuOOH-induced formation of Syn-cyt c aggregates in HeLa cells using Western blot analyses after electrophoresis in native agarose gel or SDS-PAGE (Fig. 6b). The presence and co-localization of Syn-cyt c hetero-oligomers in t-BuOOH-treated cells was confirmed by immunoprecipitation with anti-Syn antibodies (Fig. 6c). Recent reports indicate that Syn can translocate to and form aggregates in mitochondria (33). Given that cyt c is one of the abundant proteins in the inter-membrane space of mitochondria, we reasoned that the peroxidase activity of cyt c-Syn-CL complexes could initiate the hetero-oligomerization. Indeed, we detected the accumulation of co-localized Syn-cyt c hetero-oligomers in mitochondria isolated from HeLa cells pretreated with t-BuOOH (Fig. 6d).
FIGURE 6.

Detection of inclusions and co-localization of hetero-polymerized Syn and cyt c aggregates in HeLa and SH-SY5Y cells after exposure to ActD or t-BuOOH. a, immunostaining of cyt c (green) and Syn (red) in intact HeLa cells and cyt c-deficient HeLa 1.2 cells. In control cells, cyt c appears in a punctate, perinuclear pattern of staining with little overlap with cytosolic Syn staining. ActD induces cytoplasmic blebbing, nuclear fragmentation, and transition from perinuclear to a diffuse staining pattern for cyt c. Focal regions of accentuated Syn staining, suggestive of aggregation, are observed, and this frequently co-localizes with focal regions of accentuated cyt c staining in cells with both preserved and fragmented nuclear contours. HeLa 1.2 clones engineered to express decreased levels of cyt c show a similar distribution of cyt c and Syn, although there is less cyt c immunoreactivity. HeLa 1.2 cells show preserved perinuclear cyt c and reduced propensity for clumped or aggregated Syn in response to the same doses of ActD, and there is no evidence of Syn and cyt c co-localization in these cells. b, Western blot analysis of Syn-cyt c aggregates after native agarose gel electrophoresis (left panel) and SDS-PAGE (right panel) in HeLa cells exposed to t-BuOOH. Left panel, staining with anti-Syn (Syn) and anti-cyt c antibodies (cyt c) reveals the appearance of co-localized hetero-oligomers after treatment of cells with t-BuOOH. Right panel, staining with anti-Syn (Syn) and anti-cyt c antibodies (cyt c) demonstrates disappearance of monomeric form of Syn, and accumulation of very high molecular weight Syn aggregates partially overlapping with aggregated forms of cyt c. Typical gels representative of three experiments are shown. c, detection of Syn-cyt c hetero-oligomers in HeLa cells exposed to t-BuOOH using immunoprecipitation with anti-Syn antibodies and Western blot analysis with anti-Syn and anti-cyt c antibodies after SDS-PAGE. Note that proteins immunoprecipitated with anti-Syn antibody contained high molecular weight aggregates positive for both Syn (asterisk) and cyt c (arrowhead) after treatment with t-BuOOH. Bands around 50 kDa likely correspond to heavy chains of immunoglobulins remaining after immunoprecipitation. Arrow indicates Syn monomer. Typical gels representative of three experiments are shown. d, Western blot analysis of Syn-cyt c aggregates after SDS-PAGE in mitochondria t-BuOOH from HeLa cells pretreated with t-BuOOH. Staining with anti-Syn (Syn) and anti-cyt c antibodies (cyt c) demonstrates accumulation of very high molecular weight Syn aggregates partially overlapping with aggregated forms of cyt c. Note that mitochondria from control cells contain significant amounts of monomeric cyt c but hardly detectable levels of monomeric Syn. After treatment with t-BuOOH, polymerized forms of Syn and cyt c are readily detectable, and the monomeric form of cyt c is decreased. Typical gels representative of three experiments are shown. e, immunostaining of cyt c (green) and Syn (red) in SH-SY5Y cells. Note that both proapoptotic agents, t-BuOOH and ActD, caused the appearance of LB-like inclusions revealed as yellow color in merged images (arrows). In normal SH-SY5Y cells, cyt c had a punctate mitochondrial distribution, and Syn was localized in the cytoplasm and perinuclear area. Following ActD or t-BuOOH treatment, Syn displayed aggregated pattern of distribution and overlapped with anti-cyt c staining (yellow spots on merged images).
Dopaminergic SH-SY5Y cells triggered to apoptosis by either ActD or t-BuOOH (as evidenced by 28.1 ± 7.4 and 36.9 ± 9.7% of cells with externalized PS by annexin V binding) elicited inclusions in which Syn and cyt c were co-localized (Fig. 6e). Thus, pro-oxidant stimulation or proapoptotic stimulation accompanied by the production of reactive oxygen species resulted in the formation of Syn-cyt c covalent aggregates.
Syn Co-localizes with Cyt c in LB and LN in Vivo
We used a well established rotenone rat model of PD in vivo and followed cyt c and Syn distribution in the midbrain. After the rotenone treatment, cyt c distribution changed from punctate (presumably mitochondrial) to apparent aggregates that co-localized with Syn in dopaminergic substantia nigra neurons (Fig. 7). Moreover, using FRET protocol we were able to prove that Syn and cyt c are within molecular proximity to each other in LNs (Fig. 7b). This strongly supports our hypothesis on the closeness and molecular availability of Syn and cyt c for peroxidase-catalyzed cross-linking.
FIGURE 7.

Syn distribution and co-localization with cyt c in substantia nigra dopaminergic neurons after rotenone. a, brain sections through substantia nigra were double-labeled for cyt c (red) and Syn (green) and imaged by laser scanning confocal microscopy. Note the increased fluorescence and altered distribution of Syn after rotenone. Additionally, there is FRET between cyt c and Syn, strongly suggesting direct protein/protein interactions. b, fluorescence intensity profiles of Syn immunoreactivity in single dopaminergic neuron from control and rotenone-treated rats. Each fluorescence intensity profile is from an identical rectangular region of interest encompassing part of a single dopaminergic neuron. After rotenone, the neuronal levels of Syn increase, and its distribution changes from relatively diffuse to markedly punctate. c, quantification of the co-localization of Syn with cyt c in substantia nigra from a control animal and a rotenone-treated animal. After rotenone treatment there is marked increase in the Pearson correlation coefficient (R) and in the co-localization coefficients for both channel 1 (cyt c) and channel 2 (Syn). Data are from confocal images (×60) that each contain ∼20 dopaminergic neurons and surrounding neuropil.
To determine the extent to which Syn in synaptic terminals is capable of preventing retrograde expansion of apoptotic events to soma of neuronal cells in humans, we analyzed co-localization of Syn and cyt c in midbrain sections from Parkinson/Lewy body disease patients. Demographic characteristics of patients are presented in supplemental Table 1. By definition, control patient brains lacked Syn aggregates and were not used further for co-localization scoring. Case 1 and Case 2 showed classic PD presentation clinically, with late development of dementia prior to death (Parkinson disease dementia). Case 3 had symptoms of “incipient dementia” within a year of clinical diagnosis of Parkinson disease, and is classified as “pure” diffuse LB disease in accord with the recommendations of the Third Consortium on diffuse Lewy body disease. The neuropathology and clinical features of Parkinson disease dementia and diffuse LB disease are indistinguishable aside from the temporal sequence of symptoms (34). Case 4 represented a diffuse LB disease patient with concurrent Alzheimer-type neuropathology. Using these samples, we confirmed the co-localization of Syn and cyt c (in approximately half of Syn immunoreactive structures) and found that it was 2-fold more frequent in LNs than in the soma (Fig. 8). This suggests that Syn acts as an anti-apoptotic scavenger of cyt c likely preventing undesirable spread of apoptotic signals from neuritic processes and terminals to the soma (35).
FIGURE 8.

Co-localization of Syn and cyt c in human PD/Lewy body disease substantia nigra. Midbrain sections from patients with PD/Lewy body disease were immunolabeled for cyt c (red) and Syn (green). 4′,6-Diamidino-2-phenylindole (blue) was used to stain nuclei. Normal nigral neurons exhibit a granular cytoplasmic staining pattern for cyt c, consistent with its mitochondrial localization (A, lower neuron). In contrast, the neuron containing a LB (arrow) shows light, diffuse cytoplasmic cyt c staining and co-localization of cyt c and Syn (yellow) in the LB (A, upper neuron). Many Lewy neurites also showed co-localization of cyt c and Syn (B and C), although cyt c negative Lewy neurites (asterisk) were also observed (inset, C). Note the absence of red staining in the LB containing neuron (arrowhead) when nonimmune rabbit antiserum was substituted for antibody to cyt c as a negative staining control (D). More than 1,200 consecutive Syn immunoreactive structures from four cases were scored by location in the neuronal soma (Lewy body/pale body, LB/PB) or in neuritic processes (Lewy neurites). These were analyzed for presence or absence of cyt c co-localization. The pie chart (E) shows Lewy neurites without cyt c (light green-blue), LB/PB without cyt c (dark blue), LB/PB co-localizing with cyt c (magenta), and Lewy neurites co-localizing with cyt c (yellow). Note that nearly half of all Syn immunoreactive structures show co-localization with cyt c (yellow and red combined). Moreover, more than two-thirds of Syn-cyt c co-localizing structures involved neurites (yellow).
DISCUSSION
Oxidative stress has long been associated with the development of PD and accompanying LB formation (36). In addition to noncovalent aggregation of partially unfolded Syn molecules, covalent cross-linking and accumulation of granular material is typical of (dopaminergic) neurons of PD patients (37). Although DA oxidation products and their adducts with Syn have been suggested to contribute to fibril formation, specific mechanisms translating oxidative stress into covalent cross-links and their role in the pathogenesis of the disease have not been well understood (38). Hashimoto et al. (8), has suggested that cyt c may be involved in H2O2-dependent Syn aggregation implying contribution of a peroxidase-type mechanism. However, cyt c is a poor peroxidase (39). Therefore, aggregation of Syn by cyt c required long term incubations and high concentrations of cyt c/H2O2 to cause the aggregation (8). Thus specific catalytic mechanisms of cyt c-driven oxidative cross-linking of Syn have not been identified.
Mitochondrial impairment plays a key role in PD and its models, and several mutations that cause PD are associated with mitochondrial abnormalities (40, 41). Systemic deficits in mitochondrial complex I activity have been reported in PD patients (42). Evidence of mitochondrial oxidative stress and cyt c release are also observed in animal and cell culture models of PD (20, 43–46). Yet despite abundant evidence of mitochondrial alterations (47–49), classic morphologic changes of apoptosis are disproportionately rare (50), suggesting the possibility of additional mechanisms in dopaminergic neurons that serve to limit proapoptotic effects of mitochondrial damage.
Our results on anionic lipid-dependent conversion of cyt c into a peroxidase (12) along with the data on anionic phospholipid binding capacity of Syn (5) prompted us to hypothesize that a triple complex of Syn-cyt c-anionic phospholipid can act as a peroxidase. Previous work has demonstrated that oxidative stress-mediated damage of mitochondria can trigger proapoptotic events in synaptic terminals likely associated with synaptic remodeling and plasticity (35). Here we present evidence that the peroxidase activity of this complex utilizes Syn as a peroxidase substrate resulting in oxidative catalytic aggregation of cyt c and Syn. In this way, Syn acts as a scavenger of the key proapoptotic factor, cyt c, resulting in its covalent association with Syn.
There could be at least two possible ways through which phospholipids induce Syn-cyt c interactions. Tightly bound to the mitochondrial membranes, CL undergoes cyt c-catalyzed oxidation during apoptosis (12); oxidized CL, particularly when two or more of its fatty acid residues are oxidatively modified, has a significantly lower hydrophobicity, potentially resulting in the departure of cyt c-CL complexes from mitochondrial membranes. In this scenario, Syn scavenging of cyt c-CL complexes requires the presence of CL to mediate Syn-cyt c interaction. Alternatively, cytosolic Syn can be associated with one of the anionic phospholipids (PI, PS, PA, and free fatty acids) and is primed by this interaction to promptly bind cyt c. Clearly, both of these mechanisms may be realized in some proportions simultaneously. Previous reports showed that protofibrils and fibrils of Syn interact with lipid membranes more effectively than the soluble monomeric protein (23–25). In agreement with this, our data demonstrate that aged Syn has a significantly higher affinity for CL than intact Syn indicating that Syn oligomers with bound CL may act as substrates for peroxidase activity of cyt c. The presence of β-sheets in fibrillated Syn may facilitate hydrophobic interactions with cyt c. It has been shown that, after interaction with CL, cyt c forms fibrils containing β-sheets (51).
Oxidative stress-dependent production of H2O2 acts as a source of oxidizing equivalents for the activation of the peroxidase complex resulting in cross-linking of Syn and cyt c and the formation of their high molecular weight aggregates. The peroxidase activity of triple complexes formed in the presence of cyt c, TOCL, and Syn mutants, A30P and A53T/A30P, was higher than in the presence of wild-type Syn. The significance of this for the PD process is that peroxidase activity of cyt c-CL-Syn complexes may act as a persistent source of oxidative stress. The low level of this activity compared with cyt c/CL alone defines a slow oxidative stress “regimen” and is compatible with the known pro-disease role of synuclein.
In separate experiments, we utilized three other proteins, lactoglobulin (not present in LBs), 14-3-3 (known to be in included in LBs), and β-amyloid-peptide (Aβ-(1–42)), and we tested their ability to undergo hetero-oligomerization catalyzed by the cyt c-CL complex. We found that, similarly to Syn, the peroxidase activity of the complex effectively cross-linked 14-3-3 (data not shown) and Aβ-peptide (see supplemental Fig. 1). In contrast, lactoglobulin incubated with cyt c/CL in the presence of H2O2 was not polymerized (data not shown). Hetero-oligomerization of cyt c with anionic lipid-binding proteins and formation of triple peroxidase complexes may not be uniquely selective toward Syn. Other proteins, particularly those with significant binding affinity toward CL, may get involved in cyt c-driven covalent cross-linking. The likelihood of the cross-linking should be to a large extent dependent on the concentration and affinity of a given protein and its molecular proximity to CL and cyt c. Although Syn is the major component of LBs, Aβ is the main structural component of senile plaques of Alzheimer disease (52). However, it is also possible that some specificity can be observed (e.g. because of the lack of reactive and accessible Tyr residues in a protein). Notably, the presence of Tyr residues is essential for aggregation of Syn and LB formation as well as for its involvement in cross-linking through a catalytic peroxidase process (53), in line with our findings on protein-derived (Tyr˙) radicals in oligomers and high molecular weight aggregates.
As one of the most abundant proteins in presynaptic terminals (54), Syn, in the presence of anionic phospholipids, can scavenge cyt c and hence prevent high affinity binding of the latter with Apaf-1 (55). This suggests a mechanism by which cyt c can be eliminated from its potential involvement in the execution of apoptotic caspase activation. Syn has been reported to play a beneficial role in preventing neuronal death (56). For example, forced expression of Syn diminished apoptotic response in dopaminergic cells (32). We demonstrated that siRNA knockdown of cyt c in HeLa cells with high endogenous levels of Syn essentially eliminated protein oligomerization. Reportedly, anti-apoptotic effects of Syn can also be realized through its interactions with other proapoptotic proteins, protein kinase Cδ and BAD, as has been demonstrated by co-immunoprecipitation studies in mesencephalic dopaminergic neuronal cells challenged with 1-methyl-4-phenylpyridinium (57).
There is a significant body of literature supporting the notion that expression of normal Syn is essential for survival of neuronal cells. A recent communication (58) states, based on antisense oligonucleotide Syn silencing, that the protein has an essential pro-survival role toward primary neurons. Previous work has demonstrated that wild-type but not mutated Syn delayed death of neuronal cells after serum withdrawal (59), and protected cells by preventing activation of cell death-inducing proteins (caspases, p53, and Bad) (57, 60). An anticonvulsant drug, valproic acid, protected cerebellar granule cells from glutamatergic excitotoxicity via induction of Syn (62). However, this hypothesis may be in disagreement with several other important reports demonstrating that elevated levels of Syn lead to the increased likelihood of PD (63–65), whereas experimental deletion of Syn is protective against standard models such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrridine (66, 67).
We believe that a beneficial role for protein expressed at normal levels does not translate into the assumption that more must be better. Indeed, there are many precedents for over-activation of adaptive responses proving to be harmful. Many biological phenomena show bell-shaped curves, such that either too little or too much is harmful. Our proposal that a normal role for Syn includes sequestration of cyt c in LB-like aggregates does not at all contradict observations that mutation or overexpression due to gene multiplication leads to human disease. Indeed, the data in our paper offer an explanation as one of the possible mechanisms for this type of trade off; although apoptosis is prevented, increased peroxidase activity and/or excessive aggregation may interfere with cellular health if not effectively cleared. In agreement with this interpretation, wild-type Syn was shown to protect neurons from apoptosis by inhibiting caspase-3 (68). Aggregation of wild-type and PD-related mutated Syn was reported to be associated with enhanced cell death (69). This process could be caused by the loss of an anti-apoptotic function. The latter possibility is supported by the evidence that Syn lowers p53-dependent apoptotic response of neurons (68) and activates MAPK (mitogen-activated protein kinase) survival pathways (70). Although a number of studies have focused on toxic gain-of-function effects of Syn in PD, such as its aggregation, other dominantly inherited diseases such as spinocerebellar ataxia 1 are caused by concomitant gain-of-function and loss-of-function mechanisms (71). Most misfolding diseases involve both toxic gain of function and loss of normal protective functions (72). Furthermore, the functions of Syn might be dependent on the cellular milieu and neuronal cell type. For example, in vitro, Syn is proapoptotic in dopaminergic neurons, with toxicity requiring dopamine and the production of reactive oxygen species, but it is neuroprotective in nondopaminergic cortical neurons (73); however, in vivo, tyrosine hydroxylase promoter-driven expression of wild-type or mutant human Syn is not toxic to mouse dopaminergic neurons (61).
In conclusion, our results demonstrate that the covalent conjugation of Syn with cyt c in LB-like aggregates prevents signaling effects of the cyt c as a death signal in the cytosol but contributes to the formation of LB and LN. Thus Syn acts as a sacrificial scavenger of cyt c and prevents acute cell death. However, this protection from acute neuronal cell death does not come without a price. Because the peroxidase activity is retained in aggregates of cyt c with Syn and anionic lipids, they could act as an additional source of oxidative stress in affected neurons, contributing to chronic neurodegeneration. This explains, at least in part, the conflicting protective and damaging roles of Syn and LB reported to date in PD. Manipulation of components of the Syn-cyt c-anionic phospholipid complexes and/or their peroxidase activity might lead to development of mechanism-based therapies for neurodegenerative diseases associated with LB/LN formation.
Supplementary Material
Acknowledgment
Post-mortem brain tissues were obtained from the University of Pittsburgh brain bank, supported in part by National Institutes of Health Grant AG05133.
This work was supported, in whole or in part, by National Institutes of Health Grants U19 AIO68021, NS061817, HD057587, AG026389, and ES012068 (NIEHS). This work was also supported by Pennsylvania Department of Health Grant SAP 4100027294 and The Picower Foundation.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Fig. S1.
- LB
- Lewy body
- Syn
- synuclein
- cyt c
- cytochrome c
- LN
- Lewy neuritis
- PD
- Parkinson disease
- DA
- dopamine
- CL
- cardiolipin
- TOCL
- tetraoleoyl-CL
- TLCL
- tetralinoleoyl-CL
- PI
- phosphatidylinositol
- PA
- phosphatidic acid
- PC
- phosphatidylcholine
- PS
- phosphatidylserine
- NAO
- 10-N-nonyl acridine orange
- DMPO
- 5,5-dimethyl-1-pyrroline-N-oxide
- MEC
- mouse embryonic cells
- ActD
- actinomycin D
- t-BuOOH
- tert-butyl hydroperoxide
- DOPC
- dioleoylphosphatidylcholine
- DTPA
- diethylenetriaminepentaacetic acid
- DTT
- dithiothreitol
- MALDI-TOF
- matrix-assisted laser desorption/ionization time-of-flight
- LC-ESI-MS
- liquid chromatography-electrospray ionization-tandem mass spectrometry
- HPLC
- high performance liquid chromatography
- DMSO
- dimethyl sulfoxide
- NBD-CL
- 1,1′,2-trioleoyl-2′-[12-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-cardiolipin
- FRET
- fluorescence resonance energy transfer
- siRNA
- small interfering RNA
- FBS
- fetal bovine serum.
REFERENCES
- 1.Betarbet R., Sherer T. B., Di Monte D. A., Greenamyre J. T. ( 2002) Brain Pathol. 12, 499– 510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Irizarry M. C., Growdon W., Gomez-Isla T., Newell K., George J. M., Clayton D. F., Hyman B. T. ( 1998) J. Neuropathol. Exp. Neurol. 57, 334– 337 [DOI] [PubMed] [Google Scholar]
- 3.Baba M., Nakajo S., Tu P. H., Tomita T., Nakaya K., Lee V. M., Trojanowski J. Q., Iwatsubo T. ( 1998) Am. J. Pathol. 152, 879– 884 [PMC free article] [PubMed] [Google Scholar]
- 4.Dickson D. W. ( 2001) Curr. Opin. Neurol. 14, 423– 432 [DOI] [PubMed] [Google Scholar]
- 5.Shults C. W. ( 2006) Proc. Natl. Acad. Sci. U. S. A. 103, 1661– 1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellis C. E., Murphy E. J., Mitchell D. C., Golovko M. Y., Scaglia F., Barcelo-Coblijn G. C., Nussbaum R. L. ( 2005) Mol. Cell. Biol. 25, 10190– 10201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giasson B. I., Duda J. E., Murray I. V., Chen Q., Souza J. M., Hurtig H. I., Ischiropoulos H., Trojanowski J. Q., Lee V. M. ( 2000) Science 290, 985– 989 [DOI] [PubMed] [Google Scholar]
- 8.Hashimoto M., Takeda A., Hsu L. J., Takenouchi T., Masliah E. ( 1999) J. Biol. Chem. 274, 28849– 28852 [DOI] [PubMed] [Google Scholar]
- 9.Burkitt M., Jones C., Lawrence A., Wardman P. ( 2004) Biochem. Soc. Symp. 71, 97– 106 [DOI] [PubMed] [Google Scholar]
- 10.Qian S. Y., Chen Y. R., Deterding L. J., Fann Y. C., Chignell C. F., Tomer K. B., Mason R. P. ( 2002) Biochem. J. 363, 281– 288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang J., Kini V., Belikova N., Serinkan B. F., Borisenko G. G., Tyurina Y. Y., Tyurin V. A., Kagan V. E. ( 2004) Lipids 39, 1133– 1142 [DOI] [PubMed] [Google Scholar]
- 12.Kagan V. E., Tyurin V. A., Jiang J., Tyurina Y. Y., Ritov V. B., Amoscato A. A., Osipov A. N., Belikova N. A., Kapralov A. A., Kini V., Vlasova I. I., Zhao Q., Zou M., Di P., Svistunenko D. A., Kurnikov I. V., Borisenko G. G. ( 2005) Nat. Chem. Biol. 1, 223– 232 [DOI] [PubMed] [Google Scholar]
- 13.Bayır H., Fadeel B., Palladino M., Witasp E., Kurnikov I., Tyurina Y. Y., Tyurin V. A., Amoscato A., Jiang J., Kochanek P. M., DeKosky S., Greenberger J., Shvedova A. A., Kagan V. E. ( 2006) Biochim. Biophys. Acta 1757, 648– 659 [DOI] [PubMed] [Google Scholar]
- 14.Li K., Li Y., Shelton J. M., Richardson J. A., Spencer E., Chen Z. J., Wang X., Williams R. S. ( 2000) Cell 101, 389– 399 [DOI] [PubMed] [Google Scholar]
- 15.Castner T. G. J. ( 1959) Phys. Rev. 115, 1506– 1515 [Google Scholar]
- 16.Petit J. M., Maftah A., Ratinaud M. H., Julien R. ( 1992) Eur. J. Biochem. 209, 267– 273 [DOI] [PubMed] [Google Scholar]
- 17.Folch J., Lees M., Sloane Stanley G. H. ( 1957) J. Biol. Chem. 226, 497– 509 [PubMed] [Google Scholar]
- 18.Tyurin V. A., Tyurina Y. Y., Kochanek P. M., Hamilton R., DeKosky S. T., Greenberger J. S., Bayır H., Kagan V. E. ( 2008) Methods Enzymol. 442, 375– 393 [DOI] [PubMed] [Google Scholar]
- 19.Liu X., Kim C. N., Yang J., Jemmerson R., Wang X. ( 1996) Cell 86, 147– 157 [DOI] [PubMed] [Google Scholar]
- 20.Betarbet R., Sherer T. B., MacKenzie G., Garcia-Osuna M., Panov A. V., Greenamyre J. T. ( 2000) Nat. Neurosci. 3, 1301– 1306 [DOI] [PubMed] [Google Scholar]
- 21.Kenworthy A. K. ( 2001) Methods (San Diego) 24, 289– 296 [DOI] [PubMed] [Google Scholar]
- 22.Eliezer D., Kutluay E., Bussell R., Jr., Browne G. ( 2001) J. Mol. Biol. 307, 1061– 1073 [DOI] [PubMed] [Google Scholar]
- 23.Smith D. P., Tew D. J., Hill A. F., Bottomley S. P., Masters C. L., Barnham K. J., Cappai R. ( 2008) Biochemistry 47, 1425– 1434 [DOI] [PubMed] [Google Scholar]
- 24.Giannakis E., Pacifico J., Smith D. P., Hung L. W., Masters C. L., Cappai R., Wade J. D., Barnham K. J. ( 2008) Biochim. Biophys. Acta 1778, 1112– 1119 [DOI] [PubMed] [Google Scholar]
- 25.Zhu M., Li J., Fink A. L. ( 2003) J. Biol. Chem. 278, 40186– 40197 [DOI] [PubMed] [Google Scholar]
- 26.Borisenko G. G., Kapralov A. A., Tyurin V. A., Maeda A., Stoyanovsky D. A., Kagan V. E. ( 2008) Biochemistry 47, 13699– 13710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramirez D. C., Mejiba S. E., Mason R. P. ( 2005) J. Biol. Chem. 280, 27402– 27411 [DOI] [PubMed] [Google Scholar]
- 28.Vlasova I. I., Tyurin V. A., Kapralov A. A., Kurnikov I. V., Osipov A. N., Potapovich M. V., Stoyanovsky D. A., Kagan V. E. ( 2006) J. Biol. Chem. 281, 14554– 14562 [DOI] [PubMed] [Google Scholar]
- 29.Chen Y. R., Chen C. L., Chen W., Zweier J. L., Augusto O., Radi R., Mason R. P. ( 2004) J. Biol. Chem. 279, 18054– 18062 [DOI] [PubMed] [Google Scholar]
- 30.Lassmann G., Odenwaller R., Curtis J. F., DeGray J. A., Mason R. P., Marnett L. J., Eling T. E. ( 1991) J. Biol. Chem. 266, 20045– 20055 [PubMed] [Google Scholar]
- 31.Tyurina Y. Y., Kini V., Tyurin V. A., Vlasova I. I., Jiang J., Kapralov A. A., Belikova N. A., Yalowich J. C., Kurnikov I. V., Kagan V. E. ( 2006) Mol. Pharmacol. 70, 706– 717 [DOI] [PubMed] [Google Scholar]
- 32.Machida Y., Chiba T., Takayanagi A., Tanaka Y., Asanuma M., Ogawa N., Koyama A., Iwatsubo T., Ito S., Jansen P. H., Shimizu N., Tanaka K., Mizuno Y., Hattori N. ( 2005) Biochem. Biophys. Res. Commun. 332, 233– 240 [DOI] [PubMed] [Google Scholar]
- 33.Lee S. J. ( 2003) Antioxid. Redox. Signal. 5, 337– 348 [DOI] [PubMed] [Google Scholar]
- 34.McKeith I. G., Dickson D. W., Lowe J., Emre M., O'Brien J. T., Feldman H., Cummings J., Duda J. E., Lippa C., Perry E. K., Aarsland D., Arai H., Ballard C. G., Boeve B., Burn D. J., Costa D., Del Ser T., Dubois B., Galasko D., Gauthier S., Goetz C. G., Gomez-Tortosa E., Halliday G., Hansen L. A., Hardy J., Iwatsubo T., Kalaria R. N., Kaufer D., Kenny R. A., Korczyn A., Kosaka K., Lee V. M., Lees A., Litvan I., Londos E., Lopez O. L., Minoshima S., Mizuno Y., Molina J. A., Mukaetova-Ladinska E. B., Pasquier F., Perry R. H., Schulz J. B., Trojanowski J. Q., Yamada M. ( 2005) Neurology 65, 1863– 1872 [DOI] [PubMed] [Google Scholar]
- 35.Mattson M. P. ( 2000) Brain Pathol. 10, 300– 312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Norris E. H., Giasson B. I. ( 2005) Antioxid. Redox. Signal. 7, 672– 684 [DOI] [PubMed] [Google Scholar]
- 37.Souza J. M., Giasson B. I., Chen Q., Lee V. M., Ischiropoulos H. ( 2000) J. Biol. Chem. 275, 18344– 18349 [DOI] [PubMed] [Google Scholar]
- 38.Conway K. A., Rochet J. C., Bieganski R. M., Lansbury P. T., Jr.( 2001) Science 294, 1346– 1349 [DOI] [PubMed] [Google Scholar]
- 39.Radi R., Turrens J. F., Freeman B. A. ( 1991) Arch. Biochem. Biophys. 288, 118– 125 [DOI] [PubMed] [Google Scholar]
- 40.Abou-Sleiman P. M., Muqit M. M., Wood N. W. ( 2006) Nat. Rev. Neurosci. 7, 207– 219 [DOI] [PubMed] [Google Scholar]
- 41.Greenamyre J. T., Hastings T. G. ( 2004) Science 304, 1120– 1122 [DOI] [PubMed] [Google Scholar]
- 42.Swerdlow R. H., Parks J. K., Davis J. N., II, Cassarino D. S., Trimmer P. A., Currie L. J., Dougherty J., Bridges W. S., Bennett J. P., Jr., Wooten G. F., Parker W. D. ( 1998) Ann. Neurol. 44, 873– 881 [DOI] [PubMed] [Google Scholar]
- 43.Sherer T. B., Betarbet R., Stout A. K., Lund S., Baptista M., Panov A. V., Cookson M. R., Greenamyre J. T. ( 2002) J. Neurosci. 22, 7006– 7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dauer W., Przedborski S. ( 2003) Neuron 39, 889– 909 [DOI] [PubMed] [Google Scholar]
- 45.Callio J., Oury T. D., Chu C. T. ( 2005) J. Biol. Chem. 280, 18536– 18542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu C. T., Zhu J. H., Cao G., Signore A., Wang S., Chen J. ( 2005) J. Neurochem. 94, 1685– 1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu J. H., Guo F., Shelburne J., Watkins S., Chu C. T. ( 2003) Brain Pathol. 13, 473– 481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Horbinski C., Chu C. T. ( 2005) Free Radic. Biol. Med. 38, 2– 11 [DOI] [PubMed] [Google Scholar]
- 49.Chu C. T. ( 2006) J. Neuropathol. Exp. Neurol. 65, 423– 432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anglade P., Vyas S., Hirsch E. C., Agid Y. ( 1997) Histol. Histopathol. 12, 603– 610 [PubMed] [Google Scholar]
- 51.Alakoskela J. M., Jutila A., Simonsen A. C., Pirneskoski J., Pyhajoki S., Turunen R., Marttila S., Mouritsen O. G., Goormaghtigh E., Kinnunen P. K. ( 2006) Biochemistry 45, 13447– 13453 [DOI] [PubMed] [Google Scholar]
- 52.Kang J., Lemaire H. G., Unterbeck A., Salbaum J. M., Masters C. L., Grzeschik K. H., Multhaup G., Beyreuther K., Muller-Hill B. ( 1987) Nature 325, 733– 736 [DOI] [PubMed] [Google Scholar]
- 53.Olteanu A., Pielak G. J. ( 2004) Protein Sci. 13, 2852– 2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kahle P. J., Neumann M., Ozmen L., Muller V., Jacobsen H., Schindzielorz A., Okochi M., Leimer U., van Der Putten H., Probst A., Kremmer E., Kretzschmar H. A., Haass C. ( 2000) J. Neurosci. 20, 6365– 6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Purring-Koch C., McLendon G. ( 2000) Proc. Natl. Acad. Sci. U. S. A. 97, 11928– 11931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seo J. H., Rah J. C., Choi S. H., Shin J. K., Min K., Kim H. S., Park C. H., Kim S., Kim E. M., Lee S. H., Lee S., Suh S. W., Suh Y. H. ( 2002) FASEB J. 16, 1826– 1828 [DOI] [PubMed] [Google Scholar]
- 57.Kaul S., Anantharam V., Kanthasamy A., Kanthasamy A. G. ( 2005) Brain. Res. Mol. Brain Res. 139, 137– 152 [DOI] [PubMed] [Google Scholar]
- 58.Monti B., Polazzi E., Batti L., Crochemore C., Virgili M., Contestabile A. ( 2007) J. Neurochem. 103, 518– 530 [DOI] [PubMed] [Google Scholar]
- 59.Lee M., Hyun D., Halliwell B., Jenner P. ( 2001) J. Neurochem. 76, 998– 1009 [DOI] [PubMed] [Google Scholar]
- 60.da Costa C. A., Ancolio K., Checler F. ( 2000) J. Biol. Chem. 275, 24065– 24069 [DOI] [PubMed] [Google Scholar]
- 61.Matsuoka Y., Vila M., Lincoln S., McCormack A., Picciano M., LaFrancois J., Yu X., Dickson D., Langston W. J., McGowan E., Farrer M., Hardy J., Duff K., Przedborski S., Di Monte D. A. ( 2001) Neurobiol. Dis. 8, 535– 539 [DOI] [PubMed] [Google Scholar]
- 62.Leng Y., Chuang D. M. ( 2006) J. Neurosci. 26, 7502– 7512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hsu L. J., Sagara Y., Arroyo A., Rockenstein E., Sisk A., Mallory M., Wong J., Takenouchi T., Hashimoto M., Masliah E. ( 2000) Am. J. Pathol. 157, 401– 410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith W. W., Jiang H., Pei Z., Tanaka Y., Morita H., Sawa A., Dawson V. L., Dawson T. M., Ross C. A. ( 2005) Hum. Mol. Genet. 14, 3801– 3811 [DOI] [PubMed] [Google Scholar]
- 65.Masliah E., Rockenstein E., Veinbergs I., Mallory M., Hashimoto M., Takeda A., Sagara Y., Sisk A., Mucke L. ( 2000) Science 287, 1265– 1269 [DOI] [PubMed] [Google Scholar]
- 66.Fornai F., Schluter O. M., Lenzi P., Gesi M., Ruffoli R., Ferrucci M., Lazzeri G., Busceti C. L., Pontarelli F., Battaglia G., Pellegrini A., Nicoletti F., Ruggieri S., Paparelli A., Sudhof T. C. ( 2005) Proc. Natl. Acad. Sci. U. S. A. 102, 3413– 3418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Klivenyi P., Siwek D., Gardian G., Yang L., Starkov A., Cleren C., Ferrante R. J., Kowall N. W., Abeliovich A., Beal M. F. ( 2006) Neurobiol. Dis. 21, 541– 548 [DOI] [PubMed] [Google Scholar]
- 68.Alves Da Costa C., Paitel E., Vincent B., Checler F. ( 2002) J. Biol. Chem. 277, 50980– 50984 [DOI] [PubMed] [Google Scholar]
- 69.Giasson B. I., Duda J. E., Quinn S. M., Zhang B., Trojanowski J. Q., Lee V. M. ( 2002) Neuron 34, 521– 533 [DOI] [PubMed] [Google Scholar]
- 70.Iwata A., Maruyama M., Kanazawa I., Nukina N. ( 2001) J. Biol. Chem. 276, 45320– 45329 [DOI] [PubMed] [Google Scholar]
- 71.Lim J., Crespo-Barreto J., Jafar-Nejad P., Bowman A. B., Richman R., Hill D. E., Orr H. T., Zoghbi H. Y. ( 2008) Nature 452, 713– 718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Winklhofer K. F., Tatzelt J., Haass C. ( 2008) EMBO J. 27, 336– 349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu J., Kao S. Y., Lee F. J., Song W., Jin L. W., Yankner B. A. ( 2002) Nat. Med. 8, 600– 606 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.