

Abstract
N2-Oxopropenyldeoxyguanosine (2) forms in duplex DNA by modification of dG residues with base propenal or malondialdehyde. The pKa of 2 was estimated to be 6.9 from the pH dependence of its ring-closing to the pyrimidopurinone derivative 1. The acidity of 2 may be an important determinant of its miscoding properties and its reactivity to nucleophiles in DNA or protein. To test this hypothesis, analogous N-oxopropenyl derivatives of dA (4), dC (5), and N1-methyl-dG (6) were synthesized and their pKa’s determined by optical titration. The N-oxopropenyl derivatives of dA and dC both exhibited pKa’s of 10.5 whereas the N-oxopropenyl derivative of N1-methyldG exhibited a pKa of 8.2. Crosslinking of 2, 4, 5, and 6 to Nα-acetyl-lysine was explored at neutral pH. Adduct 2 did not react with Nα-acetyl-lysine whereas 4-6 readily formed crosslinks. The structures of the crosslinks were elucidated and their stabilities investigated. The results define the acidity of oxopropenyl deoxynucleosides and highlight its importance to their reactivity toward nucleophiles. This study also identifies the structures of a potential novel class of DNA-protein cross-links.
Introduction
3-(2-Deoxy-β-D-erythro-pentofuranosyl)pyrimido[1,2-α]purin-10(3H)-one (M1dG, 1) is a mutagenic product of oxidative DNA damage generated endogenously in humans.1 It is derived from the bifunctional aldehydes, base propenal and malondialdehyde, and induces transversions to dT, transitions to dA, and frameshift mutations when replicated in bacterial or mammalian cells.2-6 Compound 1 undergoes reversible hydrolytic ring-opening in duplex DNA when it is opposite dC residues but not when opposite dT residues (eq 1).7 The product of ring-opening, 3-(2-deoxy-β-D-erythro-pentofuranosyl)N2-oxopropenylguanine (2), hydrogen-bonds to dC residues, is bypassed with high fidelity when replicated by DNA polymerase in vitro, and appears to be less mutagenic in vivo.5,8 Compound 2 is stable in duplex DNA but rapidly cyclizes to 1 upon denaturation to single-stranded DNA.7,9
 |
eq 1 |
Adducts 1 and 2 represent potential reactive electrophiles in the genome and their ability to condense with nucleophiles has been reported.10,11 Of particular interest is the potential for 1 and 2 to form interstrand or intrastrand DNA-DNA cross-links or DNA-protein cross-links. Such cross-links are of increasing interest because of their potential to block DNA replication or to induce mutations. The ability of the structurally-related N2-(3-oxopropyl)deoxyguanosine to form DNA-DNA and DNA-protein cross-links is well-documented.12-15 Previous model studies indicate that 1 reacts with tris-hydroxymethylaminomethane to form an imine conjugate that a model cross-link whereas 2 does not.16 A clue to the unanticipated lower reactivity represents of 2 is provided by recent studies of the ring-opening and ring-closing of 1 and 2 at different pH’s.17,18 Hydrolytic ring-opening of 1 occurs by reversible addition of hydroxide to form the anion of 2.18 At pH’s below neutrality, the anion of 2 protonates and rapidly cyclizes to a hydroxypropeno intermediate, 3, that slowly dehydrates to 1 (Scheme 1).17 The pH dependence of cyclization suggests that the pKa of 2 is 6.9, which correlates to the results of DFT calculations indicating an N2oxopropenyl derivative of dG has a pKa ∼ 4 pH units lower than dG.17 The low pKa of 2 indicates that at physiological pH, 2 exists mainly as its conjugate base, which is anticipated to be less reactive to nucleophiles than its protonated form or, apparently, than 1.
Scheme 1.
To test this hypothesis, we synthesized a series of oxopropenyl adducts to the exocyclic amines of deoxynucleosides and determined their pKa’s and ability to form cross-links with the ε-amino group of Nα-acetyllysine. The results define the acid-base chemistry of oxopropenyl-deoxynucleoside adducts and their cross-linking potential and they are consistent with the hypothesis that the poor reactivity of 2 is due to its acidity. The results also have important implications for the characterization of a novel class of DNA damage products.
Materials and Methods
Synthesis of 1, 2, 4-6
The synthesis of the MDA-modified nucleosides were accomplished as previously described.19
HPLC
HPLC analyses and purifications were performed on a Beckman Coulter solvent delivery system with a diode-array UV detector operated by Karat 32 software (v. 6.0). Separations were performed on a YMC ODS-AQ S-5 column (120□ 4.6 × 250 mm) with a 1.5 mL/min flow rate. The solvent gradient was as follows: (A: water, B: acetonitrile): initial conditions were 1 % B; a linear gradient to 10 % B over 15 min; a linear gradient to 20 % B over 5 minutes; isocratic at 20% B for 5 minutes; 3 minute linear gradient to 80 % B; isocratic at 80 % B for two minutes; B, 28-30 min: a linear gradient to the initial conditions over three minutes.
Mass Spectrometry
LC-MS data were collected on a ThermoFinnigan LCQ instrument using a YMC HPLC ODS-AQ narrow-bore HPLC column (S-5 120□ 1.0 × 250 mm) with a 0.1 mL/min flow gradient (A: 1% acetonitrile in 10 mM ammonium acetate, B: 99% acetonitrile in 10 mM ammonium acetate without pH adjustment, gradient as described above). Samples were prepared in 0.1% acetic acid solution in deionized water accordingly: 0.1 mg material was dissolved in 1 mL solution and 50 μL of this solution was placed into a glass insert (150 μL) with silica-coating of which, 10 μL of the solution was injected onto the column. The ESI source was set up in the positive ion mode to the following initial conditions: capillary temperature 325 °C, source spray voltage, 4.0 kV, source current 6.5 μA, sheath, 28 units and auxiliary gas, 23 For each analyte, an automatic tuning was applied to optimize peak size of the [M+1]+ or units. the [M+Na]+ peak. Automatic tuning resulted in an average increase of 200-400 % in signal size. The data were qualitatively analyzed with the X-calibur software package.
N1-methyl-N2-(3-oxopropenyl)-2′-deoxyguanosine (6)
A solution of 1 (30 mg, 0.1 mmol) and K2CO3 (28 mg, 0.2 mmol) in dimethylacetamide (0.5 mL) and water (0.5 mL) was stirred at room temperature for 3 h, then iodomethane (142 mg, 1 mmol) was added dropwise via syringe. HPLC analysis of the reaction mixture showed the presence of a new product. The product 6 (14.1 mg, 43 % yield) was isolated by semi-preparative reversed-phase HPLC using a gradient as described above at a flow-rate of 5 mL/min on a YMC ODS column (120 □ 9.1 × 250 mm). 1H NMR (DMSO-d6): δ: 10.20 (1H, br, N2H); 9.35 (1H, d, CHO); 8.35 (1H, d, α-oxopropenyl); 8.10 (1H, s, C8); 6.25 (1H, m, β-oxopropenyl); 5.90 (1H, br, H1′′); 5.30 (1H, s, 3′-OH); 4.90 (1H, s, 5′-OH); 4.40 (1H, s, H4′); 3.90 (1H, d, H5′′); 3.60 (1H, m, H5′); 3.50 (3H, s, Methyl); 2.65 (1H, m, H2′); 2.25 (1H, m, H2′′). 13C NMR (DMSO-d6): δ 190 (CHO); 157 (C6); 150 (C2); 148 (C4); 142 (β-oxopropenyl); 132 (C8); 118 (C5); 88 (C4′); 87 (C1′); 71 (C3′); 62 (C5′); 40 (C2′); LC-ESI-MS calcd for C14H17N5O5Na [M+Na], 358.11, found, 358.08
Hydrolysis of 6 to 1-methyl-dG
A solution of 6 (0.5 mg) in sodium phosphate buffer (500 μL, 50 mM, pH 7.4) was stirred at room temperature for 10 h. A new product was observed by reversed-phase HPLC analysis, which had a retention time, UV spectrum and mass spectrum that was identical to an authentic sample of N1-methyl-dG (Berry and Associates Medicinal Chemistry Co.). The half-life for the hydrolysis of 6 was estimated to be ∼ 12 h.
pKa measurements
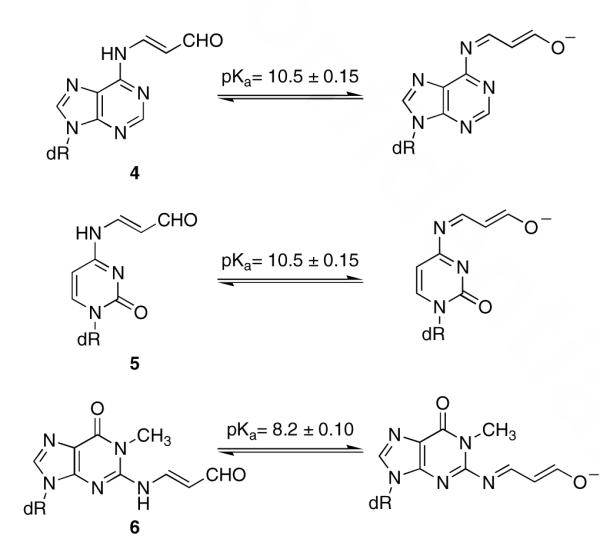
Stock buffer solutions were prepared from the following stocks: 1 M stock solutions of H3PO4 and NaH2PO4 (buffering range: pH 1.12 - 3.12), 1 M stock solutions of citric acid and NaOH (buffering range: 2.10 − 7.40), 1M stock solutions of NaH2PO4 and Na2HPO4 (buffering range: pH 6.21 − 8.21), 1 M stock solutions of bicine buffer with NaOH (buffering range: pH 7.35 − 9.35); 1 M stock solutions of NaHCO3 and Na2CO3 (buffering range: pH 9.30 − 11.30); 1 M stock solutions of Na2HPO4 and Na3PO4 (buffering range: pH 11.70 − 13.70). Buffers were prepared at selected pH’s using a Ag/AgCl electrode. Buffers were diluted to 200 mM. An aqueous stock-solution of 4 was dissolved in 50 mM sodium phosphate or sodium carbonate buffers at the selected pH’s to a final concentration of 40 μM. A 50 mM buffer solution was used to zero the instrument at each pH. The UV spectrum was then recorded from 220-420 nm at each pH. The intensities at the major absorbances (324 and 372 nm for 4, 322 and 372 nm for 5, 314 and 354 nm for 6) were plotted vs pH, then fit to a sigmoidal function using Kaleidagraph (v. 4.03). The midpoint of the curve was assigned as the pKa of the dissociable proton. Measurements were performed in duplicate and the error was calculated to a 67% confidence level. The pKa’s were 10.5 ± 0.15 for 4 and 5, and 8.3 ± 0.2 for 6.
Stock solutions of cross-linked nucleosides 7 and 8 (1 mM) were diluted 100-fold in the stock buffer solutions (20 mM) at the desired pH’s and the UV spectra were recorded from 220-420 nm. The intensities of the UV absorbances at at 344 and 380 nm for 7 and 338 and 365 for 8 were plotted versus pH. The data were fit as described above to obtain a pKa value of 8.8 ± 0.2 and 6.3 ± 0.2 for 7 and 8, respectively.
Reaction of 4 and 6 with Nα-acetyllysine
Stock solutions of Nα-acetyllysine (200 mM in 200 mM sodium phosphate buffer), 4 (4 mM in deionized water), and 6 (4 mM in deionized water) were prepared. To a stock solution of Nα-acetyllysine (200 μL) was added the stock solution of modified nucleoside 4 or 6 (200 μL). The final concentrations of the reactants were 100 mM Nα-acetyllysine and 2 mM of nucleoside 4 or 6 in 100 mM sodium phosphate buffer (200 μL). The cross-linking reaction was monitored by HPLC for 24 h. New products were isolated by reversed-phase HPLC using conditions described above. 7: 1H-NMR (DMSO-d6) δ: 8.15 (1H, s, H2); 8.10 (1H, s, H8); 8.02 (2H, m,); 7.8 (1H, s, NH-dA); 6.40 (1H, m, H1′′); 6.10 (1H, dd,); 4.40 (1H, m, H3′); 4.05 (1H, m); 3.85 (1H, m, H4′); 3.60 (1H, m, H5′′); 3.45 (1H, m, H5′); 3.34 (2H, m); 2.7 (1H, m, H2′); 2.3 (1H, m, H2′); 1.8 (3H, s, CH3). 13C NMR (DMSO-d6) δ: 162, 152, 142, 109, 89, 85, 71, 61, 57, 53, 40, 32, 23; LC-ESI-MS: m/z calcd for C21H30N7O6 [M+H], 476.23; found, 476.17. 8: 1H-NMR (DMSO-d6) δ: 8.68 (1H, d, imine), 7.59 (1H, m, C1-oxopropenyl vinyl), 7.20 (1H, dd, C2-oxopropenyl vinyl), 5.57 (1H, m, C1), 4.53 (1H, m, C4), 3.70 (2H, q, C4 Lys), 3.82 (1H, q, C5), 3.76 (1H, q, C5), 3.62 (1H, C3′), 3.3 (3H, s, acetyl CH3), 3.1 (1H, s, Cα-Lys), 2.63 (1H, pp, C2′), 2.69 (2H, pp, C2′′), 1.6-1.2 (6H, m, (CH2)3 Lys); 13C NMR (DMSO-d6) 172 (C1-oxopropenyl vinyl), 167 (C2-oxopropenyl vinyl), 159 (C-imine), 99 (C1), 88 (C5), 71 (C4), 62 (C3), 54 (C4 Lys), 44 (Cα Lys), 39 (C2), 33 (CH2 Lys). LC-ESI-MS: m/z calcd for C22H32N7O7 [M+H], 506.24; found, 506.08
Hydrolytic stability of 7 and 8
The UV spectrum of a solution of 7 or 8 (10 μM) in sodium phosphate buffer (20 mM, pH 7.4) was recorded from 220-450 nm at 15 min intervals. A spectral shift from 344 to 324 nm for 7 and 365 to 314 nm for 8 was observed, corresponding to the hydrolysis of the cross-link to modified nucleosides 4 and 6, respectively. A plot of the absorbance versus time was fit to an exponential function and the rates of hydrolysis were calculated to be 0.14 ± 0.013 h-1 for 7 and 0.029 ± 0.02 h-1 for 8.
Reduction of cross-links 7 and 8
Cross-link 7 (1 mg, 2.0 μmols) was dissolved in sodium phosphate buffer (1.3 mL, 100 mM, pH 7.4) and vortexed for 30 s. A stock solution of NaBH4 (100 mM) was made by adding NaBH4 (50 μL of a 2M solution in diglyme) to 950 μL of deionized water. Equal volumes of the solutions of 7 and NaBH4 were mixed; 7 was completely consumed within 15 min resulting in five products in additions to dA. The two major products were isolated by reversed-phase HPLC using the conditions described above and characterized by 1H and 13C NMR and LC-ESI-MS. The minor products were only characterized by LC-ESI-MS. 9: 1H-NMR (DMSO-d6): δ 8.20 (1H, s, H2), 8.00 (1H, s, H8), 7.60 (1H, s, dA-NH), 6.40 (1H, t, H1′′), 5.40 (2H, m, C1 and C2 oxopropenyl vinyls), 4.45 (1H, m, H3′), 3.95 (1H, m, H4′), 3.90 (1H, m, H5′′), 3.65 (2H, Cα OpdA), 3.53 (1H, m, H5′), 2.90 (2H, Cγ OPdA); 2.8 (1H, m, H2′), 2.3 (1H, m, H2′′), 1.95 (2H), 1.65 (1H, m, Lys-CH2), 1.60 (1H, m, Lys-CH2), 1.50 (2H, m, Lys-CH2), 1.35 (1H, m, Lys-CH2), 1.25 (1H, m, Lys-CH2). 13C-NMR (DMSO-d6): 153 (C2 and C4), 139 (C8), 88 (C4′), 84 (C1′), 72 (C3′), 62 (C5′), 58 (C4-lys), 53 (Cα-lys), 48 (CH2-oxopropenyl), 41 (CH2-oxopropenyl), 40 (C2′), 32 (Lys-CH2), 28 (CH2-oxopropenyl), 23 (Lys-CH2), 22 (Lys-CH2). LC-ESI-MS calcd for C21H34N7O6 [M+H], 480.26; found, 480.10. 10: 1H NMR (DMSO-d6): δ 8.49 (1H, s, H2), 8.31 (1H, s, H8), 7.60 (1H, s, dA-NH), 6.50 (1H, t, H1′′), 5.50 (2H, m, Cα and Cβ OpdA), 4.52 (1H, m, H3′), 3.95 (1H, m, H4′), 3.91 (1H, m, Cα-H lys), 3.86 (1H, m, H5′′), 3.53 (1H, m, H5′), 3.32 (2H, m, C4-H lys), 3.30 (2H, m, cγ-OPdA), 2.8 (1H, m, H2′), 2.4 (1H, m, H2′′), 1.9 (3H, s, CH3), 1.70 (2H, m, lys sidechain), 1.60 (2H, m, lys sidechain), 1.45 (1H, m, lys sidechain), 1.35 (1H, m, lys sidechain). 13C NMR (DMSO-d6): 153 (C2 and C4), 141 (C8), 88 (C4′), 84 (C1′), 72 (C3′), 70 (enamine), 60 (C5′), 58 (C4-lys), 54 (CH2-oxopropenyl), 53 (Cα-lys), 40 (C2′), 32 (lys sidechain), 23 (lys sidechain), 22 (lys sidechain). LC-ESI-MS calcd for C21H32N7O6 [M+H], 478.24; found, 478.11.
An identical procedure was used for the reduction of cross-link 8. A single product was observed and was isolated by reversed-phase HPLC using the conditions described above. 11: LC-ESI-MS calcd for C22H36N7O7 [M+H] and C22H35N7O7Na [M+Na], 510.27 and 532.25; found, 510.05 and 532.14.
Results
Synthesis of MDA-modified Nucleosides
Nucleosides 1, 4 and 5 were prepared as previously described by our laboratories.19 N1-Methyl-N2-(3-oxopropenyl)-dG (6), which serves a model for 2 with a non-dissociable N1-group, was prepared from M1dG. M1dG was treated with base to affect ring-opening to the anion of 2, then quenched with iodomethane resulting in 6. The site of methylation was determined by HMBC (Figure 1) and chemical correlation. A three-bond correlation between the N1-methyl protons (δ = 3.50 ppm) and the C2 (δ = 156 ppm) and C6 (δ = 150 ppm) of the guanine ring was observed. Alkylation at O6 would be expected to give correlation between the methyl group and C6 only. The C2-resonance was unambiguously assigned by the observation of a three-bond correlation to the β-vinyl proton of the N2-oxopropenyl group (δ = 8.35 ppm). Furthermore, no correlation was observed between the methyl carbon resonance and the oxopropenyl vinyl protons, therefore ruling out alkylation at N2. Above pH 10, M1dG undergoes ring-opening and hydrolysis of the oxopropenyl group to dG.18 Base hydrolysis of 6 resulted in N1-methyl-dG, which was correlated to an authentic standard by its HPLC retention time and mass spectrometric fragmentation. The observation that 6 is hydrolyzed to N1-methyl-dG provides further support that M1dG undergoes methylation at N1.
Figure 1.
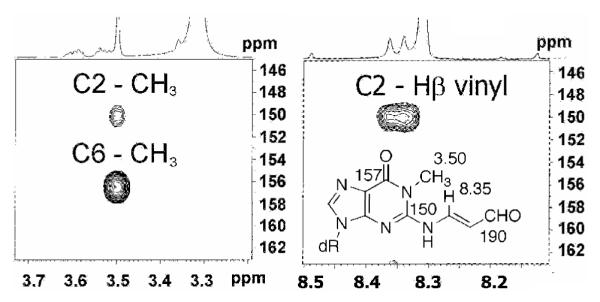
HMBC spectra of 6. 1H-13C correlations between the N1-methyl group and C6 and N2 (right). 1H-13C correlation between the β-proton of the N2-oxopropenyl group and C2.
pKa Measurements
The pKa’s of nucleosides 4-6 were determined by monitoring the shift in the UV absorbance versus pH. Buffers (50 mM) were chosen to maintain proper buffering capacity at the given pH. The λmax’s of nucleosides 4-6 were 324 nm (ε = 60,000 M-1cm-1), 322 nm (ε = 45,000 M-1cm-1), and 314 nm (ε = 30,000 M-1cm-1), respectively, in pH 6.0 buffer. Deprotonation resulted in a hypochromic shift of the λmax to 372 nm for 4 and 5 and 354 nm for 6 (Figure 2). An isosbetic point at 342 nm was clearly observed for the titration of 4 and 5, and at 354 nm for 6. Additional isosbestic points were observed at 234, 244, and 266 nm for 4, and 234, 244 and 266 nm for 5. The pKa’s were determined by plotting the intensity of the UV absorbance versus pH, then fitting the data to a sigmodial function (Figure 2, insets). The pKa’s of 4, 5, and 6 are 10.5 ± 0.15, 10.5 ± 0.15, and 8.2 ± 0.10, respectively (Scheme 2). Nucleosides 1 and 2 did not show similar optical transitions over a pH range of 3.0 – 13.0, suggesting that the exocyclic N2-proton does not dissociate under these conditions. This is presumably the result of 2 already being negatively charged, which is likely to increase the pKa of the N2-proton.17
Figure 2.

UV titration of: A. 4, B. 5, and C. 6 from pH 8.0 to 13.0 in the appropriate buffer (50 mM). The pKa’s were determined by plotting the UV absorbances of the two major peaks vs the pH, then fitting to a sigmoidal function (insets). The pKa’s of 4-6 are pH: 10 ± 0.15, 10.5 ± 0.15, and 8.2 ± 0.10, respectively.
Scheme 2.
Structures of the 2′-deoxynucleoside-malondialdehyde adducts (4-6) used in our investigations.
Reaction of MDA-modified Nucleosides 4 and 6 with Nα-acetyllysine
Compounds 4 and 6 were reacted individually with excess Nα-acetyllysine in pH 7.4, phosphate buffer (Figures 3A and 4A) resulting in cross-links 7 and 8. In addition, oxopropenyl transfer was observed to afford Nε-(3-oxopropenyl)-Nα-acetyllysine. Cross-links 7 and 8 were relatively stable following HPLC purification. Cross-link 7 hydrolyzed back to 4 with a first-order rate of 0.14 ± 0.013 h-1 in pH 7.4, phosphate buffer; the rate of hydrolysis of 8 to 6 was slower, 0.029 ± 0.002 h-1. The alternative hydrolytic pathway resulting in overall transfer of the oxopropenyl group to the ε-amino group of Nα-acetyllysine was very slow, but essentially irreversible under these conditions. The methyl ester of Nε-(3-oxopropenyl)-Nα-acetyllysine is modestly mutagenic in Salmonella typimurium, which may be reflective of its low reactivity with DNA bases.3 As anticipated, compound 2 did not form cross-links with Nα-acetyllysine under these conditions.
Figure 3.

A. Reactivity of cross-link 7. B. UV titration of cross-link 7, pKa = 8.8 ± 0.2. C. UV spectrophotometric analysis of the hydrolysis of 7. D. Determination of the first order rate of hydrolysis of 7, 0.14 ± 0.013 h-1.
Figure 4.

A. Reactivity of cross-link 8. B. UV titration of cross-link 7, pKa = 6.3 ± 0.2. C. UV spectrophotometric analysis of the hydrolysis of 8. D. Determination of the first order rate of hydrolysis of 8, 0.029 ± 0.002 h-1.
The structures of 7 and 8 were initially assigned by LC-ESI-MS. For the reaction of 4, a new product with an m/z of 475.2 Da. was observed, consistent with 7. A daughter ion at m/z 360.2 Da corresponding to the neutral loss of deoxyribose (-116 Da) was also observed. Similarly, the reaction of 6 gave a new product of m/z 506.1 Da with a daughter ion at 390.2 Da, consistent with cross-link 8. Two-dimensional NMR was used to confirm the structures of cross-links 7 and 8. Proton resonances were assigned by COSY spectra based on chemical shifts and scaler couplings. The carbon resonances were then assigned by a one-bond 1H-13C correlation (HMQC) spectra. A three-bond 1H-13C correlation was observed between the imine carbon and the ε-methylene protons of the lysine moiety confirmed the structures of 7 and 8.
The pKa’s for 7 and 8 were determined by UV titration as described above and found to be 8.8 ± 0.2 and 6.3 ± 0.2, respectively (Figures 3B and 4B); the dissociable proton is presumably the protonated imine. The hydrolytic stability of 7 and 8 are reflective of their pKa’s. Hydrolysis back to the oxopropenyl nucleosides and Nα-acetyllysine is predicted to occur through their respective protonated imine. The hydrolysis of the cross-links is associated with a spectral shift from 343 nm to 323 nm for 7, and 362 nm to 316 nm for 8 (Figures 3C and 4C). The rates of hydrolysis were determined by plotting the UV intensities versus time, then fitting the data to an exponential function (Figures 3D and 4D). Cross-link 7, is predicted to be largely protonated at neutral pH while 8 is largely unprotonated in pH 7.4 medium. The rate of hydrolysis of cross-link 7 (k = 0.14 ± 0.013 h-1) is nearly five-fold faster than for 8 (k = 0.029 ± 0.002 h-1).
Treatment of the cross-link 7 in water with excess NaBH4 (in diglyme) gave two major products and three minor ones, in addition to a small amount of dA (Scheme 3). The major products were consistent with the addition of one and two equivalents (9, 10) of hydride to the α,β-unsaturated imine. LC-ESI-MS and NMR were used to determine the structures of the major reduction products. The minor products were analyzed by LC-ESI-MS; their masses were consistent with being derived from hydrolysis of the imine and subsequent reduction of 4. Reduction of cross-link 8 gave a single product, which was identified as the fully reduced cross-link (11).
Scheme 3.

Discussion
The present experiments, taken with the results of a previous study, define the acidity of the exocyclic N-oxopropenyl adducts of dG, N1-Me-dG, dA, and dC as 6.9. 8.2, 10.5, and 10.5, respectively.17 The oxopropenyl group is an effective electron-withdrawing group that lowers the pKa of 2 by ∼ 4 pKa units relative to dG and the pKa of 4 by > 2 pKa units relative to dA. The significant difference in pKa between 2 and 4 or 5 (6.9 vs 10.5) suggests that the dissociable proton in 2 is the N1-imino proton rather than the exocyclic N2 proton. Indirect confirmation of this is provided by the finding that the reaction of methyl iodide with deprotonated 2 occurs exclusively on N1. The position of alkylation was established by multi-dimensional NMR experiments and by base hydrolysis of the product to N1-Me-dG. The pKa of 8.2 for 6 is significantly lower than those of 4 and 5.
Reaction of 4 and 6 with Nα-acetylysine at neutral pH readily formed the imine conjugates, 7 and 8. These conjugates are models for cross-links that could form between the oxopropenyl DNA adducts and lysine residues of proteins.20 In contrast to 4 and 6, 2 did not form cross-links even on prolonged exposure to an excess of Nα-acetylysine. This is consistent with our hypothesis that the inability of 2 to form cross-links is due to its low pKa, which renders it predominantly anionic and, therefore, much less reactive at neutral pH. Hypothetically, 2 might be able to react with Nα-acetyllysine at pHs below its pKa of 6.9. However, it is unlikely that even a large excess of Nα-acetylysine could compete with the intramolecular cyclization of 2 to 3. Cyclization occurs very rapidly following protonation of the anion of 2 and 3 ultimately dehydrates to 1.17 Dehydration is general acid-catalyzed and can also be rapid depending on pH and buffer concentration. Adduct 1 is actually significantly more reactive to nucleophiles than 2, undoubtedly because it is neutral and because it possesses an α,β-unsaturated imine group.16 Nucleophiles add at C-8 and hydride reducing agents add at C-6.10,21
The N-oxopropenyl group is a vinylogous formamide and is expected to have low reactivity toward nucleophiles due to resonance delocalization of the nitrogen lone pair into the aldehyde. However, the nitrogen lone pairs of 2, 4 and 6 are likely to have strong resonance interactions with the electron-withdrawing nucleoside bases, making the aldehyde more reactive toward nucleophiles. It is likely that the electron-withdrawing nature of guanine is significantly reduced when deprotonated at N1 (Scheme 4). Consequently, the N2-lone pair would be free for strong resonance interactions with the oxopropenyl aldehyde, making the anion of 2 much less reactive toward cross-linking reactions.
Scheme 4.

The pKas of 7 and 8 were determined to be 8.8 and 6.3, respectively. This implies that 7 exists primarily as the protonated derivative whereas 8 exists as the deprotonated derivative at neutral pH. The higher pKa of 7 is consistent with its higher rate of hydrolysis at neutral pH compared to 8. Hydrolysis of 7 and 8 occurred primarily to yield 4 and 6, respectively. A small percentage of the alternative hydrolysis occurred to produce dA or N1-Me-dG plus oxopropenyl-Nα-acetyllysine. The net result of the latter transformation is oxopropenyl transfer from 4 or 6 to Nα-acetylysine.
The differential in the ability of 2 and 4 (and presumably 5) to form cross-links is striking and suggests that under physiological conditions, the principal source of base propenal- or malondialdehyde-induced DNA-DNA or DNA-protein cross-links is 4. Adduct 4 is generated in amounts corresponding to 20% of 1 + 2 although a detailed comparison of their levels has not been conducted under different conditions or from in vivo samples.22 The ability to reduce 7 or 8 to dihydro or tetrahydro derivatives provides a strategy that should be useful in developing analytical methods with which to quantify these cross-links.
DNA-DNA and DNA-protein cross-links are of increasing interest from a biological point of view because of their production from bifunctional electrophiles and their ability to block DNA replication and induce mutation.14,15,20,23 γ-Hydroxy-propanodeoxyguanosine ring-opens in duplex DNA to form N2-oxopropyl-dG in a reaction that is analogous to the ring-opens opening of 1 to 2.24 N2-Oxopropyl-dG condenses with the α-amino groups of amino acids at the N-termini of peptides or with the ε-amino group of Lys residues within peptides to form imine conjugates.25 However, these imine conjugates are in rapid equilibrium with the carbinolamine and aldehyde precursors. The imines are stabilized for detection and biological evaluation by hydride reduction. In contrast, conjugates 7 and 8 are relatively stable even as the deoxynucleosides and can be isolated by HPLC and characterized spectroscopically. Thus, it seems likely that if cross-links form between N-oxopropenyl adducts in duplex DNA (i.e., 4 and 5) and Lys residues of DNA-binding proteins, they will be quite stable especially if the enamino-imine linkage is protected from hydrolysis by the association of the protein molecule with DNA. Our results further predict that the major-groove oxopropenyl adduct, 4, is a better candidate for cross-link formation. The most common DNA-protein binding motif involves interactions of α-helices of transcription factors and restriction enzymes with the major groove.26
Supplementary Material
Acknowledgements
This work was supported by a research grant from the National Cancer Institute (CA87819).
References
- (1).Marnett LJ, Plastaras JP. Trends Genet. 2001;17:214–221. doi: 10.1016/s0168-9525(01)02239-9. [DOI] [PubMed] [Google Scholar]
- (2).Dedon PC, Plastaras JP, Rouzer CA, Marnett LJ. Proc.Natl.Acad.Sci.USA. 1998;95:11113–11116. doi: 10.1073/pnas.95.19.11113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Plastaras JP, Riggins JN, Otteneder M, Marnett LJ. Chem.Res.Toxicol. 2000;13:1235–1242. doi: 10.1021/tx0001631. [DOI] [PubMed] [Google Scholar]
- (4).Zhou X, Taghizadeh K, Dedon PC. J.Biol.Chem. 2005;280:25377–25382. doi: 10.1074/jbc.M503079200. [DOI] [PubMed] [Google Scholar]
- (5).Fink SP, Reddy GR, Marnett LJ. Proc Natl Acad Sci U S A. 1997;94:8652–7. doi: 10.1073/pnas.94.16.8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).VanderVeen LA, Hashim MF, Shyr Y, Marnett LJ. Proc.Natl.Acad.Sci.USA. 2003;100:14247–14252. doi: 10.1073/pnas.2332176100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Mao H, Schnetz-Boutaud NC, Weisenseel JP, Marnett LJ, Stone MP. Proc.Natl.Acad.Sci.USA. 1999;96:6615–6620. doi: 10.1073/pnas.96.12.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hashim MF, Riggins JN, Schnetz-Boutaud N, Voehler M, Stone MP, Marnett LJ. Biochemistry. 2004;43:11828–11835. doi: 10.1021/bi049360f. [DOI] [PubMed] [Google Scholar]
- (9).Mao H, Reddy GR, Marnett LJ, Stone MP. Biochemistry. 1999;38:13491–13501. doi: 10.1021/bi9910124. [DOI] [PubMed] [Google Scholar]
- (10).Schnetz-Boutaud N, Daniels JS, Hashim MF, Scholl P, Burrus T, Marnett LJ. Chem.Res.Toxicol. 2000;13:967–970. doi: 10.1021/tx000135i. [DOI] [PubMed] [Google Scholar]
- (11).Jeong YC, Sangaiah R, Nakamura J, Pachkowski BF, Ranasinghe A, Gold A, Ball LM, Swenberg JA. Chem.Res.Toxicol. 2005;18:51–60. doi: 10.1021/tx049853l. [DOI] [PubMed] [Google Scholar]
- (12).Kozekov ID, Nechev LV, Moseley MS, Harris CM, Rizzo CJ, Stone MP, Harris TM. J.Am.Chem.Soc. 2003;125:50–61. doi: 10.1021/ja020778f. [DOI] [PubMed] [Google Scholar]
- (13).Cho YJ, Wang H, Kozekov ID, Kozekova A, Kurtz AJ, Jacob J, Voehler M, Smith J, Harris TM, Rizzo CJ, Lloyd RS, Stone MP. Chem.Res.Toxicol. 2006;19:1019–1029. doi: 10.1021/tx0600604. [DOI] [PubMed] [Google Scholar]
- (14).Sanchez AM, Minko IG, Kurtz AJ, Kanuri M, Moriya M, Lloyd RS. Chem.Res.Toxicol. 2003;16:1019–1028. doi: 10.1021/tx034066u. [DOI] [PubMed] [Google Scholar]
- (15).Minko IG, Zou Y, Lloyd RS. Proc.Nat.Acad.Sci.USA. 2002;99:1905–1909. doi: 10.1073/pnas.042700399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Niedernhofer LJ, Riley M, Schnetz-Boutaud N, Sanduwaran G, Chaudhary AK, Reddy GR, Marnett LJ. Chem.Res.Toxicol. 1997;10:556–561. doi: 10.1021/tx960191c. [DOI] [PubMed] [Google Scholar]
- (17).Riggins JN, Pratt DA, Voehler M, Daniels JS, Marnett LJ. J.Am.Chem.Soc. 2004;126:10571–10581. doi: 10.1021/ja040010q. [DOI] [PubMed] [Google Scholar]
- (18).Riggins JN, Daniels JS, Rouzer CA, Marnett LJ. J.Am.Chem.Soc. 2004;126:8237–8243. doi: 10.1021/ja040009r. [DOI] [PubMed] [Google Scholar]
- (19).Wang H, Marnett LJ, Harris TM, Rizzo CJ. Chem.Res.Toxicol. 2004;17:144–149. doi: 10.1021/tx034174g. [DOI] [PubMed] [Google Scholar]
- (20).Voitkun V, Zhitkovich A. Mutat.Res. 1999;424:97–106. doi: 10.1016/s0027-5107(99)00011-1. [DOI] [PubMed] [Google Scholar]
- (21).VanderVeen LA, Druckova A, Riggins JN, Sorrells JL, Guengerich FP, Marnett LJ. Biochemistry. 2005;44:5024–5033. doi: 10.1021/bi0472898. [DOI] [PubMed] [Google Scholar]
- (22).Chaudhary AK, Reddy GR, Blair IA, Marnett LJ. Carcinogenesis. 1996;17:1167–1170. doi: 10.1093/carcin/17.5.1167. [DOI] [PubMed] [Google Scholar]
- (23).Minko IG, Kozekov ID, Kozekova A, Harris TM, Rizzo CJ, Lloyd RS. Mutat.Res. 2007 doi: 10.1016/j.mrfmmm.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).de los Santos C, Zaliznyak T, Johnson F. J.Biol.Chem. 2001;276:9077–9082. doi: 10.1074/jbc.M009028200. [DOI] [PubMed] [Google Scholar]
- (25).Kurtz AJ, Lloyd RS. J.Biol.Chem. 2003;278:5970–5976. doi: 10.1074/jbc.M212012200. [DOI] [PubMed] [Google Scholar]
- (26).Patikoglou G, Burley SK. Annu.Rev.Biophys.Biomol.Struct. 1997;26:289–325. doi: 10.1146/annurev.biophys.26.1.289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.