

Abstract
Gastrin-releasing peptide (GRP), the mammalian equivalent of bombesin (BBS), is an autocrine growth factor for neuroblastoma; its receptor is up-regulated in undifferentiated neuroblastomas. The phosphatidylinositol 3-kinase (PI3K) is a critical cell survival pathway; it is negatively regulated by the PTEN tumor suppressor gene. We have recently found that poorly-differentiated neuroblastomas express decreased PTEN protein levels. Moreover, overexpression of the GRP receptor, a member of G-protein coupled receptor family, downregulates PTEN expression, resulting in increased neuroblastoma cell growth. Therefore, we sought to determine whether GRP or BBS activates PI3K in neuroblastoma cells (BE(2)-C, LAN-1, SK-N-SH). GRP or BBS treatment rapidly increased phosphorylation of Akt and GSK-3β in neuroblastoma cells. Inhibition of GRP receptor, with antagonist GRP-H2756 or siRNA, attenuated BBS-induced phosphorylation of Akt. LY294002, a PI3K inhibitor, also abrogated BBS-stimulated phospho-Akt as well as its cell cycle targets. GRP increased G1/S phase progression in SK-N-SH cells. BBS-mediated BrdU incorporation was blocked by LY294002. Our findings identify PI3K as an important signaling pathway for GRP-mediated neuroblastoma cell growth. A novel therapy targeted at GRP/GRP receptor may prove to be an effective treatment option to inhibit PI3K in neuroblastomas.
Keywords: PI3K, Akt, Cell Cycle, GRP, Bombesin, Neuroblastoma
1. Introduction
Neuroblastoma is the most common extracranial solid tumor in infants and children, accounting for more than 8-10% of all childhood cancers [1-3]. Many advances have been made in understanding the biology of this enigmatic tumor; however, despite current multimodal therapy, the overall prognosis for all stages of tumor is still dismal. There have been numerous studies directed towards known prognostic factors, such as MYCN amplification. However, neuroblastoma is a heterogeneous tumor with an unpredictable course, in spite of prognostic factors [2, 3]. Therefore, in regards to therapeutic agents, it is necessary to target entities that are generalized to tumor progression. The programs that direct cell survival in tumors are common to a wide range of tumor tissues, including the well-described phosphatidylinositol 3-kinase (PI3K) pathway [4].
The PI3K pathway regulates cell growth in normal and cancer cells by inducing phosphorylation of its downstream effector, Akt [5, 6]. Cell cycle progression is one of the many survival pathways modulated by PI3K; this regulation involves PI3K/Akt inhibition of GSK-3β leading to the rescue and nuclear accumulation of cyclin D, an inducer of G1/S phase progression [5, 6]. PI3K can also regulate tumor suppressors p21 and p27, two negative regulators of the cell cycle, by promoting their phosphorylation and translocation to the cytoplasm; additionally, PI3K regulates the degradation of p27 [4, 6]. We have previously shown that PTEN (phosphatase and tensin homologue deleted on chromosome ten), a negative regulator of the PI3K pathway, is down-regulated in poorly differentiated neuroblastomas [7], which may contribute to a malignant phenotype.
Since neuroblastomas are neuroendocrine tumors, they secrete and respond to various hormones including gastrin-releasing peptide (GRP) [8, 9]. We have found that GRP, the mammalian equivalent of bombesin (BBS), stimulates neuroblastoma cell growth by an autocrine and/or paracrine effect [10]. We also found that the GRP receptor, a member of the G-protein coupled receptor (GPCR) family, is significantly increased in more undifferentiated neuroblastomas [10], and that overexpression of the GRP receptor down-regulates PTEN expression, resulting in increased neuroblastoma cell growth [7]. However, the intracellular signaling mechanisms involved in these GRP-mediated proliferative processes are not clear.
In this study, we sought to elucidate the cell survival mechanisms involved in GRP-induced neuroblastoma cell growth and whether the PI3K pathway is involved. Since the PI3K pathway is a positive regulator of cell cycle progression, we determined whether GRP activates this pathway and its downstream cell cycle regulators, thereby, amplifying the pro-growth effects of GRP. Our findings demonstrate that inhibition of the GRP receptor or PI3K leads to significant decreases in Akt phosphorylation and modulates G1/S phase regulators cyclin D, p21, and p27.
2. Materials and methods
2.1. Reagents
GRP, BBS, and GRP-H2756 were purchased from Bachem (Torrance, CA). BME was a gift from Dr. David H. Coy (Tulane University, New Orleans, LA). SB216763 and SB415286 were purchased from Tocris Bioscience (Ellisville, MO). LY294002 and antibodies against phospho-Akt, Akt, phospho-GSK-3α/β, phospho-Rb, Rb were purchased from Cell Signaling (Beverly, MA). Antibodies against GSK-β, cyclin D, p21, and p27 were purchased from BD Biosciences (San Jose, CA). Antibodies against GRP receptor and β-actin were from Abcam (Cambridge, MA) and Sigma-Aldrich (St. Louis, MO), respectively. All secondary antibodies against mouse and rabbit IgG were purchased from Santa Cruz Inc. (Santa Cruz, CA). Cellular DNA Flow Cytometric Analysis and Cell Proliferation BrdU ELISA kits were obtained from Roche Applied Science (Indianapolis, IN). Small-interference (si) RNA directed to the GRP receptor was purchased from Dharmacon (Lafayette, CO), along with non-targeting scrambled sequences as controls.
2.2. Cell culture, transfection and treatment
The human neuroblastoma cell lines SK-N-SH and BE(2)-C were purchased from American Type Culture Collection (Manassas, VA) and LAN-1 was a gift from Dr. Robert C. Seeger (University of Southern California, Los Angeles, CA). Cells were cultured in RPMI 1640 medium (Cellgro Mediatech Inc., Herndon, VA) supplemented with 10% fetal bovine serum (FBS) (Sigma Aldrich), in a humidified atmosphere of 5% CO2 at 37°C. For siRNA transfection assays, 6 × 106 cells/400 μl (SK-N-SH, BE(2)-C) or 9 × 106 cells/200 μl (LAN-1) were transfected with siRNA (100 nM) targeted to the GRP receptor or a non-targeting control by electroporation using Gene Pulser Xcell System (Bio-Rad, Hercules, CA) and seeded onto a 100mm dish. Setup conditions were 400V, 500 μF for SK-N-SH, BE(2)-C and 300V, 150 μF for LAN-1. The next day the cells were replated onto a 6-well plate (2-5 × 105 cells/well). In order to clearly understand the effects of GRP on signaling transduction and to eliminate the effects of serum in neuroblastoma cells, all GRP and BBS treatments were performed using serum–free medium. The cells were seeded on culture plates and serum-starved overnight, then treated with GRP-H2756/BME (1 μM), LY294002 (20 μM), and/or GSK-3β inhibitors, SB216763 (10 μM) or SB415286 (30 μM), for 30 min prior to GRP/BBS (100 nM) stimulation for the indicated time periods. Cells were then harvested for immunoblot or cell cycle analysis. Experiments were repeated on at least 2 separate occasions.
2.3. Cell cycle analysis
Cell cycle distribution was analyzed using flow cytometry. 1 × 106 cells were trypsinized, washed once with PBS, and fixed in 70% ethanol. Fixed cells were washed with PBS, incubated with 100 μg/ml RNAase for 30 min at 37°C, stained with propidium iodide (50 μg/ml), and analyzed on a FACScan flow cytometer (Becton-Dickinson Instruments, San Jose, CA). The percentages of cells in different cell cycle phases were analyzed using Cell-FIT software (Becton-Dickinson Instruments).
2.4 BrdU Incorporation Assay
Cells were seeded in 96-well plates at a density of 1.2 × 104 cells per well and BrdU incorporation was determined by Cell Proliferation ELISA. Briefly, SK-N-SH cells were treated with either BBS and/or LY294002 for 24-72 h. BrdU was added to the cell culture for 16 h prior to detection and BrdU incorporation was measured according to the manufacturer's instruction. The values, corresponding to the amount of DNA synthesis, were read at OD450 with EL808 Ultra Microplate Reader (BioTek Instrument Inc., Winooski, VT).
2.5. Immunoblots
Cell lysis buffer (Cell Signaling) containing 1 mM PMSF and protein inhibitors cocktail (Roche Applied Science) was used to extract total protein. The protein concentrations were quantified using Bio-Rad Protein Assay kit. Equivalent amounts of protein (50-100 μg) were electrophoresed on NOVEX NuPAGE 4-12% Bis-Tris gels (Invitrogen, Carlsbad, CA), electro-transferred to polyvinylidene diflouride membranes (Bio-Rad), and probed with primary antibodies followed by HRP-conjugated secondary antibody (1:5000 dilution). Protein levels were visualized by an enhanced chemiluminescence (ECL) system (Amersham Bioscience, Arlington, IL).
3. Results
3.1. GRP/BBS induces cell cycle progression and PI3K-regulated BrdU incorporation
GRP is an autocrine growth factor for neuroblastomas [9]. We determined whether the effects of GRP are associated with cell cycle progression in neuroblastoma cells by flow cytometry analysis using SK-N-SH cells treated with GRP for 16 h (Fig. 1A). GRP treatment resulted in a significant increase in the S phase, compared to control cells. BBS treatment also increased BrdU incorporation in SK-N-SH cells (Fig. 1B); increase was statistically significant at 48 and 72 h timepoints. LY294002 treatment with or without BBS strongly reduced BrdU incorporation, in comparison to control cells (Fig. 1B). Interestingly, control cells showed marked decrease in BrdU incorporation over a time course, likely as a result of serum-starvation. These data are consistent with the findings that GRP/BBS stimulates neuroblastoma cell growth by regulation of the cell cycle.
Figure 1. GRP/BBS treatment induces S-phase progression and PI3K-regulated BrdU incorporation in neuroblastoma cells.

(A) SK-N-SH cells were serum starved for 24 h prior to treatment with GRP for 16 h. GRP caused an increase in the S and G2/M phases, in comparison to control cells. (B) SK-N-SH cells were serum starved for 24 h prior to treatment with BBS and/or LY294002 over a time course (24-72 h). BBS induced BrdU incorporation at 48 and 72 h; this response was blocked significantly with LY294002. Data represent mean ± SEM; * p < 0.05 vs. control.
3.2. PI3K/Akt/GSK-3β pathway is activated by BBS treatment
PI3K is a potent regulator of the cell cycle [5, 6]. In order to determine whether there is interaction between GRP and PI3K signaling, cells were treated with BBS or GRP over a time course and immunoblots were performed to assess phosphorylation of Akt and GSK-3α/β, downstream effectors for the PI3K pathway. Treatment with BBS or GRP rapidly increased phosphorylation of Akt and GSK-3β (up to 5-fold); the maximal stimulation was noted at 5 min (SK-N-SH, BE(2)-C) (Figs. 2, 3) and 15 min (LAN-1) after treatment (Fig. 3). To further confirm GRP activation of PI3K, cells were pretreated with GRP receptor antagonists GRP-H2756 or BME, a PI3K inhibitor LY294002, or GSK-3β inhibitors SB216763 or SB415286, for 30 min prior to BBS stimulation. Akt phosphorylation was decreased by GRP receptor antagonists and completely abrogated by LY294002 in BE(2)-C and SK-N-SH cells (Fig. 2). BBS-induced GSK-3β phosphorylation was also inhibited by SB216763 and SB415286 in SK-N-SH cells (Fig. 2B). Collectively, these results demonstrate that GRP activates the PI3K pathway.
Figure 2. BBS treatment causes activation of PI3K/Akt/GSK-3β pathway in neuroblastoma cells.
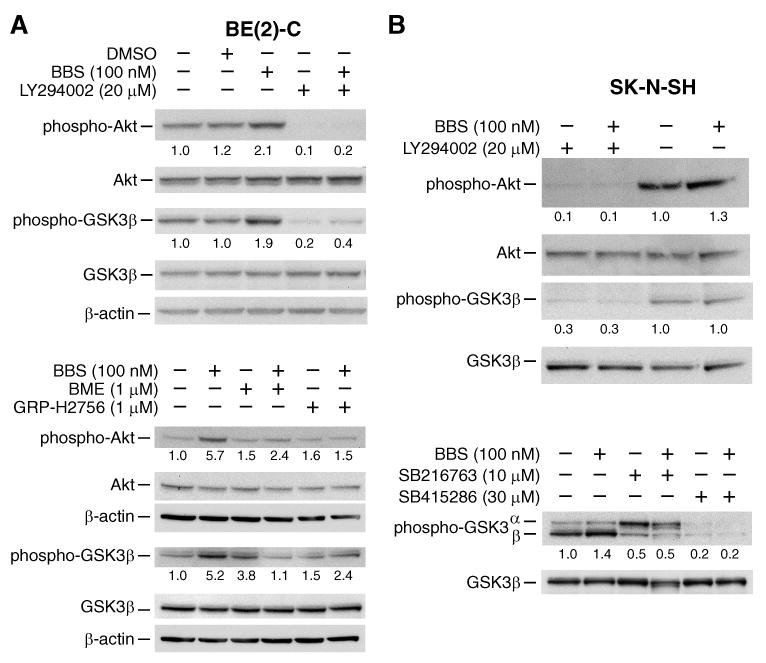
(A, B) BBS treatment caused rapid phosphorylation of Akt and GSK-3β, which was decreased by GRP receptor antagonists and completely abrogated by LY294002. (B) BBS-induced GSK-3β phosphorylation was also inhibited by SB216763 and SB415286. Values under blots represent protein fold-change levels (relative to respective β-actin) in comparison to control lane.
Figure 3. GRP/BBS modulates G1/S-phase regulators in neuroblastoma cells by the PI3K pathway.
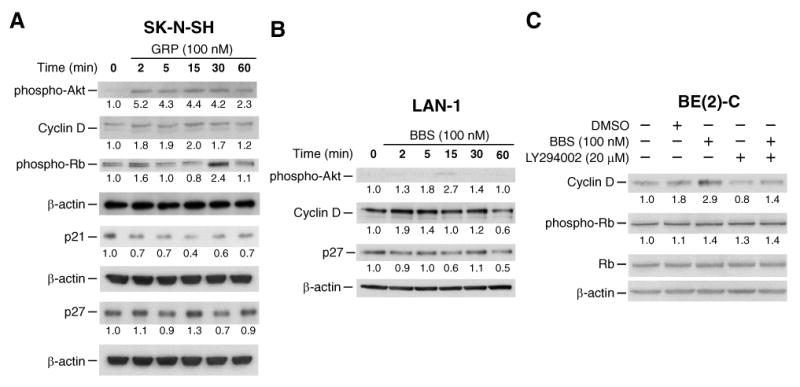
(A) SK-N-SH cells demonstrated increased phospho-Akt, cyclin D and phospho-Rb expression, while displaying decreased p21, and p27, with GRP treatment. (B) LAN-1 cells exhibited increased phospho-Akt and cyclin D and decreased p27 with BBS treatment. (C) BBS-induced changes to G1/S-phase regulator cyclin D in BE(2)-C cells were blocked with LY294002 (membrane in Figure 2A was stripped and reprobed). Values under blots represent protein fold-change levels (relative to respective β-actin) in comparison to control lane.
3.3. Modulation of G1/S-phase regulators by GRP-mediated PI3K stimulation
To determine a possible mechanism of cross-talk between GRP and cell cycle regulation, we next examined the effects of GRP stimulation on G1/S-phase cell cycle regulators cyclin D, Rb, p21, and p27. As shown in Figure 3A, GRP treatment increased expression of cyclin D (approximately 2-fold) and phosphorylated Rb (approximately 2-fold), and decreased expression of the cyclin-dependent kinase inhibitors p21 (2-60 min) and p27 (5 and 30 min) in SK-N-SH cells. Similarly, increased expression of cyclin D (2 and 5 min) and decreased expression of p27 (2, 15, and 60 min) was noted in LAN-1 cells after BBS treatment (Fig. 3B). These results are consistent with the GRP-mediated increase of the G1/S phase noted by flow cytometry and DNA synthesis assessed by BrdU incorporation (Fig. 1). BBS-induced increases of the G1/S-phase regulator cyclin D in BE(2)-C cells were blocked with LY294002 (Fig. 3C). These data further confirm the critical role of the PI3K pathway in GRP-mediated cell cycle changes in neuroblastoma cells.
3.4. Inhibition of the PI3K pathway by GRP receptor knockdown
GRP-mediated PI3K activation was further confirmed using siRNA targeted to the GRP receptor. BE(2)-C, SK-N-SH, and LAN-1 cells were transfected with siRNA directed to the GRP receptor (siGRPR) or a non-targeting control (siNTC). Protein was collected 72 h post-transfection, immunoblotted, and probed for phospho-Akt, Akt, and GRP receptor. β-actin was used as a loading control. GRP receptor siRNA decreased BBS-induced Akt phosphorylation in all three neuroblastoma cell lines (Fig. 4). These data further support the finding that PI3K is activated by GRP.
Figure 4. Knockdown of GRP receptor inhibits PI3K activation.
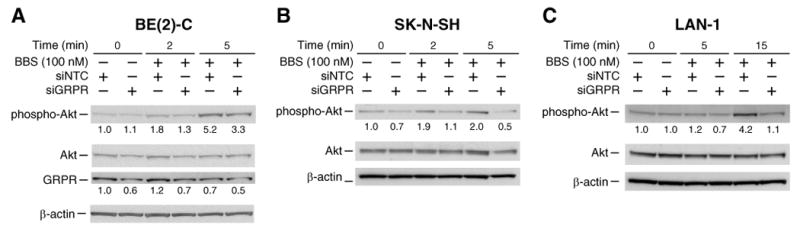
(A) BE(2)-C cells were transfected with siGRPR or siNTC. Protein was collected 72 h post-transfection. Knockdown of GRP receptor caused a decrease in BBS-induced Akt phosphorylation. (B, C) SK-N-SH and LAN-1 cells also demonstrated decrease in phospho-Akt by siGRPR. Values under blots represent protein fold-change levels (relative to respective β-actin) in comparison to control lane.
4. Discussion
The signaling mechanisms involved in GRP-induced neuroblastoma cell growth are not fully understood. Based on our previous study demonstrating a relationship between undifferentiated neuroblastomas and deregulation of both GRP and PI3K pathways, we sought to determine whether these two pathways converge to regulate the cell cycle in neuroblastoma cells. We found that GRP activates the PI3K pathway to induce cell cycle progression by modulating G1/S-phase regulators. Moreover, inhibition of PI3K blocked the GRP/BBS-mediated alterations to cell cycle proteins and inhibited BBS-induced BrdU incorporation. Therefore, the growth stimulatory properties of GRP may be due, in part, to PI3K regulation of the cell cycle.
PI3K pathway regulation of the G1/S-phase is well established; therefore, we analyzed the effect of PI3K activation on the expression of cyclin D, p21, and p27. Upon phosphorylation by GSK-3β, cyclin D translocates to the cytosol where it undergoes protein degradation [4, 6]. PI3K-mediated phosphorylation of GSK-3β, and resultant inhibition, leads to the nuclear accumulation of cyclin D which promotes cell cycle progression [6]. PI3K/Akt has also been shown to be important in regulating cyclin D gene transcription and translation [6]. Our results confirmed that cyclin D protein expression is increased with PI3K activation by GRP treatment and decreased upon addition of its pathway inhibitor (Fig. 3). Akt phosphorylation of cyclin-dependent kinase inhibitors p21 and p27 also induces the progression of G1/S phase. Once phosphorylated, p21 and p27 are translocated and sequestered in the cytoplasm; p27 may also undergo proteolysis by indirect Akt stimulation [4, 6]. Here, we show that GRP/BBS stimulation decreased levels of p21 and p27 in SK-N-SH cells and decreased p27 in BE(2)-C and LAN-1 cells (Fig. 3). These findings were further corroborated by the increase in G1/S phase (Fig. 1A), as recorded by flow cytometry, and BrdU incorporation (Fig. 1B), a marker for DNA synthesis. Notably, levels of p21 could not be detected in BE(2)-C and LAN-1 cells; we speculate that this is most likely due to their p53-/- genotype [11, 12]. Since p53 regulates p21 gene transcription [13, 14]; the absence of p53 in these cell lines may explain the lack of p21 expression. LY294002 inhibition of BrdU incorporation (Fig. 1B) reinforces the role of PI3K in this GRP-mediated process.
We have shown that GRP promotes rapid activation of the PI3K pathway by Akt phosphorylation (Fig. 2). The GRP receptor is a member of the GPCR family and has been associated with phospholipase C and Ca2+ second messenger signaling [10, 15]. In lymphocytes, PI3K is coupled to GPCRs through direct interaction with the Gβγ protein subunits [16]. It is unclear whether the GRP receptor activates the PI3K pathway directly or through indirect cross-talk mechanisms involving other signal transduction pathways. In a study of head and neck squamous cancer cells, Lui et al. [17] found that the stimulatory effects of GRP were mediated by the epidermal growth factor receptor (EGFR)/MAPK pathway. Sumitomo et al. [18] also found that the mitogenic effects of BBS could be attributed to the insulin-like growth factor receptor (IGFR) leading to PI3K activation in prostate cancer cells. This implicates GRP signaling in the transactivation and indirect stimulation of major proliferation and survival pathways. Neuroblastoma cells also express EGFR and IGFR [19, 20]. Further studies of GRP receptor signaling would help delineate the downstream targets regulating neuroblastoma growth; we speculate that both direct and indirect mechanisms are possible.
In summary, we have shown that GRP/BBS rapidly activated the PI3K pathway; this was associated with cell cycle progression. Inhibition of the GRP receptor prevented BBS-induced activation of the PI3K pathway, further suggesting that PI3K is an important signaling mechanism for GPCR-mediated stimulation of neuroblastoma cell growth. A novel therapy targeted to the GRP receptor may prove to be an effective combinational treatment option to inhibit PI3K activation in neuroblastomas.
Acknowledgments
The authors thank Karen Martin for manuscript preparation and Lan Pang for assistance with the experiments. This work was supported by grants RO1 DK61470, RO1 DK48498, RO1 CA104748 and PO1 DK35608 from the National Institutes of Health and an Institutional Research Grant (IRG-110376) from the American Cancer Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van Noesel MM, Versteeg R. Pediatric neuroblastomas: genetic and epigenetic ‘danse macabre’. Gene. 2004;325:1–15. doi: 10.1016/j.gene.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 2.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:203–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 3.Schwab M, Westermann F, Hero B, Berthold F. Neuroblastoma: biology and molecular and chromosomal pathology. Lancet Oncol. 2003;4:472–480. doi: 10.1016/s1470-2045(03)01166-5. [DOI] [PubMed] [Google Scholar]
- 4.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 5.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 6.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 7.Qiao J, Kang J, Cree J, Evers BM, Chung DH. Gastrin-releasing peptide-induced down-regulation of tumor suppressor protein PTEN (phosphatase and tensin homolog deleted on chromosome ten) in neuroblastomas. Ann Surg. 2005;241:684–691. doi: 10.1097/01.sla.0000161173.47717.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17:2264–2279. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- 9.Gustafson WC, De Berry BB, Evers BM, Chung DH. Role of gastrointestinal hormones in neuroblastoma. World J Surg. 2005;29:281–286. doi: 10.1007/s00268-004-7815-4. [DOI] [PubMed] [Google Scholar]
- 10.Kim S, Hu W, Kelly DR, Hellmich MR, Evers BM, Chung DH. Gastrin-releasing peptide is a growth factor for human neuroblastomas. Ann Surg. 2002;235:621–629. doi: 10.1097/00000658-200205000-00003. discussion 629-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carr J, Bell E, Pearson AD, Kees UR, Beris H, Lunec J, Tweddle DA. Increased frequency of aberrations in the p53/MDM2/p14(ARF) pathway in neuroblastoma cell lines established at relapse. Cancer Res. 2006;66:2138–2145. doi: 10.1158/0008-5472.CAN-05-2623. [DOI] [PubMed] [Google Scholar]
- 12.Wallick CJ, Gamper I, Thorne M, Feith DJ, Takasaki KY, Wilson SM, Seki JA, Pegg AE, Byus CV, Bachmann AS. Key role for p27Kip1, retinoblastoma protein Rb, and MYCN in polyamine inhibitor-induced G1 cell cycle arrest in MYCN-amplified human neuroblastoma cells. Oncogene. 2005;24:5606–5618. doi: 10.1038/sj.onc.1208808. [DOI] [PubMed] [Google Scholar]
- 13.Liu G, Lozano G. p21 stability: linking chaperones to a cell cycle checkpoint. Cancer Cell. 2005;7:113–114. doi: 10.1016/j.ccr.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 14.Yu J, Zhang L. The transcriptional targets of p53 in apoptosis control. Biochem Biophys Res Commun. 2005;331:851–858. doi: 10.1016/j.bbrc.2005.03.189. [DOI] [PubMed] [Google Scholar]
- 15.Sorrentino G, Singh IN, Massarelli R, Kanfer JN. Stimulation of phospholipase C activity by norepinephrine, t-ACPD and bombesin in LA-N-2 cells. Eur J Pharmacol. 1996;308:81–86. doi: 10.1016/0014-2999(96)00246-4. [DOI] [PubMed] [Google Scholar]
- 16.Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3:317–330. doi: 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- 17.Lui VW, Thomas SM, Zhang Q, Wentzel AL, Siegfried JM, Li JY, Grandis JR. Mitogenic effects of gastrin-releasing peptide in head and neck squamous cancer cells are mediated by activation of the epidermal growth factor receptor. Oncogene. 2003;22:6183–6193. doi: 10.1038/sj.onc.1206720. [DOI] [PubMed] [Google Scholar]
- 18.Sumitomo M, Milowsky MI, Shen R, Navarro D, Dai J, Asano T, Hayakawa M, Nanus DM. Neutral endopeptidase inhibits neuropeptide-mediated transactivation of the insulin-like growth factor receptor-Akt cell survival pathway. Cancer Res. 2001;61:3294–3298. [PubMed] [Google Scholar]
- 19.Ho R, Minturn JE, Hishiki T, Zhao H, Wang Q, Cnaan A, Maris J, Evans AE, Brodeur GM. Proliferation of human neuroblastomas mediated by the epidermal growth factor receptor. Cancer Res. 2005;65:9868–9875. doi: 10.1158/0008-5472.CAN-04-2426. [DOI] [PubMed] [Google Scholar]
- 20.Leventhal PS, Randolph AE, Vesbit TE, Schenone A, Windebank A, Feldman EL. Insulin-like growth factor-II as a paracrine growth factor in human neuroblastoma cells. Exp Cell Res. 1995;221:179–186. doi: 10.1006/excr.1995.1365. [DOI] [PubMed] [Google Scholar]