

Abstract
We studied the effect of pH on ligand binding in wild-type lactose permease or mutants in the four residues—Glu-269, Arg-302, His-322, and Glu-325—that are the key participants in H+ translocation and coupling between sugar and H+ translocation. Although wild-type permease or mutants in Glu-325 and Arg-302 exhibit marked decreases in affinity at alkaline pH, mutants in either His-322 or Glu-269 do not titrate. The results offer a mechanistic model for lactose/H+ symport. In the ground state, the permease is protonated, the H+ is shared between His-322 and Glu-269, Glu-325 is charge-paired with Arg-302, and substrate is bound with high affinity at the outside surface. Substrate binding induces a conformational change that leads to transfer of the H+ from His-322/Glu-269 to Glu-325 and reorientation of the binding site to the inner surface with a decrease in affinity. Glu-325 then is deprotonated on the inside because of rejuxtaposition with Arg-302. The His-322/Glu-269 complex then is reprotonated from the outside surface to reinitiate the cycle.
Lactose permease (lac permease), the product of the lac Y gene (1), transduces free energy stored in an electrochemical H+ gradient into a sugar concentration gradient by catalyzing the coupled stoichiometric translocation of galactosides and H+ (lactose/H+ symport) (reviewed in refs. 2 and 3). As such, the permease is a paradigm for energy-transducing transmembrane proteins, which are a highly significant fraction of the genomes sequenced thus far and represent targets for some of the most widely prescribed drugs in the world.
Site-directed mutagenesis of wild-type permease and Cys–scanning mutagenesis of all of the residues in a functional mutant devoid of Cys residues has allowed delineation of functionally important amino acids (reviewed in ref. 4). Furthermore, application of a battery of site-directed methods to an extensive library of mutants has led to a structure at a level of helix packing, as well as dynamic information to help unravel the transport mechanism (reviewed in ref. 5). Of the 417 residues in lac permease, only six side chains are irreplaceable for active transport. Glu-126 (helix IV) and Arg-144 (helix V) are directly involved in substrate binding and specificity (6–9). Furthermore, the two residues form a salt bridge (7, 10, 11). In contrast, Glu-325 (helix X) is directly involved in H+ translocation, whereas Glu-269 (helix VIII), Arg-302 (helix IX), and His-322 (helix X) participate in coupling H+ and substrate translocation (reviewed in ref. 12). In addition, Glu-269, Glu-325, Arg-302, and His-322 are in close proximity (Fig. 1) and at about the same depth in the membrane as Glu-126/Arg-144 (5).
Figure 1.

Model for proposed ground-state conformation of lac permease, which binds substrate with high affinity. For clarity, 6 of the 12 helices in the permease are shown. Glu-126 (helix IV) and Arg-144 (helix V) are charge-paired and with Cys-148 (helix V) comprise the major components of the substrate binding site. Arg-302 (helix IX) is charge-paired with Glu-325 (helix X), while protonated His-322 (helix X) interacts with Glu-269 (helix VIII). Thus, the relevant H+ is shared by His-322 and Glu-269. Also shown are the charge pair between Asp-240 (helix VII) and Lys-319 (helix X), which are not essential for the mechanism. See text for further details regarding the transport mechanism.
Structural information in addition to differences in the functional properties of mutants in the six irreplaceable residues led to the notions that the protonation state of Glu-325 indirectly controls binding affinity and that this could form the basis for coupling between substrate and H+ translocation (see ref. 5). To test the latter notion, the pH dependence of ligand binding by mutants in Glu-325, Arg-302, His-322, or Glu-269 were compared. These studies involve a unique binding assay in which substrate protection of Cys–148 against alkylation by N-ethylmaleimide is quantified (8, 9, 13). Cys–148 is a component of the substrate binding site, interacting hydrophobically with the β face of the galactosyl moiety (9, 14, 15). By this means, affinities ranging from submicromolar to high millimolar can be measured accurately. This binding assay is crucial for our study because some of the mutants tested have such sufficiently low affinities for substrate that it would not be possible to measure binding affinities by traditional methods. The observations indicate that the permease must be protonated to bind ligand with high affinity on the outside surface and that the site of protonation is formed by His-322 and Glu-269. The results provide insight into the mechanism of lactose/H+ symport.
Experimental Procedures
Materials.
N-[ethyl-1-14C]maleimide (40 mCi/mmol) was purchased from DuPont NEN.
Construction of Mutants.
Site-specific labeling of Cys-148 with [14C]NEM (N-ethylmaleimide) is carried out with a permease mutant in which Cys-148 is the only Cys residue (single-Cys-148 permease), and all other native Cys residues are replaced with Ser or Val (16). Single-Cys-148 permease binds ligand with the same affinity as wild-type permease. Construction of mutants E325D, R302A, R302K, H322A, and E269D in single-Cys-148 permease with a biotin-acceptor domain in the middle cytoplasmic loop has been described (17). Mutants E325Q, H322N, and H322Q were constructed by overlap-extension PCR-mutagenesis (18) and cloned into single-Cys-148 lacY carrying a C-terminal biotin-acceptor domain by using KpnI and SpeI restriction sites (19).
Growth of Cells and Preparation of Right-Side-Out (RSO) Membrane Vesicles.
E. coli T184 (20) were grown in Luria-Bertani broth, and RSO membrane vesicles were prepared as described (21, 22) with the following modification (8): to prevent Cys oxidation during vesicle preparation, 5.0 mM DTT was included in all buffers. At the end of the preparation, the vesicles were washed with 100 mM potassium phosphate (pH 7.5) to remove DTT, resuspended in the same buffer at a protein concentration of 13–18 mg/ml, frozen in liquid N2, and stored at −80°C until use.
NEM Labeling.
Reactivity of Cys-148 with [14C]NEM was determined in situ in the absence or presence of given concentrations of β-galactopyranosyl 1-thio-β-d-galactopyranoside (TDG) as described (8, 13). Permease mutants used for these assays contain a biotin-acceptor domain in the middle cytoplasmic loop or at the C terminus and are biotinylated in vivo. Labeling at pH 5.5 or 7.5 was initiated by addition of 12 μl of [14C]NEM to a final concentration of 0.5 mM (40 mCi/mmol), and the vesicles were incubated for 15 or 5 min, respectively, at 25°C, as indicated. Labeling at pH 9.5 was carried out by adding 5 μl of [14C]NEM to a final concentration of 0.22 mM for 2 min. Reactions were quenched by addition of 10 mM DTT. The vesicles then were solubilized by 2% dodecyl maltoside (DDM; final concentration), and the samples were mixed with immobilized monomeric avidin (avidin-Sepharose) equilibrated with 50 mM NaPi, pH 7.5/0.1 M NaCl/0.02% DDM (wt/vol). The resin was washed with 5 ml of equilibration buffer, and biotinylated permease then was eluted with equilibration buffer containing 5 mM d-biotin. Eluates were analyzed electrophoretically on a SDS/12% polyacrylamide gel. The gel was dried and exposed to a PhosphorImager screen for 2–5 days. Incorporation of [14C]NEM was visualized and quantitated by a Storm 860 PhosphorImager (Molecular Dynamics).
Data Analysis.
The calculation of the apparent equilibrium dissociation constant, KD, of substrate was based on the equation
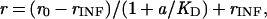 |
1 |
where r is the extent of reaction at protecting ligand concentration a, r0 is the extent of reaction at ligand concentration 0, and rINF is the extent of reaction extrapolated to infinite ligand concentration. r, r0, and a are known, and the nonlinear least-squares fit of the equation yields KD and rINF.
To analyze the pH dependence of ligand binding, we assumed that there is one protonation site in the permease with an acid dissociation constant, KA, controlling ligand binding, that the ligand dissociation constant of the deprotonated permease is K0, and that the ligand dissociation constant of the protonated permease is K1. The apparent equilibrium dissociation constant, KD, determined above, is the concentration of ligand at which the permease is half-saturated and the degree of reaction is midway between r0 and rINF. Simple algebraic manipulation of the equilibrium binding equations gives
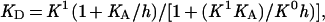 |
2 |
where h = [H+].
When the protonated permease binds ligand with much higher affinity (i.e., lower dissociation constant) than the deprotonated permease, as is the case here (i.e., when K1 ≪ K0, and when K1/K0 ≪ h/KA), then Eq. 2 can be approximated by
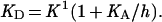 |
3 |
The nonlinear least-squares fit of Eq. 3 to KD versus h yields the parameters KA and K1. We thus estimated KA and hence pKa for wild-type permease and each mutant.
Results
NEM Reactivity of Single-Cys-148 Permease at pH 5.5, 7.5, and 9.5.
Ligand-dependent protection of single-Cys-148 permease against alkylation by [14C]NEM is useful for measuring substrate binding to lac permease, particularly when affinity is low (7–9, 13, 23). At 25°C and pH 7.5, labeling with 0.5 mM NEM is linear for up to 10 min, and when ligand protection is tested within this time frame, quantitative determination of KD can be obtained (8, 9, 13). At pH 5.5, reactivity is significantly decreased, and linearity is observed for up to 20 min. When labeling is performed at pH 9.5 at a lower NEM concentration (0.22 mM), the reaction is linear for ca. 2 min and reaches completion by 3 min (data not shown). Moreover, Cys-less permease does not label to any extent whatsoever. Therefore, NEM labeling for 15, 5, or 2 min, respectively, was used to determine KD at pH 5.5, 7.5, or 9.5.
Effect of pH on Ligand Binding by Single-Cys-148 Permease with Replacements for Glu-325.
NEM labeling was studied at increasing TDG concentrations in single-Cys-148 permease without or with mutations E325Q or E325D (Fig. 2). Protection against alkylation at each TDG concentration was quantified, and the KD (i.e., the ligand concentration at which 50% protection is observed) was determined (Table 1). The KD for single-Cys-148 or E325Q/Cys-148 permease is similar and does not change significantly between pH 5.5 and 7.5, exhibiting values of 27 and 16 μM, respectively, for single-Cys-148, or 20 and 50 μM, for E325Q/Cys-148 permease. However, KD increases (i.e., affinity decreases) by about 30-fold at pH 9.5 with both single-Cys-148 (523 μM) or E325Q/Cys-148 permease (632 μM). Remarkably, conservative replacement of Glu-325 with Asp (Fig. 2; Table 1) causes a marked increase in KD at pH 5.5 and 7.5 (139 and 512 μM, respectively) relative to single-Cys-148 permease, and at pH 9.5, a marked relative increase in KD also is observed (6,234 μM; ca. a 45-fold decrease in affinity). Notwithstanding the changes in KD, in each case, the apparent pKa with respect to TDG binding is estimated to be 8.1–8.2.
Figure 2.

Effect of TDG on NEM labeling of Cys-148 in single-Cys-148 permease or mutants E325Q/Cys-148, E325D/Cys-148, R302A/Cys-148, R302K/Cys-148, H322A/Cys-148, H322N/Cys-148, H322Q/Cys-148, and E269D/Cys-148 at pH 5.5, pH 7.5, and pH 9.5. (Left) Right-side-out membrane vesicles were incubated with 0.5 mM (at pH 5.5 and 7.5) or 0.22 mM (at pH 9.5) [14C]NEM at 25°C, for 15 min (pH 5.5), 5 min (pH 7.5), or 2 min (pH 9.5) in the absence or presence of given concentrations of TDG. Reactions were quenched with DTT, and biotinylated permease was solubilized and purified by affinity chromatography on monomeric avidin. Aliquots of protein were separated on a SDS/12% polyacrylamide gel, and [14C]-labeled permease was visualized by autoradiography. Although not shown, a fraction of the protein was analyzed by Western blotting to determine the amount of permease in each sample; no significant differences were observed. (Right) Incorporation of [14C]NEM was quantitated by a Storm 860 PhosphorImager, and labeling in the presence of various concentrations of TDG is expressed as % labeling observed in the absence of TDG. ○, pH 5.5; ■, pH 7.5; ●, pH 9.5.
Table 1.
Effect of pH on the apparent binding affinity constants (KD) of single-Cys-148 permease and replacements for Glu-325, Arg-302, His-322, and Glu-269
|
KD,
μM
|
pKa | |||
|---|---|---|---|---|
| pH 5.5 | pH 7.5 | pH 9.5 | ||
| Cys-148 | 27 | 16 | 523 | 8.1 |
| E325Q/Cys-148 | 20 | 50 | 632 | 8.1 |
| E325D/Cys-148 | 139 | 512 | 6,234 | 8.2 |
| R302A/Cys-148 | 47 | 76 | 707 | 8.5 |
| R302K/Cys-148 | 125 | 436 | 1,001 | 9.1 |
| H322A/Cys-148 | 1,678 | 1,410 | 2,151 | |
| H322N/Cys-148 | 945 | 861 | 1,195 | |
| H322Q/Cys-148 | 1,189 | 1,109 | 1,475 | |
| E269D/Cys-148 | 14,483 | 4,550 | 7,575 | |
NEM-labeling experiments were carried out at pH 5.5, 7.5, and 9.5 (see Fig. 2), and KD and pKa values were determined as described in Experimental Procedures. The values shown represent the average of two or three determinations, with standard errors of 20% or less.
Replacements for Arg-302.
Relative to single-Cys-148 permease, R302A/Cys-148 permease exhibits a small increase in KD from pH 5.5 to 7.5 (47 and 76 μM, respectively), whereas at pH 9.5, about a 15-fold increase (707 μM) is observed relative to pH 5.5 (Fig. 2; Table 1). A less pronounced, but marked pH dependence is observed with the conservative Lys replacement, and KD increases from 125 μM at pH 5.5 to 436 μM at pH 7.5 to 1 mM at pH 9.5 (Fig. 2; Table 1). It is also noteworthy that the apparent pKa for ligand binding is shifted to ca. 8.5 and 9.1 in R302A/Cys-148 and R320K/Cys-148 permease, respectively.
Replacements for His-322.
Replacement of His-322 with Ala, Asn, or Gln causes an increase in KD to 1.2–2.2 mM (Fig. 2; Table 1), but surprisingly, no significant change is observed from pH 5.5 to 9.5. Thus, KD at pH 9.5 with each mutant is only 1.3 to 1.5 times higher than at pH 5.5 or 7.5, which stands in stark contrast to the changes observed with single-Cys-148 or the Glu-325 and Arg-302 mutants.
Asp Replacement for Glu-269.
The only replacement for Glu-269 that exhibits any transport activity (24, 25) or ligand binding (17) (P. Venkatesan and H.R.K., data not shown) whatsoever is Asp. With respect to the effect of pH on binding affinity, mutant E269D/Cys-148 behaves in a manner similar to that observed for the His-322 mutants. Thus, the KD is increased, and there is no significant effect of pH from 7.5 to 9.5 (Fig. 2). The relatively high KD observed at pH 5.5 may be caused by partial neutralization of the negative charge at position 269, as single-Cys-148 permease with neutral replacements for Glu-269 exhibit no significant protection against alkylation at very high ligand concentrations (17) (P. Venkatesan and H.R.K., unpublished observations).
Discussion
Because lac permease in the absence of substrate does not translocate H+, and a substrate concentration gradient in and of itself generates a proton electrochemical gradient, it seems clear that the primary trigger for turnover must be binding and dissociation of substrate on opposite sides of the membrane. Therefore, to understand the mechanism, it is essential to determine the relationship between substrate affinity and the protonation state of the residues directly involved in H+ translocation and coupling. In the present paper, the effect of pH on TDG binding was examined in single-Cys-148 permease without or with conservative or neutral replacements of Glu-325, Arg-302, His-322, or Glu-269, four residues that are irreplaceable for H+ translocation and/or coupling between sugar and H+ translocation (see ref. 12). Although affinity per se is compromised in certain mutants, it remains relatively constant from pH 5.5 to 7.5. However, at alkaline pH, distinct patterns emerge. (i) Glu-325 replacements (Asp or Gln) exhibit a 30- to 40-fold decrease in affinity at pH 9.5 with a pKa of about 8, as observed with single-Cys-148 permease. (ii) Arg-302 replacements (Lys or Ala) exhibit a 10- to 15-fold decrease in affinity with a pKa of 8.5–9.0. (iii) Surprisingly, His-322 replacements (Ala, Asn, or Gln) or Glu-269 replacement with Asp show essentially no change in affinity as a function of pH. The results argue strongly that the permease must be protonated to bind ligand with high affinity and that His-322 and Glu-269 mediate the effects of pH on ligand affinity. Furthermore, because the apparent pKa of His-322, as judged from titrations with the Glu-325 and Arg-302 mutants, is perturbed (i.e., >8.0), it seems reasonable to conclude that the H+ is shared between His-322 and Glu-269 (Fig. 1). In conjunction with the notion that Glu-269 also might be involved in sugar binding (9) and evidence that the face of helix VIII with Glu-269 undergoes a ligand-induced conformational change (26), the important corollary of this conclusion is that His-322/Glu-269 might couple H+ translocation to substrate binding and dissociation during the transport cycle.
Previous observations (reviewed in ref. 27) demonstrate that lac permease mutants with neutral replacements for Glu-325 are specifically defective in all translocation modes that involve net H+ movement, but bind ligand and catalyze equilibrium exchange and counterflow as well or better than wild type. Therefore, Glu-325 must play a direct role in H+ translocation, although the present findings demonstrate clearly that the protonation state of this carboxylate is not involved with the affinity of the permease for substrate.
Taken together, the observations offer a mechanistic model for lactose/H+ symport. In the ground state (Fig. 1), the permease is protonated, and the H+ is shared between His-322 and Glu-269, and Glu-325 is charge-paired with Arg-302. In this conformation, the permease binds ligand at the interface between helices IV (Glu-126) and V (Arg-144 and Cys-148) at the outer surface of the membrane with relatively high affinity. Substrate binding induces a conformational change that leads to transfer of the H+ from His-322/Glu-269 to Glu-325 and reorientation of the binding site to the inner surface with a decrease in affinity. Glu-325 then is deprotonated on the inside because of rejuxtaposition with Arg-302 as the conformation relaxes. The His-322/Glu-269 complex then is reprotonated from the outside surface to reinitiate the cycle.
Acknowledgments
We acknowledge Pushpa Venkatesan for initiating this work and give special thanks to Joseph Runner for technical assistance. This work was supported in part by National Institutes of Health Grant DK51131 to H.R.K. and National Institute of Neurological Disorders and Stroke Grant NS07065 to A.K.
Abbreviations
- lac permease
lactose permease
- TDG
β,d-galactopyranosyl 1-thio-β,d-galactopyranoside
- NEM
N-ethylmaleimide
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.200351797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.200351797
References
- 1.Müller-Hill B. The lac Operon: A Short History of a Genetic Paradigm. Berlin: de Gruyter; 1996. [Google Scholar]
- 2.Kaback H R. J Cell Physiol. 1976;89:575–593. doi: 10.1002/jcp.1040890414. [DOI] [PubMed] [Google Scholar]
- 3.Kaback H R. J Membr Biol. 1983;76:95–112. doi: 10.1007/BF02000610. [DOI] [PubMed] [Google Scholar]
- 4.Frillingos S, Sahin-Tóth M, Wu J, Kaback H R. FASEB J. 1998;12:1281–1299. doi: 10.1096/fasebj.12.13.1281. [DOI] [PubMed] [Google Scholar]
- 5.Kaback H R, Wu J. Acc Chem Res. 1999;32:805–813. [Google Scholar]
- 6.Frillingos S, Gonzalez A, Kaback H R. Biochemistry. 1997;36:14284–14290. doi: 10.1021/bi972314d. [DOI] [PubMed] [Google Scholar]
- 7.Venkatesan P, Kaback H R. Proc Natl Acad Sci USA. 1998;95:9802–9807. doi: 10.1073/pnas.95.17.9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sahin-Tóth M, le Coutre J, Kharabi D, le Maire G, Lee J C, Kaback H R. Biochemistry. 1999;38:813–819. doi: 10.1021/bi982200h. [DOI] [PubMed] [Google Scholar]
- 9.Sahin-Tóth M, Akhoon K M, Runner J, Kaback H R. Biochemistry. 2000;39:5097–5103. doi: 10.1021/bi0000263. [DOI] [PubMed] [Google Scholar]
- 10.Zhao M, Zen K-C, Hubbell W, Kaback H R. Biochemistry. 1999;38:7407–7412. doi: 10.1021/bi9906524. [DOI] [PubMed] [Google Scholar]
- 11.Wolin C D, Kaback H R. Biochemistry. 2000;39:6130–6135. doi: 10.1021/bi0001269. [DOI] [PubMed] [Google Scholar]
- 12.Kaback H R. Proc Natl Acad Sci USA. 1997;94:5539–5543. doi: 10.1073/pnas.94.11.5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frillingos S, Kaback H R. Biochemistry. 1996;35:3950–3956. doi: 10.1021/bi952601m. [DOI] [PubMed] [Google Scholar]
- 14.Jung H, Jung K, Kaback H R. Biochemistry. 1994;33:12160–12165. doi: 10.1021/bi00206a019. [DOI] [PubMed] [Google Scholar]
- 15.Wu J, Kaback H R. Biochemistry. 1994;33:12166–12171. doi: 10.1021/bi00206a020. [DOI] [PubMed] [Google Scholar]
- 16.van Iwaarden P R, Pastore J C, Konings W N, Kaback H R. Biochemistry. 1991;30:9595–9600. doi: 10.1021/bi00104a005. [DOI] [PubMed] [Google Scholar]
- 17.He M, Kaback H R. Biochemistry. 1997;36:13688–13692. doi: 10.1021/bi9715324. [DOI] [PubMed] [Google Scholar]
- 18.Ho S N, Hunt H D, Horton R M, Pullen J K, Pease L R. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 19.Consler T G, Persson B L, Jung H, Zen K H, Jung K, Prive G G, Verner G E, Kaback H R. Proc Natl Acad Sci USA. 1993;90:6934–6938. doi: 10.1073/pnas.90.15.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teather R M, Bramhall J, Riede I, Wright J K, Furst M, Aichele G, Wilhelm V, Overath P. Eur J Biochem. 1980;108:223–231. doi: 10.1111/j.1432-1033.1980.tb04715.x. [DOI] [PubMed] [Google Scholar]
- 21.Kaback H R. Methods Enzymol. 1971;XXII:99–120. [Google Scholar]
- 22.Short S A, Kaback H R, Kohn L D. J Biol Chem. 1975;250:4291–4296. [PubMed] [Google Scholar]
- 23.Frillingos S, Kaback H R. Protein Sci. 1997;6:438–443. doi: 10.1002/pro.5560060221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ujwal M L, Sahin-Tóth M, Persson B, Kaback H R. Mol Membr Biol. 1994;1:9–16. doi: 10.3109/09687689409161024. [DOI] [PubMed] [Google Scholar]
- 25.Franco P J, Brooker R J. J Biol Chem. 1994;269:7379–7386. [PubMed] [Google Scholar]
- 26.Frillingos S, Ujwal M L, Sun J, Kaback H R. Protein Sci. 1997;6:431–437. doi: 10.1002/pro.5560060220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaback H R. Biochemistry. 1987;26:2071–2076. doi: 10.1021/bi00382a001. [DOI] [PubMed] [Google Scholar]