

Abstract
The Old Yellow Enzyme has been shown to catalyze efficiently the NADPH-linked reduction of nitro-olefins. The reduction of the nitro-olefin proceeds in a stepwise fashion, with formation of a nitronate intermediate that is freely dissociable from the enzyme. The first step involves hydride transfer from the enzyme-reduced flavin to carbon 2 of the nitro-olefin. The protonation of the nitronate at carbon 1 to form the final nitroalkane product also is catalyzed by the enzyme and involves Tyr-196 as an active site acid/base. This residue also is involved in aci-nitro tautomerization of nitroalkanes, the first example of a nonredox reaction catalyzed by the enzyme.
Old Yellow Enzyme (OYE; EC 1.6.99.1), isolated from brewer's bottom yeast, was the first enzyme where the requirement of a prosthetic group (cofactor) for catalysis was demonstrated (1). OYE is a mixture of homodimers and a heterodimer arising from two yeast genes, with each monomer binding a molecule of flavin mononucleotide. Although OYE's exact physiological function is still unknown, many tight binding ligands such as phenols and steroids have been identified, raising the possibility that this enzyme might be involved in yeast sterol metabolism (2). Upon binding with phenols and heteroaromatic compounds containing ionizable hydroxyl groups OYE produces characteristic long wavelength charge transfer complexes (3).
The crystal structure of OYE has been solved, revealing several amino acid residues around the active site of the enzyme to effect catalysis and ligand binding (4). Phenolic compounds are held parallel to the flavin by hydrogen bonding of the phenolate oxygen with His-191 and Asn-194. The long wavelength absorbance band arises from parallel stacking interaction between the phenolate anion and the flavin. Several mutants of OYE have been constructed and confirmed the importance of His-191, Asn-194, Tyr-196, and Thr-37 residues in ligand binding interactions and catalysis (5–7). We also have discovered that this enzyme catalyzes the reduction of the olefinic bond of α,β-unsaturated carbonyl compounds by using NADPH as a reductant (2, 8) and also catalyzes slow aromatization of cyclic enones like cyclohexenone by a novel dismutation reaction (8). Using R- (2H) NADPH in D2O it was unequivocally established that reduction of α,β-unsaturated carbonyl compounds proceeds via stereospecific transfer of hydride to the carbon atom β to the carbonyl function and transfer a proton to the α position, resulting in trans addition to the double bond (8). Another class of compounds similar to α,β-unsaturated carbonyls is that of unsaturated nitro compounds. Like a carbonyl group a nitro group exerts a strong electron attracting influence within the molecule, enhancing the acidity of the hydrogen atoms attached to the carbon α to the substituent group. Nitro compounds also exhibit a tautomerism exactly analogous to keto-enol tautomerism. Chiral nitro compounds are an important class of synthetically useful compounds, which can be easily converted into chiral amines, and chiral amines are useful intermediates in organic synthesis (9). In this study we have undertaken a systematic study of the reaction of OYE with unsaturated nitro compounds.
During the study of the reduction of unsaturated nitro compounds we observed an intermediate product that was characterized as a nitronate. We have established that in the case of unsaturated nitro compounds hydride transfer from reduced flavin occurs before proton transfer, resulting in the intermediate nitronate. The uptake of a proton by the intermediate nitronate also is catalyzed by OYE. The formation of nitronate from a saturated nitro compound also is catalyzed by OYE, and both proton abstraction by nitronate and proton release to form the nitronate are mediated by tyrosine residue 196 of OYE acting as a general acid/base catalyst. The pKa of this process is ≈9.1, independent of the nature of the substrate nitro compound, and is ascribed to that of the active site tyrosine residue.
Materials and Methods
Preparation of OYE1.
Wild-type and mutant enzyme forms were prepared as described (2, 5, 7). NADPH, NADP, glucose-6-phosphate, and glucose-6-phosphate dehydrogenase (from Leuconostoc mesenteroides) were from Sigma. 1-Nitro-1-cyclohexene, trans-2-(2-nitrovinyl)thiophene, trans-β-nitrostyrene, nitrocyclohexane, and nitrocyclopentane were from Aldrich. Dihydronitrostyrene (nitroethylbenzene) was synthesized according to the published procedure (10).
Spectroscopic Studies.
UV-visible absorbance spectra were recorded with a Hewlett–Packard Diode Array spectrophotometer or a Cary model 219 double-beam spectrophotometer. 1H NMR spectra were recorded with a Bruker 500-Mhz NMR instrument. Chemical shift values are reported in parts per million relative to tetramethylsilane.
Steady-State Kinetics Experiments.
To determine the steady-state kinetic parameters it is necessary to exclude oxygen from the system. We therefore used the capabilities of a stopped-flow spectrophotometer to mix together both NADPH and nitro-olefins and follow anaerobically the course of the reaction. With nitrocyclohexene as substrate, 3.91 μM OYE1 (all concentrations given are those after mixing) was mixed in one set of experiments with 64 μM NADPH and 150, 200, 250 and 350 μM nitrocyclohexene, and the oxidation of NADPH was followed at 340 nm. Turnover numbers were estimated from the time interval changes during the reaction. In a second set of experiments the concentration of nitrocyclohexene was limiting (initially 50 μM) and the NADPH was in excess (213 and 320 μM). Under these conditions the enzyme is largely in the oxidized form during turnover and becomes reduced as the nitrocyclohexene is exhausted. The reaction was followed at 340 and 462 nm, and traces at the latter wavelength were analyzed as described (11). The traces at both NADPH concentrations were identical, because the NADPH concentrations were >20 times the Km value. For nitrostyrene and nitrovinylthiophene, similar enzyme-monitored turnover experiments were carried out, with an initial nitro-olefin concentration of 50 μM and concentrations of NADPH of 213, 267, and 320 μM. All reactions were carried out in 0.05 M KPi buffer, pH 7.0 at 25°, using a HiTech SF-61 stopped-flow spectrophotometer. Anaerobiosis was achieved as described (7).
Rapid Reaction Studies of Oxidative Half Reaction.
OYE1 was made anaerobic in a tonometer and reduced slowly with an NADPH-generating system as described (7). Reoxidation of the reduced enzyme was followed at 462 nm and other selected wavelengths on mixing with different concentrations of nitrostyrene and nitrovinylthiophene that had been bubbled in syringes with O2-free argon before introduction into the stopped-flow spectrophotometer. Reaction traces were analyzed as described (7). All reactions were carried out in 0.05 M KPi buffer, pH 7.0, at 25°C.
Kinetics of Nitronate Formation and Decay from Nitro-Olefins.
These reactions were carried out in 0.05 M KPi, pH 7.0 at 25°C, using a Hewlett–Packard 8452A Diode Array spectrophotometer. In a typical experiment the reaction cuvette contained buffer, 2 mM glucose 6-phosphate, 8 μM NADP, 10 units glucose 6-phosphate dehydrogenase, and approximately 0.1 μM OYE. After equilibration at 25°C the cuvette was set as the spectophotometric blank, and the time course of the reaction was followed by introduction of 10 μl of nitro-olefin. The reduction of the nitro-olefin was followed by the disappearance of its absorbance spectrum (λmax = 270 nm for nitrocyclohexene, ɛ270 ≈ 6,100 M−1⋅cm−1; λmax 320 nm for nitrostyrene, ɛ320 = 15,500 M−1⋅cm−1; λmax = 364 nm for nitrovinylthiophene, ɛ364 = 17,900 M−1⋅cm−1), and the formation and decay of the intermediate nitronate was followed at its λmax (230 nm for cyclohexane nitronate, ɛ230 ≈ 13,700 M−1⋅cm−1; λmax 236 nm for dihydrostyrene nitronate, ɛ236 ≈ 15,000 M−1⋅cm−1; λmax 236 nm for thiophene ethylnitronate, ɛ236 ≈ 28,000 M−1⋅cm−1). For experiments involving solvent kinetic isotope effects, the standard buffer was evaporated to dryness and redissolved in 99.9% D2O. The stock glucose 6-phosphate was also in D2O, but the other components were in H2O.
Kinetics of Aci-Nitro Tautomerization.
Three saturated nitro compounds were used in these studies, nitrocyclohexane, nitrocyclopentane, and nitroethylbenzene. Typically, 1.0 ml of a 10 μM solution of the compound in H2O was adjusted to pH ≈11.5–12 by the addition of 5 μl 1 M NaOH, and the nonenzymatic formation of the nitronate was followed by the appearance of its typical spectrum with λmax in the 230- to 240-nm region until no further change occurred. Then the pH was readjusted to the desired value by the addition of 5–6 μl 1 M acetic acid plus 50 μl of the appropriate buffer (e.g., 1 M KPi, pH 7.0). The nonenzymatic return to the initial nitroalkane was followed for an appropriate time to establish a reliable rate and then a low concentration of OYE (typically 1–10 nM) was added and the spectra were recorded until the reaction came to an end. The difference in absorbance from the end point vs. time was plotted on semilog paper and uniformly resulted in linear plots, from which kobs values for both the nonenzymatic and the enzyme-catalyzed reactions could be calculated. The kobs values for the enzyme-catalyzed reaction were directly proportional to the enzyme concentration, permitting the estimation of a second-order rate constant for the enzyme reaction.
For the enzyme-catalyzed formation of the nitronate, a solution of the nitro compound was made up in the appropriate buffer, and the nonenzymatic reaction was followed for 10–20% of the total change, followed by the addition of 1–10 nM enzyme. Again, after correction for the noncatalyzed reaction, a second-order rate constant for the enzyme-catalyzed reaction could be determined.
Results and Discussion
Detection of an Intermediate in the OYE-Catalyzed Reduction of Nitro-Olefins.
By use of an NADPH-generating system consisting of NADP, glucose 6-phosphate and glucose 6-phosphate dehydrogenase, it was possible to monitor the spectral changes undergone by nitrocyclohexene (λmax 270 nm, ɛ ≈ 6,100 M−1⋅cm−1) during catalytic turnover (Fig. 1A). In the initial stages of the reaction the reduction of nitrocyclohexene, shown by the decrease in absorbance at 270 nm, is accompanied by an increase in absorbance in the 230-nm region, representing the formation of a transient intermediate, identified as the nitronate form of nitrocyclohexane (Scheme S1). The nature of the intermediate was shown in various ways, the most dramatic being companion experiments using the Y196F mutant form of the enzyme. Now the disappearance of the nitrocyclohexene absorbance at 270 nm is accompanied solely by the appearance of product with maximal absorbance at 230 nm (Fig. 1B). In this experiment, after full reduction of nitrocyclohexene there was a very slow further conversion of the 230-nm intermediate to the final product, the normal protonated form of nitrocyclohexane, with maximal absorbance in the far UV region, λmax 205 nm. It is evident from comparison of Fig. 1 A and B that whereas wild-type enzyme and the Y196F form catalyze the reduction of nitrocyclohexene at approximately the same rate, the Y196F mutant enzyme is very inefficient in catalyzing the further reaction of the intermediate. Indeed, the slow disappearance of the intermediate observed with Y196F is the same as that of the nonenzymatic protonation of the nitronate. Similar results were obtained in the NADPH-nitro-olefin reductase reactions with nitrostyrene and nitrovinylthiophene, except that with these compounds the nitronate intermediates undergo much faster nonenzymatic decay to the saturated nitro compounds (results not shown).
Figure 1.

Formation and decay of the nitronate intermediate of nitrocyclohexane from nitrocyclohexene. See text for details. (A) In H2O solution, selected spectra after the addition of 9.5 × 10−8 M OYE1 to a 74 μM solution of nitrocyclohexene in the presence of an NADPH-generating system. (B) Same as A, except that 1 × 10−7 M OYE1 Y196F was used. (C) Same as A, except that the reaction was in 99% D2O.
Scheme 1.

Solvent Isotope Effect on the Decay of the Nitronate Intermediate.
The inability of Y196F to catalyze the protonation of the nitronate intermediate (Scheme S1) clearly implicates Tyr-196 as the active site acid responsible for providing the second hydrogen involved in the overall reaction, with the primary addition arising from hydride transfer from the reduced flavin, and ultimately originating from NADPH, as shown previously in the stereo-specific reduction of α,β-unsaturated carbonyl compounds (8). The hydroxyl residue of Tyr-196 would be expected to exchange readily with solvent, because the previous stereochemical analysis had shown that deuterium was incorporated specifically at the α-carbon when reduction of cinnamaldehyde was carried in D2O (8). In accord with this expectation the disappearance of the 230-nm intermediate was slowed down very significantly when the reaction was carried out in 99% D2O, as illustrated in Fig. 1C. In accord with the reactions shown in Scheme S1, the rate of reduction of nitrocyclohexene is little affected, but the disappearance of the intermediate is slowed down by a factor of ca 3.5, resulting in its greater accumulation before its ultimate decay.
Kinetics of the Oxidative Half Reaction of Reduced OYE by Unsaturated Nitro Compounds.
The reaction kinetics of enzyme reoxidation and nitronate formation can be followed conveniently in anaerobic stopped-flow experiments by monitoring the reoxidation of reduced OYE at 462 nm by unsaturated nitro compounds. Results are summarized in Table 1 for three such compounds, nitrocyclohexene, nitrostyrene, and nitrovinylthiophene. The reactions are very rapid, with the observed rate constant for nitrocyclohexene being within experimental error directly proportional to the nitrocyclohexene concentration (6). With the other compounds saturation kinetics are observed, with the limiting rate constants listed in Table 1 together with the apparent dissociation constants for the reduced enzyme-substrate complex. Because the limiting rate constant for reduction of OYE1 by NADPH is 5.1 ± 0.1 s−1 (5) it is clear that under most conditions the rate of catalytic reduction of unsaturated nitro compounds will be limited by the reduction of the enzyme by NADPH.
Table 1.
Kinetic and thermodynamic constants for the reoxidation of reduced OYE1 by nitro-olefins

Steady-State Kinetics.
That the rate-limiting step in the catalytic reduction of the nitro compounds was indeed the reduction of the enzyme flavin by NADPH was confirmed by the results of enzyme-monitored turnover experiments, analyzed as described (11). The kinetic constants obtained are shown (Table 2) and are in satisfactory agreement with values predicted from the rate constants for reduction of the enzyme-bound flavin mononucleotide by NADPH (5) and those for the reoxidation of the reduced flavin by the nitro compound, as listed in Table 1.
Table 2.
Steady-state kinetics
| Nitro compound | kcat, s−1 | Km(NADPH), μM | Km(nitro), μM |
|---|---|---|---|
| Nitrocyclohexene | 4.4 ± 0.2 | 12 ± 2 | 10 ± 1 |
| Nitrostyrene | 5.0 ± 0.1 | ND | 4.2 ± 0.3 |
| Nitrovinylthiophene | 4.3 ± 0.2 | ND | 7.7 ± 0.5 |
The kinetic constants were obtained from enzyme-monitored turnover experiments carried out in 0.05 M KPi buffer, pH 7.0, 25°C, and analyzed as described (11). The Km(NADPH) values with nitrostyrene and nitrovinylthiophene were in the low μM region, but could not be determined accurately because of the overlapping spectra of the nitrocompounds with that of the NADPH. In the case of nitrocyclohexene there was essentially no overlap, and the Km(NADPH) was determined from analysis of the absorbance changes at 340 nm.
For the NADPH-nitrocyclohexene reductase reaction, the reoxidation of reduced enzyme by nitrocyclohexene is a second-order reaction, so the kinetic mechanism is modified ping-pong:
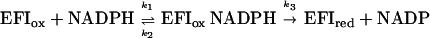 |
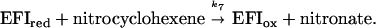 |
For such a mechanism, kcat = k3, Km NADPH) = (k2 + k3)/k1 and Km (nitro) = k3/k7. Thus, the predicted value of kcat is 5.1 ± 0.1 s−1, and the predicted value for Km (nitro) is 8.6 ± 0.2 μM, both in reasonable agreement with the experimentally determined values.
For nitrostyrene and nitrovinyl thiophene, limiting rate constants for reoxidation of the reduced flavin and Kd values for binding could be determined (Table 1). Thus, the kinetic mechanism for these compounds is standard ping pong:
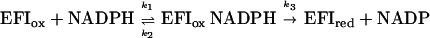 |
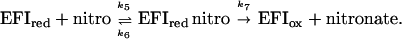 |
For such a mechanism, kcat = k3k7/(k3 + k7), Km (NADPH) = k7(k2 + k3)/k1(k3 + k7) and Km (nitro) = k3(k6 + k7)/k5 (k3 + k7). The steady-state kcat values are again in reasonable agreement with prediction, as are the Km values for the nitro compound.
Catalysis of Aci-Nitro Tautomerization.
The nature of the 230-nm absorbing intermediate as the nitronate form of nitrocyclohexane (see Scheme S1) and the catalysis of its formation and protonation by OYE was shown by independent experiments as described below. It is well known that nitro compounds exhibit aci-nitro tautomerization (Scheme S2), whose chemistry has been reviewed extensively (12). Nitrocyclohexane and similar compounds undergo slow spontaneous deprotonation in basic media. Thus it is possible to prepare the nitronate species on adjusting the pH of a nitrocyclohexane solution to a value of 11, and the process can be followed by the appearance of the characteristic absorbance maximum at 230 nm. This process is complete in ≈30 min at 25°C. On readjusting the pH to a lower value, e.g., 7.0, the nitronate is protonated rapidly to give the aci-nitro form (nitronic acid), but equilibration back to the nitrocyclohexane (λmax ≈ 205 nm) occurs extremely slowly, with a t1/2 value of ca. 200 min in the presence of 0.05 M phosphate buffer, equivalent to a second-order rate constant of 5.8 × 102 M−1⋅s−1 with respect to hydrogen ion concentration. Addition of OYE1 to such a solution results in rapid conversion to nitrocyclohexane, also in an exponential reaction strictly proportional to the enzyme concentration, with a rate constant of 1 ± 0.1 × 105 M−1⋅s−1 with respect to enzyme concentration. When such an experiment was carried out under identical conditions in 99% D2O, the nonenzymic formation of nitronate under basic conditions was unaffected, but on reneutralization both the nonenzymic reprotonation and that catalyzed by OYE1 showed a H2O/D2O isotope effect of 5.5 ± 0.3. Addition of Y196F enzyme instead of OYE resulted in no increase in rate beyond that of the spontaneous conversion. Similarly, the H191N and apoenzyme forms did not catalyze the decay of the nitronate.
Scheme 2.

The nitronate forms of nitrocyclopentane and nitroethylbenzene were formed in a similar fashion to that described above, and again OYE1, but not the mutant enzyme forms, was found to catalyze the decay. Rate constants for the reactions are given in Table 3.
Table 3.
OYE-catalyzed aci-nitro tautomerization
| Substrate | Formation of nitronate, 105 M−1⋅s−1 | Decay of nitronate, 105 M−1⋅s−1 |
|---|---|---|
| Nitrocyclohexane | 1.25 | 0.98 (pH 7.5) |
| Nitrocyclopentane | 1.45 | 0.056 (pH 6.6) |
| Nitroethylbenzene | 7.0 | 1.50 (pH 7.5) |
The rate constants for formation of the nitronates were the limiting values obtained at high pH as illustrated in Fig. 3. The values for the decay of the nitronates back to the original protonated forms were obtained at the pH values shown.
OYE also was found to catalyze the deprotonation of nitrocyclohexane to the nitronate. This can be demonstrated conveniently by following the reaction at pH 8.5, close to the pKa of nitrocyclohexane (pKa = 8.65), where the spontaneous deprotonation reaction is very slow. The addition of OYE results in rapid formation of the nitronate, again in an exponential progress curve. The observed rate constants depended on pH (see next section).
pKa Values of Nitro/Nitronate Forms and Enzyme Catalysis.
The pKa values for the aci-nitro tautomerization of nitro compounds can be determined from the absorbance in the 230- to 240-nm region at equilibrium. This may be done laboriously by following the sometimes very slow spontaneous conversions at various pH values, but is made much simpler in the presence of catalytic concentrations of OYE, where equilibrium is reached rapidly. The pKa for the aci-nitro tautomerization of nitrocyclopentane was not available from the literature and therefore was determined by measuring the concentration of the nitronate at 228 nm in the presence of OYE. Fig. 2 shows the pH vs. absorbance plot, from which the pKa of 7.8 was ascertained. Table 4 summarizes the results of such titrations also with nitrocyclohexane and nitroethylbenzene, with values in substantial agreement with literature values (13, 14).
Figure 2.

Determination of the pKa of the nitronate form of nitrocyclopentane. Solutions of nitrocyclopentane (100 μM) were prepared in different buffer solutions, and the absorbance difference from that of the buffer that was obtained after the addition of ≈1 × 10−8 M OYE1 was plotted vs. the measured pH. The curve is a theoretical one for a pKa of 7.8.
Table 4.
pKa values for nitronates and for OYE1-catalyzed nitronate formation
| Substrate | pKa | pKoye |
|---|---|---|
| Nitrocyclohexane | 8.65 | 9.15 |
| Nitrocyclopentane | 7.80 | 9.10 |
| Nitroethylbenzene | 8.75 | 9.10 |
The rates of formation of the nitronate species also were followed by recording the absorbance increase at 230 nm with fixed concentrations of enzyme and nitro compound. Fig. 3 shows the determination of the pKa of the enzyme-catalyzed reaction for the formation of cyclopentane nitronate. Values for the similar reactions with nitrocyclohexane and nitroethylbenzene also are shown in Table 4. Although the determined pKa values for OYE-catalyzed nitronate formation for nitrocyclohexane and nitroethylbenzene are similar to the pKa values for the uncatalyzed reaction, the low pKa value of aci-nitro tautomerization of nitrocyclopentane is clearly much lower than that of the enzyme-catalyzed reaction. We therefore ascribe the pKa of approximately 9.1 as that of tyrosine residue 196, in keeping with the requirement for an unprotonated enzyme residue, and the fact that the Y196F mutant enzyme does not catalyze these reactions.
Figure 3.

The effect of pH on the OYE1-catalyzed formation of cyclopentane nitronate from nitrocyclopentane. The second-order rate constants for the enzyme-catalyzed reaction were those calculated after subtraction of the uncatalyzed blank reactions. See text for details.
NMR Analysis of Reaction Products.
The reaction products of the OYE-catalyzed reduction of nitrocyclohexene, nitrostyrene, and nitrovinylthiophene clearly showed the formation of nitrocyclohexane, nitroethylbenzene, and nitroethylthiophene, respectively, as judged from the agreement of the obtained 1H NMR spectra with those of published values (results not shown). To establish the stereochemistry of the reaction by NMR analysis, the OYE-catalyzed reduction of nitrostyrene was carried out in H2O- and D2O-buffered solution. The 1H NMR spectrum of the product, nitroethylbenzene, from reaction in H2O shows that both carbon 1 and carbon 2 are associated with two protons (triplet at 4.65–4.62 ppm and triplet at 3.36–3.33 ppm, respectively). In D2O-buffered solution, 1H NMR data show that carbon 2 is associated with two protons (doublet at 3.35–3.32 ppm) and that carbon 1 is associated with one proton (multiplet at 4.65–4.58 ppm). These experiments clearly show that in D2O-buffered solution a deuterium atom is abstracted from the solvent by the intermediate nitronate after hydride transfer from the reduced flavin at the carbon 2 position. Similar results were obtained for the enzyme-catalyzed reprotonation of the nitronate of nitroethylbenzene, demonstrating that carbon 1 of the product is the only position receiving a proton from the solvent, and consistent with the previously demonstrated lack of solvent exchange with the N(5)H of the reduced flavin of the enzyme (8).
Conclusions
Although we have not specifically addressed the stereochemistry of the reduction of unsaturated nitrocompounds in this work, it would seem reasonable to conclude from previous work (8) that reduction proceeds via a trans addition across the double bond, as outlined in Scheme S3. In the crystal structure of OYE1, if nitrocyclohexene is modeled into the active site in the same way as was found experimentally with p-hydroxy-benzaldehyde (4), with a nitro oxygen hydrogen bonded to His-191 and Asn-194 (Fig. 4), carbon 2 of the nitrocyclohexene can be located ideally to receive a hydride from the N-5 of the reduced flavin, and carbon 1 positioned ideally to receive a proton from the overlying Tyr-196 hydroxyl, to give a trans-addition across the double bond, analogous to the reactions with α,β-unsaturated carbonyl compounds.
Scheme 3.

Figure 4.

The active site of OYE1 is displayed with nitrocyclohexene positioned such that the nitro group is H-bonded to His-191 and Asn-194 and the hydroxyl of Tyr-196 positioned to protonate carbon 1 of the cyclohexene ring. The coordinates of the residues were taken from the crystal structure of OYE1 with p-hydroxybenzaldehyde bound (Protein Databank ID code 1OYB). Nitrocyclohexene replaces p-hydroxybenzaldehyde in the active site. FMN, Flavin mononucleotide. The graphic was constructed by using the Insight II Molecular Modeling System from Biosym Technologies, San Diego.
In the present study the role of Tyr-196 as a proton donor to carbon 1 of unsaturated nitro compounds has been shown unequivocally. The Y196F mutant enzyme catalyzes the NADPH-nitrocyclohexene reductase reaction just as efficiently as the wild-type enzyme, but unlike the latter is not able to catalyze the protonation of the nitronate to nitrocyclohexane. The details of the reaction are illustrated in Scheme S1. The first step is the transfer of a hydride equivalent from the N(5)H of the reduced flavin to carbon 2 of nitrocyclohexene, resulting in the formation of the nitronate. Binding of the nitronate is quite weak, so that it is released readily into the solution. The second step involves protonation of the nitronate at carbon 1 and requires the protonated form of Tyr-196 for efficient catalysis. OYE thus has been found to participate also in nonredox reactions, the reversible deprotonation of saturated nitrocompounds to the nitronate forms and reprotonation of the latter.
Acknowledgments
We are indebted to Dr. Bette J. Brown and Mr. Rahul Kohli for gifts of OYE1 mutant forms, H191N and Y196F, and to Drs. Sumita Chakraborty, Bruce A. Palfey, and Bette J. Brown for valuable discussion. We acknowledge Dr. Y. S. V. N. Murthy for the initial finding that nitro-olefins were substrates for OYE. The research was supported by a grant from the U. S. Public Health Service, GM-11106.
Abbreviation
- OYE
Old Yellow Enzyme
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.190345597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.190345597
References
- 1.Theorell H. Biochem Z. 1935;275:344–346. [Google Scholar]
- 2.Stott K, Saito K, Thiele D J, Massey V. J Biol Chem. 1993;268:6097–6106. [PubMed] [Google Scholar]
- 3.Abramovitz A S, Massey V. J Biol Chem. 1976;251:5321–5326. [PubMed] [Google Scholar]
- 4.Fox K M, Karplus P A. Structure (London) 1994;2:1089–1105. [PubMed] [Google Scholar]
- 5.Brown B J, Deng Z, Karplus P A, Massey V. J Biol Chem. 1998;273:32753–32762. doi: 10.1074/jbc.273.49.32753. [DOI] [PubMed] [Google Scholar]
- 6.Kohli R M, Massey V. J Biol Chem. 1998;273:32763–32770. doi: 10.1074/jbc.273.49.32763. [DOI] [PubMed] [Google Scholar]
- 7.Xu D, Kohli R M, Massey V. Proc Natl Acad Sci USA. 1999;96:3556–3561. doi: 10.1073/pnas.96.7.3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaz A D N, Chakraborty S, Massey V. Biochemistry. 1995;34:4246–4256. doi: 10.1021/bi00013a014. [DOI] [PubMed] [Google Scholar]
- 9.Smith M B. Methods of Non-α-Amino Acid Synthesis. New York: Dekker; 1995. [Google Scholar]
- 10.Schechter H, Ley D E, Roberson E B. J Am Chem Soc. 1956;78:4984–4991. [Google Scholar]
- 11.Gibson Q H, Swoboda B E P, Massey V. J Biol Chem. 1964;239:3927–3934. [PubMed] [Google Scholar]
- 12.Nielson A T. In: The Chemistry of Nitro and Nitroso Groups, Part 1. Feuer H, editor. New York: Interscience; 1969. pp. 349–383. [Google Scholar]
- 13.Turyan Y I, Tyurin Y M, Zaitsev P M. Doklady Akad Nauk S S S R. 1960;134:850–852. [Google Scholar]
- 14.Bordwell F G, Bartness J E. J Org Chem. 1978;43:3101–3106. [Google Scholar]