

Abstract
A novel glycolipid was synthesized that corresponds to cholesteryl palmitoyl-galactopyranoside 1 found in the spirochete Borrelia burgdorferi, the causative agent of Lyme disease. In order to fashion 1 in a conjugatable form, the palmitoyl residue was modified to include a terminal aldehydo moiety that anchored the glycolipid to aminooxypropylated serum albumin using oxime chemistry. The glycolipoprotein so obtained incorporates an average of 18 glycolipid moieties per albumin molecule. The novel glycolipoprotein constructs are soluble in water and are candidates towards developing a semisynthetic vaccine against Lyme disease.
Keywords: Aminooxy, Borrelia burgdorferi, Conjugation, Glycolipid, Glycolipoprotein, Lyme disease, Oxime, Vaccine
Lyme disease (LD) is caused by the spiral-shaped Gram-negative bacterium Borrelia burgdorferi which belongs to the Spirochaetaceae family.1 LD was first reported in the United States in 1977 when a large number of children in and around the townships Lyme and Old Lyme in Connecticut were diagnosed as having juvenile rheumatoid arthritis. The disease can be manifested as a multi-system disorder affecting the skin, eyes, the joints, internal organs and the nervous system. LD has been recognized in Europe since the 1880's under various names such as erythema migrans (EM) and in 1948 EM biopsies revealed the presence of spirochetes.2 That LD is caused by B. burgdorferi was firmly established in 1982. While the progression of the symptoms of LD can be successfully treated with antibiotics, there is no vaccine available after the withdrawal from medical use of a vaccine (LYMErix, Smith Kline Beecham) in 2002 that was based on the outer surface lipoprotein of B. burgdorferi which is a single polypeptide chain consisting of 257 amino acids having covalently bound lipids at its N-terminus.
Borrelia are lacking lipooligo- and lipopolysaccharides characteristic of the cell-surface of most Gram-negative bacteria. A recently published work identified cholesteryl 6-O-palmitoyl-β-d-galactopyranoside 1 as a surface component of B. burgdorferi.3 Compound 1 was shown to elicit specific immune response when administered in Freund's adjuvant or in PBS.3 Because of this propensity, glycolipid 1 became a target for vaccine development. We reported chemical synthesis of 1 and that of a congener in which the palmitoyl group is replaced by an oleoyl group.4 The availability of the synthetic glycolipids dispelled any uncertainty that arose from the fact that the structural studies were performed on the inseparable mixture of 1 and its oleoyl congener, and paved the way towards the synthesis of a functionalized derivative suitable for covalent attachment to proteins for the purpose of enhancing its immunogenicity without the need for an adjuvant.
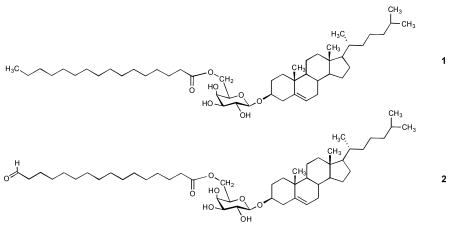
Here, we describe the synthesis of a bioconjugatable derivative (2) of glycolipid 1 and show that it can be covalently linked to aminooxypropylated bovine serum albumin (BSA)5 through oxime linkages, to yield a four-domain, water-soluble glycolipoprotein.
Our approach to the oxo derivative 2 was modeled after the reported synthesis of compound 1, relying on the triol4 3 as the starting material that was converted to the key intermediate 6 in a series of protection-deprotection steps. We reasoned that the use of a p-methoxybenzylidene acetal protection on HO-3 and 4 would optimize the overall yield because of the ease and selectivity for the construction and the cleavage of dioxolane-type benzylidene acetals.6 Thus, the triol4 3 was treated with p-methoxybenzaldehyde dimethyl acetal in the presence of camphorsulfonic acid to afford a diastereomeric mixture of the benzylidene acetals R,S-4 in 82% yield. (Scheme 1) That the dioxolane ring was, indeed, formed on O-3 and O-4 of the galactose moiety was shown by the appearance of the H-2 protons as two triplets of approximately equal intensity at 5.08 and 5.01 ppm (J ∼ 7.8 Hz for each) upon acetylation (Ac2O/C5H5N) of R,S-4. (Not described in the Experimental part.) Next, sodium hydride-mediated p-methoxybenzylaton using freshly prepared p-methoxybenzyl bromide afforded the fully protected intermediate R,S-5 in 91 % yield. We note that although the individual diastereomers were well-separated on TLC, no attempt was made to their isolation in stereochemically homogeneous form. Treatment of the silyl ether R,S-5 with tetrabutylammonium fluoride generated the alcohol R,S-6 as a diastereomeric mixture in 96 % yield, featuring the linkage site to the aldehydo palmitic acid moiety unprotected. Aldehydo palmitic acid derivative 7 was prepared from the commercially available 16-hydroxypalmitic acid as described for 6,6-dimethoxyhexanoic acid.7 Condensation of alcohol 6 with the acid 7 under promotion by dicyclohexylcarbodiimide and 4-dimethylaminopyridine afforded the fully protected key intermediate R,S-8. Following the assembly of the target, our attention turned to the removal of the protecting groups from the galactosyl and the palmitoyl moieties, respectively. Treatment of R,S-8 with trifluoroacetic acid selectively removed the anisylidene protecting group to afford compound 9 in 91 % yield. Next, the p-methoxybenzyl group was oxidatively removed by treatment with DDQ to generate the triol 10. Finally, acetic acid-mediated hydrolysis of the acetal moiety provided the targeted glycolipid derivative 2.
Scheme 1.

Reagents and conditions: (a) p-methoxybenzaldehyde dimethylacetal (6.5 eq), CSA (cat), CH2Cl2, 23 °C, 24 h, 82%; (b) p-methoxybenzyl bromide (4 eq), NaH, DMF, 0 → 23 °C, 40 min, 91%; (c) Bu4F, 23 °C, 2 h, THF, 96%; (d) 7 (1.7 eq), DCC (2.6 eq), DMAP (2.1 eq), 23 °C, 30 min, EtOAc, 93%; (e) TFA, 23 °C, 2 h, CH2Cl2, MeOH, 91%; (f) DCC (4.7 eq), 23 °C, 30 min, CH2Cl2, H2O, 82%.
Having accomplished the synthesis of the modified glycolipid derivative 2, our attention turned to its covalent attachment to proteins. We have shown that synthetic oligosaccharides containing an aldehydo or keto group in their aglyconic part can be efficiently linked to aminooxypropylated serum albumin through oxime linkages and that the conjugates so prepared induce anti-hapten antibodies in mice.5,8-10 Our initial attempts to link 2 with aminooxypropylated bovine serum albumin5 were hampered by the low solubility of 2 in most organic solvents and by its insolubility in aqueous media. Eventually, we envisaged the use of the “surfactant reversed micelle” technique in organic solvents.11,12 The method creates a microheterogeneous environment for the hapten and the albumin counterparts thus bringing the partners in close proximity for reaction. In applying this technique, a buffered aqueous solution of aminooxypropylated BSA was added to a solution of the anionic detergent sodium bis(2-ethylhexyl)sulfosuccinate in octane under stirring. A transparent, clear solution indicated the formation of reversed micelles. Next, aminooxypropyl-BSA was treated with the aldehydo-derivatized hapten 2 in pyridine to form the conjugate which was isolated by precipitation with alcohol followed by purification by size-exclusion chromatography. The solution of the conjugate so obtained was slightly opalescent but did not precipitate from the aqueous solution. In order to prove that hapten 2 was, indeed, covalently linked to BSA, the conjugate was subjected to SDS polyacrylamide gel eletrophoresis (SDS-PAGE) that confirmed the increase of the molecular mass of aminooxypropylated BSA upon treatment with the hapten 2. (Figure 1) Under our conditions, described in detail in the Experimental section, the average loading density was 18 glycolipid moieties per BSA molecule as determined by MALDI-TOF mass spectrometry. In a control experiment carried out under identical conditions, the parent glycolipid 1 failed to react with aminooxypropylated BSA as evidenced by SDS-PAGE and by mass spectrometry.
Figure 1.
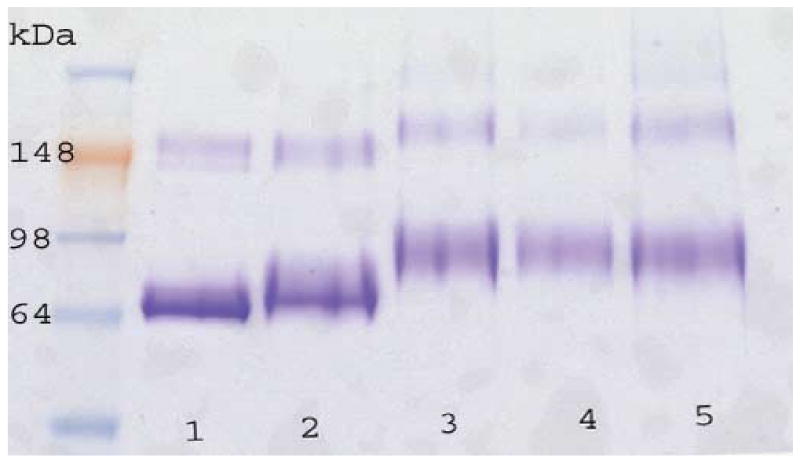
Sodium dodecylsulfate-polyacrylamide gel electrophoresis of the conjugates. Left lane: molecular weight markers; Lane 1: BSA; Line 2: aminooxypropylated BSA, MW 71.7 kDa indicating an average of 24 aminooxypropyl moieties per BSA; Lanes 3, 4, 5: BSA conjugates of glycolipid 2, average MW: 86.4 kDa indicating an average of 18 glycolipid moieties per BSA. The bands at about 150 kDa correspond to dimers. MW's were determined by MALDI-TOF.
In summary, we described a simple route to a conjugatable derivative 2 of the natural glycolipid 1 which was covalently linked to bovine serum albumin under mild conditions using oxime chemistry. The glycolipoprotein so obtained is currently being evaluated for its immunogenic properties.
4. Experimental
4.1. General Methods
All chemicals of were of commercial grade and were used without purification. Anhydrous solvents were obtained from Aldrich (St. Louis, MO). Column chromatography was performed on silica gel 60 (0.040–0.063 mm) and on dimethyloctadecylsilyl-bonded amorphous silica (C-18 adsorbent, 125 Å, Waters, MA. NMR spectra were obtained using either a Varian Mercury 300 spectrometer or a Bruker DRX-500 spectrometer at 300 K. Internal references: TMS (0.000 ppm for 1H for solutions in CDCl3), acetone (2.225 ppm for 1H and 31.07 ppm for 13C for solutions in D2O), methanol (3.358 ppm for 1H and 49.68 ppm for 13C for solution in CD3OD), and CDCl3 (77.00 ppm for 13C for solutions in CDCl3). Coupling constants are given in Hertz. The high resolution electrospray ionisation mass spectra (HRESIMS) were recorded at the Laboratory of Bioorganic Chemistry, NIDDK, NIH, Bethesda, MD. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA. For SDS-PAGE 14%, gels were used according to the manufacturer's instructions (BioRad, Hercules, CA).
4.2. Cholesteryl 6-O-(tert-butyldiphenylsilyl)-3,4-O-p-methoxybenzylidene-β-d-galactopyranoside (R,S-4)
To a stirred solution of compound4 3 (4.0 g, 5.1 mmol) in CH2Cl2 (40 mL) were added at 23 °C p-methoxybenzaldehyde dimethyl acetal (6 mL, 33.4 mmol) and a catalytic amount of camphorsulfonic acid. After 24 h, the solution was treated with Et3N followed by extraction with aq NaHCO3 and H2O. The organic phase was concentrated. Column chromatography of the residue using 6:1 → 4:1 hexanes–EtOAc gradient containing 0.05 % of Et3N afforded R,S-4 (3.75 g, 82%) as an amorphous solid: 1H NMR, partial (CDCl3): δ 5.36 (m, 1H), 4.40 (dd, 0.6H, J 5.4 Hz, J 7.6 Hz), 4.33 (dd, 1H, J 6.9 Hz, J 8.0 Hz), 4.24 (dt, 1H, J 2.6 Hz, J 6.1 Hz), 4.19 (dd, 0.4H, J 6.0 Hz, J 7.0 Hz), 3.83 (0.6H, J 1.8 Hz, J 6.9 Hz), 3.81 (s), 3.79 (s), 3.69 (dd, 0.4H, J 2.0 Hz, J 8.0 Hz); 13C NMR (CDCl3): δ 160.3, 160.1, 140.3, 135.6, 133.3, 133.2, 130.7, 129.6-127.7, 122.1, 113.63, 113.61, 104.6, 103.3, 100.6, 100.5, 79.7, 79.1, 78.9, 78.1, 75.9, 74.3, 74.0, 73.6, 73.2, 71.1, 62.9, 62.8, 56.7, 56.1, 55.3, 55.2, 50.1, 42.3, 39.7, 39.5, 38.82, 38.79, 37.2, 36.7, 36.2, 35.8, 31.9, 31.8, 29.70, 29.68, 28.2, 28.0, 26.72, 26.68, 24.3, 23.8, 22.8, 22.6, 21.0, 19.3, 19.21, 19.17, 18.7, 11.8; HRESIMS: Calcd for (C57H80O7Si + Li)+, m/z 911.5795, found, m/z 911.5833. Anal. Calcd for C57H80O7Si + 1/3 H2O: C, 75.25; H, 8.92. Found: C, 75.22; H, 8.88.
4.3. Cholesteryl 6-O-(tert-butyldiphenylsilyl)-2-O-p-methoxybenzyl-3,4-O-p-methoxybenzylidene-β-d-galactopyranoside (R,S-5)
To a stirred solution of R,S-4 (3.60 g, 4.0 mmol) in and DMF (30 mL) was added at 0 °C NaH (0.7 g of a 60% suspension in oil). After 20 min, freshly prepared p-methoxybenzyl bromide (2.7 g, 16 mmol) was added at 0 °C. The mixture was allowed to reach 23 °C. After 40 min, the mixture was re-cooled to 0 °C and was treated with MeOH (1 mL) followed by acetic acid until the pH of the mixture reached 5-6 as determined by a wet indicator paper. The mixture was concentrated at 25 °C under vacuum. Extractive work-up (CHCl3/H2O) followed by column chromatographic purification of the residue using 10:1 → 6:1 hexanes–EtOAc gradient containing 0.05 % of Et3N afforded R,S-5 (3.7 g, 91%) as a syrup: 1H NMR, partial (CDCl3): δ 5.36 (m, 1H), 4.85 (d, J 11 Hz), 4.76 (d, J 11 Hz), 4.74 (d, J 11 Hz), 4.79 (d, J 11 Hz), 4.24 (t, J 6.3 Hz), 4.22 (dd, J 1.9 Hz, J 5.5 Hz), 4.19 (dd, J 2.1 Hz, J 6.1 Hz), 3.81 (s, 1.2H), 3.79 (s, 3H), 3.78 (s, 1.8H), 3.48 (dd, J 7.2 Hz, J 7.7 Hz), 3.43 (dd, J 6.5 Hz, J 8.0 Hz; 13C NMR (CDCl3): δ 160.24, 160.21, 159.2, 159.1, 140.6, 135.6, 133.4-129.9, 113.64, 113.60, 113.5, 104.4, 103.2, 101.7, 101.4, 80.3, 79.8, 79.4, 79.3, 78.7, 75.8, 73.8, 73.6, 73.5, 73.3, 73.1, 63.1, 62.9, 56.7, 56.1, 55.31, 55.26, 55.24, 55.2, 50.1, 42.3, 39.7, 39.5, 39.1, 39.0, 37.2, 36.7, 36.2, 35.8, 32.0, 31.9, 29.91, 29.88, 28.22, 28.0, 26.72, 26.68, 24.3, 23.8, 22.8, 22.6, 21.0, 19.4, 19.2, 19.1, 15.8; HRESIMS: Calcd for (C65H88O8Si + Li)+, m/z 1031.6390, found, m/z 1031.6409. Anal. Calcd for C65H88O8Si + 1/3 H2O: C, 75.69; H, 8.66. Found: C, 75.66; H, 8.60.
4.4. Cholesteryl 2-O-p-methoxybenzyl-3,4-O-p-methoxybenzylidene-β-d-galactopyranoside (R,S-6)
To a solution of R,S-5 (3.65 g, 3.6 mmol) in THF (30 mL) was added a solution of tetrabutylammonium fluoride (5 mL of a 1 M solution in THF) at 23 °C. After 2 h, the solution was concentrated. Purification of the residue by column chromatography using 6:1 → 1:1 hexanes–EtOAc gradient containing 0.05 % of Et3N afforded R,S-6 (2.70 g, 96%) as an amorphous solid: 1H NMR, partial (CDCl3): δ 5.37 (m, 1H), 4.85 (d, J 11 Hz), 4.76 (d, J 11 Hz), 4.74 (d, J 11 Hz), 4.71 (d, J 11 Hz), 4.52-4.48 (m), 3.65-3.55 (m), 3.49 (t, 1H, J 7.8 Hz), 3.46 (dd, 1H, J 6.6 Hz, J 8.0 Hz); 13C NMR (CDCl3): δ 160.42, 160.37, 159.2, 159.1, 140.39, 140.38, 130.24, 130.2, 129.9, 128.9, 128.3, 127.8, 122.1, 122.0 113.8, 113.7, 113.6, 104.7, 103.3, 101.5, 101.3, 80.5, 79.6, 79.5, 78.8, 76.4, 76.0, 73.9, 73.6, 73.1, 73.0, 62.6, 62.3, 56.7, 56.1, 55.3, 55.2, 55.1, 50.1, 42.3, 39.7, 39.5, 39.0, 37.2, 36.7, 36.2, 35.8, 31.9, 31.8, 29.94, 29.91, 28.2, 28.0, 24.3, 23.8, 22.8, 22.5, 21.0, 19.4, 18.7, 11.8; HRESIMS: Calcd for (C49H70O8 + Li)+, m/z 793.5209, found, m/z 793.5231. Anal. Calcd for C49H70O8 + 1/3 H2O: C, 74.21; H, 8.98. Found: C, 74.36; H, 8.92.
4.5. (16,16-Dimethoxy)palmitic acid (7)
To a solution of (COCl)2 (2.74 mL, 31.3 mmol) in anh. CH2Cl2 (20 mL) was added at -62 °C a solution of DMSO (4.95 mL, 62.5 mmol) in anh. CH2Cl2 (10 mL) under stirring, in a period of 5 min. The solution was stirred for 2 min after the addition was complete. To this solution was added dropwise a solution of methyl 16-hydroxyhexadecaneoate in and CH2Cl2 [prepared by treatment of a solution of 16-hydroxydecanoic acid (7.4 g, 27 mmol) in anh CH2Cl2 with CH2N2 until a yellow color persisted]. The solution was stirred at -50 °C for 1 h and then was allowed to reach -10 °C. After 15 min, the solution was treated with Hünig's base (15 mL) and then was allowed to reach 23 °C. The solution was washed with H2O (3 × 25 mL), dried (Na2SO4), and concentrated to afford a semisolid. To a solution of the material in 2,2-dimethoxypropane (30 mL) was added a catalytic amount of 4-toluenesulfonic acid. After 30 min, approx. half of the volatiles were removed by distillation. The solution so obtained was treated with Et3N (3 mL) and then concentrated. The residue was equilibrated between CHCl3 and H2O. The organic layer was dried (Na2SO4) and concentrated to give a syrup. To a solution of this material in a dioxane (50 mL) were added H2O (5 mL) and solid KOH (1.7 g) at 23 °C. After 24 h, thin-layer chromatography (9:1 hexanes-EtOAc) indicated that most of the starting material disappeared. Water (5 mL) was added. After an additional 3 h, the solution was treated with H2O (50 mL) followed by removal of most of the organic solvent by distillation under reduced pressure. More H2O (100 mL) was added. The solution was acidified with citric acid until pH 3.5-4 was reached as detected by an indicator stripe. The mixture containing a precipitate was extracted with CHCl3 (3 × 50 mL). The combined organic layer was washed with H2O (2 × 30 mL), dried (Na2SO4) and concentrated to afford 7 (5.2 g, 64%) as an amorphous solid: 1H NMR (CDCl3): δ 4.35 (m, 1H), 3.32 (s, 3H), 3.31 (s, 3H), 2.38-2.31 (m, 2H), 1.68-1.54 (m, 4H), 1.37-1.17 (m, 22H); 13C NMR (CDCl3): δ 179.6, 104.5, 52.5, 34.0, 32.4, 29.6, 29.5, 29.44, 29.39, 29.3, 29.2, 29.0, 24.7, 24.6. Anal. Calcd for C18H36O4: C, 68.31; H, 11.47. Found: C, 68.51; H, 11.52.
4.6. Cholesteryl 6-O-(16,16-dimethoxy)palmitoyl-2-O-p-methoxybenzyl-3,4-O-p-methoxybenzylidene-β-d-galactopyranoside (8)
A solution of 6 (2.65 g, 3.4 mmol), compound 7 (1.8 g, 5.7 mmol), dicyclohexylcarbodiimide (1.8 g, 8.7 mmol), and 4-dimethylaminopyridine (0.9 g, 7.3 mmol) in EtOAc (50 mL) was stirred at 23 °C for 12 h. Water (0.5 mL) was added and stirring was continued for an other 30 min. The solids were removed by filtration and were washed with ethyl acetate. The combined filtrate and washings was concentrated. The residue was purified by chromatography using 5:1 → 3:1 hexanes–EtOAc gradient containing 0.05 % of Et3N to afford compound 8 (3.4 g, 93%) as an amorphous material: 1H NMR (CDCl3): δ 7.35-7.24 (m, 4H), 6.90-6.80 (m, 4H), 5.85 (s, 0.5H), 5.43 (s, 0.5H), 5.37 (m, 1H), 8.84, 4.75, 4.73, 4.68 (4d, J ∼ 11.3 Hz for each, 2H), 4.50-4.43 (m, 3H), 4.31 (dd, 1H, J 4.4 Hz, J 11.6 Hz), 4.27 (t, 1H, J 6.4 Hz), 4.16 (dd, 1H, J 2.4 Hz, J 6.1 Hz), 4.13 (dd, 1H, J 2.0 Hz, J 5.4 Hz), 3.82, 3.81, 3.79, 3.78 (4 s, 6H), 3.31 (s, 6H); 13C NMR (CDCl3): δ 173.52, 173.49, 160.4, 160.3, 159.2, 159.1, 154.1, 140.57, 140.55, 130.3-129.1, 121.9, 113.8-113.6, 104.7, 104.5(2C), 103.3, 101.8, 101.5, 79.3, 78.7, 77.9, 77.8, 77.6, 76.4, 75.7, 73.5, 73.1, 71.2, 70.5, 63.4, 63.1, 56.7, 56.1, 55.31, 55.26, 55.2, 52.63, 52.55, 50.2, 49.6, 42.3, 39.7, 39.5, 39.1, 37.3, 36.7, 36.1, 36.0, 35.8, 34.3, 32.8, 32.5, 31.9, 31.8, 29.9, 29.5-29.3, 28.2, 28.0, 26.4, 25.5, 25.4, 25.3, 25.0, 24.7, 24.6, 24.3, 23.8, 22.8, 22.5, 21.1, 19.4, 18.7, 11.8; HRESIMS: Calcd for (C67H104O11 + Li)+, m/z 1091.7771, found, m/z 1091.77389. Anal. Calcd for C67H104O11 + 1/2 H2O: C, 73.52; H, 9.67. Found: C, 73.29; H, 10.01.
4.7. Cholesteryl 6-O-(16,16-dimethoxy)palmitoyl-2-O-p-methoxybenzyl-β-d-galactopyranoside (9)
To a solution of 8 (3.4 g, 3.1 mmol) in CH2Cl2 (100 mL) and MeOH (25 mL) was added trifluoroacetic acid (1 mL) at 23 °C. After 2 h, the solution was treated with Et3N (3 mL). Extractive workup (CHCl3/aq NaHCO3) followed by column purification of the residue using 3:1 → 1:1 hexanes–EtOAc gradient containing 0.05 % of Et3N afforded 9 (2.75 g, 91%) as a syrup: 1H NMR, partial (CDCl3): δ 7.30-7.26 (m, 3H), 6.90-6.87 (m, 2H), 5.36 (m, 1H), 4.90 (d, 1H, J 11 Hz), 4.45 (d, 1H, J 7.7 Hz), 4.36 (t, 1H, J 5.7 Hz), 4.30 (d, 1H, J 6.5 Hz), 3.81 (s, 3H), 3.63 (t, 1H, J 7.2 Hz), 3.45 (dd, 1H, J 7.7 Hz, J 9.3 Hz), 3.31 (s, 6H); 13C NMR (CDCl3): δ 173.7, 159.4, 140.5, 130.4, 129.9, 122.0, 114.0, 104.5, 102.2, 79.8, 78.3, 74.1, 72.9, 71.8, 68.1, 62.5, 56.7, 56.1, 55.2, 52.5, 50.1, 42.3, 39.7, 39.5, 39.1, 36.7, 36.1, 35.8, 34.2, 32.5, 31.9, 31.8, 29.9, 29.66, 29.65, 29.56, 29.52, 29.48, 29.3, 29.2, 28.2, 28.0, 24.9, 24.6, 24.3, 23.8, 22.8, 22.5, 21.0, 19.4, 18.7, 11.8; HRESIMS: Calcd for (C59H98O10 + Li)+, m/z 973.7328, found, m/z 973.7320. Anal. Calcd for C59H98O10 + 1/3 H2O: C, 72.80; H, 10.22. Found: C, 72.85; H, 10.20.
4.8. Cholesteryl 6-O-(16,16-dimethoxy)palmitoyl-β-d-galactopyranoside (10)
To a solution of 8 (1.40 g, 1.4 mmol) in moist CH2Cl2 (30 mL) was added at 23 °C 4,5-dichloro-5,6-dicyano-1,4-benzoquinone (1.5 g, 6.6 mmol). After 30 min, aq NaHCO3 was added. Extractive work-up (CHCl3/H2O) followed by concentration of the organic layer afforded a syrup that was purified by column chromatography using 1:2 hexanes–EtOAc → EtOAc gradient containing 0.05 % of Et3N afforded 10 (1.0 g, 82%) as a syrup: 1H NMR (CDCl3): δ 5.35 (m, 1H), 4.36 (t, 1H, J 5.8 Hz), 4.37-4.27 (m, 3H), 3.31 (s, 6H); 13C NMR (CDCl3): δ 173.7, 140.3, 122.0, 104.5, 101.5, 79.5, 73.3, 72.3, 71.4, 68.5, 62.7, 56.7, 56.1, 52.5, 50.1, 42.3, 39.7, 39.5, 38.8, 37.2, 36.7, 36.1, 35.8, 34.2, 32.4, 31.9, 31.8, 29.73, 29.70, 29.68, 29.5, 29.43, 29.36, 29.3, 28.2, 28.0, 24.9, 24.6, 24.3, 23.8, 22.8, 22.5, 21.0, 19.3, 18.7, 11.8; HRESIMS: Calcd for (C51H90O9 + Li)+, m/z 853.6750, found, m/z 853.6745. Anal. Calcd for C51H9-O9 + 1/2 H2O: C, 71.54; H, 10.71. Found: C, 71.60; H, 10.66.
4.9. Cholesteryl 6-O-(16-oxo)palmitoyl-β-d-galactopyranoside (2)
To a stirred solution of 9 (1.9 g, 2.2 mmol) in i-PrOH were added AcOH (50 mL) and H2O (15 mL). The solution was kept at 85-90 °C for 3 h. The volatiles were removed by distillation under reduced pressure. Column chromatography of the residue using 100:4 CH2Cl2-MeOH afforded 2 (1.25 g, 84%) as an amorphous substance: 1H NMR (CDCl3): δ 9.76 (t, 1H, J 1.9 Hz), 5.34 (m, 1H), 4.45-4.17 (m, 4H), 0.99 (s, 3H), 0.91 (d, 3H, J 6.4 Hz), 0.87 (d, 3H, J 6.7 Hz), 0.86 (d, 3H. J 6.6 Hz), 0.67 (s, 3H); 13C NMR (CDCl3): δ 203.0, 173.6, 140.3, 122.0, 101.7, 79.6, 73.4, 72.4, 71.1, 68.7, 62.9, 56.7, 56.2, 50.1, 43.9, 42.3, 39.7, 39.5, 38.8, 37.3, 36.6, 36.2, 35.8, 34.2, 31.9, 31.8, 29.44, 29.41, 29.37, 29.3, 29.2, 28.2, 28.0, 24.9, 24.2, 23.8, 22.8, 22.5, 22.0, 21.0, 19.4, 18.7, 11.0; HRESIMS: Calcd for (C49H84O8 + Na)+, m/z 823.6064, found, m/z 823.6077. Anal. Calcd for C49H84O8 + H2O: C, 71.84; H, 10.58. Found: C, 72.27; H, 10.52.
4.10. Conjugation of 2 with aminooxypropylated bovine serum albumin
To a solution of aminooxypropylated BSA5 (15 mg, 0.21 μmol, having an average of 24 aminooxypropyl moieties per BSA) in PBS (3 mL, containing 0.1% glycerol, 0.005 M EDTA, and 1% octyl glucoside, at pH 7.4) was added a 0.5 M solution of sodium bis(2-ethylhexyl)sulfosuccinate in octane (18 mL) under vigorous stirring. After the formation of a transparent, clear solution, a solution of the aldehydo-derivatized hapten 2 (15 mg, 19 μmol) in pyridine (200 μL) was added. The solution was kept at 37 °C for 20 h followed by addition of 80% aqueous EtOH (80 mL). The mixture was centrifuged and the supernatant discarded. The precipitate was resuspended in 0.2 M NaCl (4 mL, containing 0.3% octyl glucoside) followed by the addition of 80% aqueous EtOH (16 mL). The precipitate obtained was isolated by centrifugation as before and was resuspended in 0.2 M NaCl (2 mL, containing 0.3% octyl glucoside). The insoluble material was removed by centrifugation and the supernatant was passed through a Sephadex G-50 column (1 × 50 cm) made in 0.2 M NaCl., using the same solution as the eluant. The void volume fractions were combined and the solution so obtained was concentrated by ultrafiltration. The average molecular mass of the conjugates was 86.4 kDa as determined by MALDI-TOF mass spectrometry. The precipitate insoluble in 0.2 M NaCl containing 0.3% octyl glucoside was solubilized in 0.5% sodium dodecylsulfate (1 mL). The solution so obtained was applied to a Sephadex G-50 column and the conjugate was isolated as described earlier. The molecular weight (MALDI-TOF) of the product eluted in the void volume was identical to that obtained above.
Acknowledgments
The authors thank Drs. J. B. Robbins and R. Schneerson for their support, Dr. J. Lloyd for the HRESIMS spectra, and Dr. G. Ekborg for technical assistance. This work was supported by the Intramural Research Program of the National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.MMWR. 2004;53:365–369. [PubMed] [Google Scholar]
- 2.European Union Concerted Action on Lyme Borreliosis. 2005 http://www.oeghmp.at/eucalb/disease-overview_1.html.
- 3.Ben-Menachem G, Kubler-Kielb J, Coxon B, Yergey A, Schneerson R. Proc Natl Acad Sci USA. 2003;100:7913–7918. doi: 10.1073/pnas.1232451100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pozsgay V, Kubler-Kielb J, Coxon B, Ekborg G. Tetrahedron. 2005;61:10470–10481. [Google Scholar]
- 5.Kubler-Kielb J, Pozsgay V. J Org Chem. 2005;70:6987–6990. doi: 10.1021/jo050934b. [DOI] [PubMed] [Google Scholar]; 2006;71:5422–5422. [Google Scholar]
- 6.Greene TW, Wuts PGM. Protecting groups in organic synthesis. John Wiley & Sons; New York: 1999. p. 217. [Google Scholar]
- 7.Pozsgay V. J Org Chem. 1998;63:5983–5999. doi: 10.1021/jo980660a. [DOI] [PubMed] [Google Scholar]
- 8.Pozsgay V, Kubler-Kielb J. ACS Symp Ser. 2006;960:238–252. [Google Scholar]
- 9.Pozsgay V, Kubler-Kielb J. ACS Symp Ser. 2007 In press. [Google Scholar]
- 10.Fekete A, Hoogerhout P, Zomer G, Kubler-Kielb J, Schneerson R, Robbins JB, Pozsgay V. Carbohydr Res. 2006;341:2037–2048. doi: 10.1016/j.carres.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 11.Yatsimirskaya EA, Gavrilova EM, Egorov AM, Levashov AV. Steroids. 1993;58:547–550. doi: 10.1016/0039-128x(93)90033-j. [DOI] [PubMed] [Google Scholar]
- 12.Pauillac S, Naar J, Mouratou B, Guesdon JL. J Immunological Methods. 2002;263:75–83. doi: 10.1016/s0022-1759(02)00039-x. [DOI] [PubMed] [Google Scholar]