

Abstract
A robust p53 cell-based assay that exploits p53’s function as a transcription factor was used to screen a small molecule library and identify bioactive small molecules with potential antitumor activity. Unexpectedly, the majority of the highest ranking hit compounds from this screen arrest cells in mitosis and most of them impair polymerization of tubulin in cells and in vitro. One of these novel compounds, JJ78:1, was subjected to structure-activity relationship studies and optimized leading to the identification of JJ78:12. This molecule is significantly more potent than the original hit JJ78:1, as it is active in cells at two-digit nanomolar concentrations and shows clear antitumor activity in a mouse xenograft model as a single agent. The effects of nocodazole, a well established tubulin poison, and JJ78:12 on p53 levels are remarkably similar, supporting that tubulin depolymerization is the main mechanism by which JJ78:12 treatment leads to p53 activation in cells. In summary, these results identify JJ78:12 as a potential cancer therapeutic, demonstrate that screening for activators of p53 in a cell-based assay is an effective way to identify inhibitors of mitosis progression and highlights p53’s sensitivity to alterations during mitosis.
Keywords: p53, tubulin polymerization, small molecule screen
Introduction
After many years of intensive research, whether p53 status influences prognosis in cancer patients is still a matter of debate. Nevertheless, irrespective of whether activation of p53 plays a role in response to treatment, it is clear that many classic cancer therapeutics (radiation, DNA interacting compounds and anti-mitotics) do activate p53’s activity as a transcription factor. Hence it is not unreasonable to think that screening for compounds that activate p53 could potentially lead to identifying novel agents with therapeutic value. With this in mind, we set out to discover small-molecules that increase the expression of a reporter construct under the control of a p53-dependent promoter. Hit compounds from this screen are not general cytotoxics as they need to increase a synthetic event, i.e., the expression of a p53-dependent reporter. Also, compounds selected through a phenotypic screen are active in cells at concentrations that are acceptable for further testing in organisms.
Screening small-molecules for their ability to activate p53 would be expected to lead to the discovery of compounds that exclusively affect the p53 pathway. Examples of this type of compounds are agents that interact directly with p53 or that like the nutlins, selectively inhibit mdm2-mediated degradation of p53.1,2 Since the p53 tumor suppressor is activated in response to alterations in a wide variety of cellular events, it is also expected that this kind of screening approach would lead to the identification of compounds that activate p53 in an indirect fashion and hence also exert p53 independent effects. Elucidating the precise mechanism of action of a hit compound of this sort can be viewed as a major task.3-5 However, due to the wealth of knowledge on the regulation of the p53 pathway there are sufficient high quality reagents and testable hypotheses to overcome this challenge.6
Here we describe that using p53 activation as a biomarker in a cell based screen of an unbiased synthetic chemical library leads to the discovery of a surprising proportion of novel anti-mitotic compounds. We also demonstrate that direct destabilization of tubulin polymers is the mechanism of action of many of these compounds and provide new insights into the mechanism by which tubulin poisons increase p53 levels. This underscores p53’s sensitivity to alterations in microtubule dynamics. Synthetic elaboration of compound JJ78:1 from this series has led to the identification of analogues with improved efficacy in cell lines and activity in a preclinical tumor model.
Results
Identification of compounds that induce cell cycle arrest at mitosis through a p53 activation assay
A cell-based p53-dependent reporter screen of the synthetic ChemBridge DIVERSet collection (consisting of 30,000 compounds) was conducted using T22-RGC-ΔFos-lacZ murine cells expressing β-galactosidase under the control of a p53-dependent promoter. Compounds were assayed in each plate at 10 μM as described.6 A total of thirty-three hit compounds from this screen were prioritized through an analysis of potency and secondary assays and selected for further characterization. Interestingly, one of the most potent compounds from this set (JJ102:1; 5175348) is the tubulin depolymerising agent nocodazole. Nocodazole inhibits spindle assembly and triggers the mitotic spindle checkpoint (independently of p53) leading to the arrest of cells in mitosis.7 Treatment of cells with two other established tubulin poisons (taxol and vincristine) under our standard screening conditions also caused an induction of the p53-dependent reporter in cells (data not shown).
Aside from nocodazole, a surprisingly high proportion of the compounds from the group of thirty-three prioritized hits, that is a total of sixteen compounds, also lead to accumulation of cells in the G2/M phase of the cell cycle (Suppl. data Fig. S1). Two of these compounds (JJ172:1 and JJ174:1) are analogues of reported tubulin interactives.8 Accumulation of cells in G2/M in response to all sixteen compounds was observed in cells with functional p53 as well as in cells where p53 function is abolished by overexpression of a dominant negative form of p53 (Suppl. data Fig. S1). Figure 4A shows an example of the two-dimensional FACS plots obtained with prototype compound JJ78:1, which is the main focus of this paper. It is important to note that cells with inactivated p53 could still replicate their DNA (as determined by BrdU incorporation) acquiring a DNA content higher than 4N, whereas cells with active p53 are precluded from entering a new DNA synthesis phase. This observation is general for all compounds tested (Suppl. data Fig. S1) and further highlights p53’s role in preventing polyploidy.9
Figure 4.
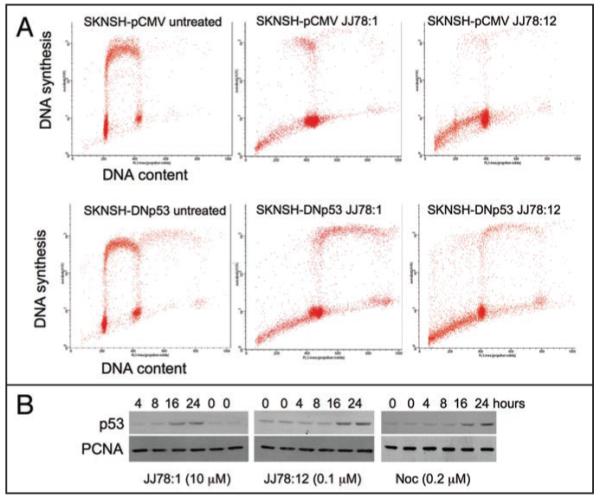
JJ78:1 and JJ78:12 have similar effects in cells. (A) SKNSH cells with either active (SKNSH-pCMV) or inactive p53 (SKNSH-DNp53) were cultured in the absence or presence of the indicated compounds (10 μM) for 48 hours. 30 minutes before harvesting cells where pulse labelled with BrdU, harvested and processed for two-dimensional FACS analysis. (B) Time course analysis of p53 levels in MCF7 cells (harbouring active p53) incubated with the indicated compounds. p53 is detected with DO1 antibody.25 PCNA detection using PC10 antibody26 is used as a loading control.
To narrow down the mechanism of action of the novel sixteen G2/M arresting hit compounds (excluding JJ102:1, i.e., nocodazole), an analysis of mitotic index was carried out. Aurora B phosphorylates histone H3 at serine 10 upon entry into mitosis.10,11 Hence, compounds that arrest cells after the onset of mitosis (like taxol) give rise to the accumulation of cells that are positive for phosphoserine H3 (P-S10-HH3) (Fig. 1A). As expected, no P-S10-HH3 was detected after incubation with compound VX-680, an inhibitor of Aurora kinases.12 An example of this assay using HeLa cells treated with compound JJ78:1 is shown in Figure 1A. JJ78:1, as well as the other fifteen compounds from the series of sixteen G2/M arresting compounds, gave rise to an increase in the number of cells with H3 phosphorylation at serine 10, demonstrating that all of the compounds from the series allow entry into mitosis and delay cell cycle progression primarily at this stage (Fig. 1B). HeLa cells are defective for p53 function due to the expression of the human papillomavirus E6 oncoprotein,13 which suggested that p53 activation is not likely to be responsible for the mitotic arrest induced by the compounds. In agreement, when we carried out the same analysis using SKNSH-pCMV cells (with active p53) and SKNSH cells expressing a dominant negative p53 form (SKNSH-DNp53)14 all compounds induced a failure in mitotic progression independently of p53 function (Fig. 1B). In general, cells with inactive p53 showed a higher mitotic index probably reflecting their faster progression through the cell cycle.
Figure 1.

A high proportion of p53-activating hit compounds cause an increase in mitotic index. (A) HeLa cells were grown in the presence of the indicated compounds for 24 hours, fixed and stained for the mitotic marker P-S10-HH3 (clone MC463, Upstate Biotech.) and DNA using Hoescht. (B) The same procedure was repeated with the 16 compounds using the SKNSH isogenic cell lines, which contain either an active (SKNSH-pCMV) or inactive p53 (SKNSH-DNp53). In this experiment, cells were treated with the compounds for 48 hours. All compounds cause an increase in the mitotic index expressed as the percentage of cells positive for P-S10-HH3 staining (1000 cells were counted in each sample). (C) In vitro inhibition of tubulin polymerisation. Purified bovine tubulin was heated to 37°C in a glycerol buffer, in the presence or absence of different compounds. The polymerisation of tubulin into microtubules was monitored by an increase in fluorescence. In mock treated samples (DMSO) the tubulin forms microtubule in a normal manner whereas in the presence of taxol polymerisation is enhanced due to the stabilising properties of this compound. In the presence of vinblastine, an inhibitor of tubulin polymerisation, no microtubules can form. JJ79:1, JJ98:1 and JJ99:1 polymerisation curves are shown as examples of hit compounds from the screen. All compounds are at 10 μM (0.5% DMSO).
Most of the compounds directly impair tubulin polymerization
Further immunofluorescence imaging of cells treated independently with the sixteen compounds showed that the microtubule network in interphase appears disordered and that there are numerous aberrant mitotic figures where cells are unable to form a mitotic spindle (Fig. 3E shows results obtained with compound JJ78:1). Given that agents that delay mitotic progression frequently bind to tubulin and perturb tubulin polymerization,15 we used an in vitro assay to test whether the compounds promote the stabilization of tubulin polymers (like taxol) or destabilize tubulin polymers (like vinca alkaloids or nocodazole). This in vitro assay demonstrated that all but four of the sixteen compounds tested (JJ58:1, JJ82:1, JJ89:1 and JJ99:1) directly target tubulin and decrease its polymerization. Figure 1C shows examples of these results.
Figure 3.

Functional analyses of JJ78:12. (A) Selected data showing levels of p53 activation in T22- RGC-ΔFos-LacZ cells as a function of compound concentration for a series of JJ78:1 analogues. (B) Comparison between JJ78:1 and JJ78:12 in the p53 activation cell based assay (T22- RGC-ΔFos-LacZ cells). (C) A selection of JJ78 derivatives produced via SAR studies were tested in the in vitro tubulin polymerisation assay. All compounds were used at 10 μM (0.5% DMSO). Polymerisation curves are the average of three independent experiments. (D) Mitotic index calculation (using the mitotic marker P-S10-HH3) for selected JJ78:1 derivatives in MCF7 cells. (E) MCF-7 cells were treated with the indicated concentrations of JJ78:1 and JJ78:12 for 24 hours, fixed and stained for γ tubulin and DNA using Hoescht dye. Images of interphase and mitotic cells are shown.
Two compounds from the series of twelve tubulin poisons were chosen for further characterization. One of these, JJ98:1, is one of the most potent compounds in the in vitro tubulin polymerization assay (Fig. 1C). Twelve other compounds with structures related to JJ98:1 were identified and purchased. These compounds were tested for their effects on mitotic index. Three of these twelve compounds had no effect in cells, ten were less potent than JJ98:1 and two were as potent as JJ98:1. The latter two compounds were not present in the original 30,000 set used in the primary screen explaining why they were not identified in the initial p53 phenotypic screen. This further establishes the validity of our p53-cell based screen to identify potent inhibitors of mitotic progression that are active in cells. No further experiments were carried out with JJ98:1 due to its poor water solubility as this limits its use for in vivo experiments.
The other compound analysed (JJ78:1) is a moderate inhibitor of tubulin polymerization in vitro (Fig. 3C) but was highly effective in delaying cell cycle progression (Figs. 1, 3A and Suppl. Fig. S1) and caused a significant increase in p53-dependent transcriptional activity at submicromolar concentrations (Fig. 3A and B). Importantly, JJ78:1’s chemical tractability made it a good candidate for optimization studies.
Synthesis and structure activity relationship studies on JJ78:1
A sample of JJ78:1 was initially obtained from a commercial source. To further confirm bioactivity, chemical structure and purity, larger quantities of JJ78:1 were synthesized from ethyl 3,5-dimethyl-1H-pyrrole-2-carboxylate (1) in line with literature precedent16 (Fig. 2A). To date, all batches of JJ78:1 prepared by us have shown identical chemical analysis and biological activity to the DMSO stock solution of JJ78:1 used in the high-throughput screen and the commercially available sample. The structure of JJ78:1 was also confirmed by small molecule x-ray crystallographic analysis (see Suppl. material).
Figure 2.
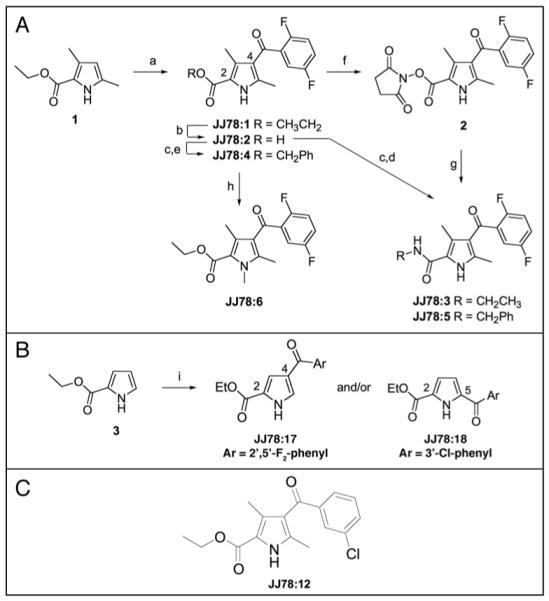
Synthesis and SAR studies on JJ78:1. (A) Details of the synthesis of JJ78:1-6: reaction conditions: (a) AlCl3, 2,5-difluorobenzoyl chloride, chlorobenzene (55%); (b) KOH, ethanol (70%); (c) SOCl2, DMF (cat.), CH2Cl2; (d) EtNH2/EtOH, CH2Cl2 (59%); (e) benzyl alcohol (70%); (f) DCC, N-hydroxysuccinimide, CH2Cl2 (33%); (g) benzyl amine, CH2Cl2 (70%); (h) K2CO3, iodomethane, CH3CN (96%). (B) Details of the synthesis of JJ78:17-18: reaction conditions: (i) AlCl3, acid chloride, chlorobenzene. (C) Structure of JJ78:12.
Inspection of the chemical structure of JJ78:1 raised questions about the role of the ester functional group. The lability of esters in cells is well documented (for example see Mizen et al.,17) and it was therefore decided to explore the contribution of this group to the observed biological activity. Saponification of JJ78:1 provided the corresponding carboxylic acid JJ78:2 (Fig. 2A) which was then converted to the ethylamide JJ78:3 and benzyl ester JJ78:4 derivatives using standard protocols. Acid JJ78:2 was also converted to the benzylamide JJ78:5 via the activated ester 2. The biological activity of all the derivatives of JJ78:1 discussed here was determined using the p53-reporter assay described above. The reasons for selecting this assay included its robust and relatively easy protocol, its ability to readout out biological activity in a cellular environment (as opposed to in vitro) and the fact that it provided a semi-quantitative measure for analysing the effects of compounds on the tubulin network in cells in contrast to the use of immunofluorescence-based approaches the results from which are viewed as qualitative. When tested in the p53 reporter assay, carboxylic acid JJ78:2 showed no activity, implying that either the compound is membrane impermeant and hence not able to access the intracellular target or alternatively that the ethyl ester makes a significant contribution to the binding of JJ78:1 to tubulin (see Table S1 for assay data on JJ78:1-6). The analysis of compounds JJ78:4 and JJ78:5 added further weight to the view that a relatively small group is required at position 2 of JJ78:1 (see Fig. 2A for numbering system). In addition the ethyl amide analogue JJ78:3 also exhibited no significant activity implying that the ester alkyl-oxygen moiety is essential. Interestingly, a molecular model of a related tubulin depolymerising agent has recently been reported.18 This compound also contains an ethyl ester functional group which has been proposed to occupy a tight binding pocket making a series of hydrogen bonding interactions with tubulin. Our observations on the essential nature of the ethyl ester is consistent with JJ78:1 and its analogues occupying a related binding mode and will be discussed in detail in subsequent reports. Evidence that the pyrrole NH functionality was essential for the activity of JJ78:1 was gained from analysis of the NMe-analogue, JJ78:6 (Fig. 2A), which was shown to be inactive.
One of the few remaining sites for potential modification in JJ78:1 was the 4-aroyl-position and it was therefore decided to explore the effect of modification at this position whilst retaining the other functionality. The trisubstituted pyrrole 1 (Fig. 2A) was shown to exhibit no biological activity highlighting the importance of the 4-substituent. Acylation of 1 was carried out in parallel employing a modified version of the protocol used to prepare JJ78:1. Initial studies focussed on the synthesis of a set of analogues substituted in the aromatic ring. Analogues JJ78:7-11 were prepared in good yield with high levels of purity using this protocol. The relative activity as well as the structures of these analogues are shown in Table 1. A spread of potencies was observed across the series and a clear trend emerged suggesting an unfavorable steric effect on 4′-substitution. The exception to this trend was JJ78:7, which despite having a large 4′-chloro substituent in addition to the 3′-chloro, exhibited the highest potency in the series. This observation led to the intriguing possibility that an analogue bearing a single 3′-chloro substituent might be significantly more active.
Table 1. p53 activation on cells by selected JJ78:1 analogues shown in terms of their relative potency order with the most potent analogue assigned as 1.
| Compound | Aryl substituents | Potency order |
|---|---|---|
| JJ78:1 | 2′,5′-F2 | 2 |
| JJ78:7 | 3′,4′-Cl2 | 1 |
| JJ78:8 | 4′-Cl | 6 |
| JJ78:9 | 4′-CH3 | 4 |
| JJ78:10 | 4′-OCH3 | 5 |
| JJ78:11 | H | 3 |
This analogue, ethyl 4-(3-chlorobenzoyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (JJ78:12) (Fig. 2C), was synthesized as part of an analogue expansion series to further investigate SAR. This third compound set (Table 2 for structures) also included JJ78:13, a derivative of JJ78:1 in which the bridging ketone had been reduced to a methylene group16 and JJ78:14, an analogue bearing a 2′-methoxybenzoyl group in the 4-position of the dimethylpyrrole ring (Table 2). A by-product was isolated during the synthesis of JJ78:14, in which the 2′-methoxy group had been cleaved giving the corresponding phenol JJ78:15, under the reaction conditions. A further aroyl analogue JJ78:16 was synthesized by displacement of the 2′-fluoro group present in JJ78:1 by morpholine. This reaction occurred in preference to the anticipated conversion of the ethyl ester to the corresponding morpholinoamide. In addition to these derivatives, a final pair of analogues was prepared with the goal of exploring the importance of the 3- and 5-methyl substituents in JJ78:1. Ethyl pyrrole-2-carboxylate (3) (Fig. 2B) was treated with two aroyl chlorides to give JJ78:17 and JJ78:18, respectively. X-ray crystallographic analysis of both of these analogues (see Suppl. material) confirmed that variation in the position of acylation occurred as a function of the acylating agent. For example, 2,5-difluorobenzoyl chloride reacted to give, as the major product, JJ78:17, the result of acylation at the 4-position of the pyrrole providing a direct analogue of JJ78:1. However, the use of 3-chlorobenzoyl chloride led predominantly to reaction at the 5-position providing JJ78:18 under parallel conditions. It has been previously reported that the proportion of 4- and 5-acyl regioisomers formed in this system is dependent on reaction conditions, catalyst and acylating reagent.19
Table 2. p53 activation by JJ78:1 analogues shown in terms of their relative potency order with the most potent analogue assigned as 1.
| Compound | C3 | C4 | C5 | Aryl substituents | Potency order |
|---|---|---|---|---|---|
| JJ78:12 | CH3 | COAr | CH3 | 3′-Cl | 1 |
| JJ78:13 | CH3 | CH2Ar | CH3 | 2′,5′-F2 | 4 |
| JJ78:14 | CH3 | COAr | CH3 | 2′-OMe | =3 |
| JJ78:15 | CH3 | COAr | CH3 | 2′-OH | =3 |
| JJ78:16 | CH3 | COAr | CH3 | 2′-(morpholin-4-yl)-5′-F | 2 |
| JJ78:17 | H | COAr | H | 2′,5′-F2 | Inactive |
| JJ78:18 | H | H | COAr | 3′-Cl | Inactive |
Reduction of the ketone functionality in JJ78:1 to a methylene group in JJ78:13 caused an increase in concentration of compound required to induce maximal p53-dependent reporter stimulation and the amplitude of the induction was also reduced (Table 2 and Fig. 3A). This supports a view that the carbonyl functionality is important for maximal biological activity. Interestingly the X-ray structural analysis of JJ78:1 suggests that, in the solid state at least, the carbonyl group in JJ78:1 is nearly co-planar with the pyrrole ring and that the aromatic ring is significantly distorted out of the plane of the pyrrole ring. More rotational flexibility in this region of the molecule would be expected following reduction of the carbonyl group, potentially consistent with a higher entropic cost on target binding resulting in reduced potency of JJ78:13 compared with JJ78:1. Further evidence that the ketone is important came from the reduced activity observed following its conversion to the oxime in JJ78:19 (p53 activation only seen above 0.5 μM, Suppl. material).
JJ78:16 showed good cellular activity being equipotent with JJ78:7 (Fig. 3A). However, JJ78:16 was less potent than the 3′-chloro containing analogue JJ78:12 which, in line with prediction, showed a significant increase in potency compared to both JJ78:7 and JJ78:1 enabling p53 transcriptional activity induction to be detectable at a final concentration of JJ78:12 of only 10 nM in the cell-based assay (Fig. 3B). A final concentration of above 0.1 μM of JJ78:1 was required to give an analogous level of p53 activation, hence JJ78:12 is at least 10-fold more active in cells than JJ78:1.
Finally, the 3,5-unsubstituted pyrrole JJ78:17, a direct analogue of JJ78:1, showed no evidence of bioactivity. This somewhat surprising result may reflect an increase in molecular planarity and rotational freedom associated with the aromatic ring of the 4-substituent in JJ78:17 compared with JJ78:1 where the pyrrole and phenyl rings are observed to be orthogonal by X-ray structure analysis (see supplementary material). The 2,5-disubstituted compound JJ78:18 which bears a 3′-chlorobenzoyl group was also found to be inactive (Fig. 3A) adding to the view that there is a very well defined pocket available for the binding of the active analogues.
Validation of JJ78:12 as a tubulin poison
As a whole, the efficacy of particular JJ78 analogues in the tubulin biochemical assay correlated with their level of p53 activation in cells (examples of these experiments are shown in Fig. 3A-C). JJ78:12 was the most potent compound in both types of assay. This supports that its inhibitory effect on tubulin polymerization is the main cause for the observed increase in p53 activation in cells. As such, JJ78:12 is also highly effective at increasing the mitotic index (Fig. 3D). Furthermore, the increase in potency of JJ78:12 was matched by the increased potency at causing microtubule disordering in cells. This was especially noticeable in mitotic cells as the mitotic spindle could not form in the presence of 50 nM JJ78:12 (Fig. 3E).
Also suggesting that the effects on cells of JJ78:12 occur primarily through its ability to destabilize tubulin polymers, this compound induces the accumulation of cells with a 4N DNA content (Fig. 4A). Another feature shared by nocodazole, JJ78:1 and JJ78:12 is that their effect on p53 levels requires that compounds are present in cell cultures for more than 8 hours (Fig. 4B). Instead, compounds that act like the nutlins to decrease p53 degradation by directly inhibiting mdm2,2 or like the tenovins preventing p53 deacetylation,6 can increase p53 levels within 30-120 minutes of incubation (not shown). Accumulation of p53 in response to other tubulin poisons such as taxol also requires incubation times above 8 hours.20 This delay in p53 induction in response to agents like taxol or nocodazole, which arrest cells in mitosis, suggests that only when a sufficient number of cells accumulates at this stage of the cell cycle for a sufficient amount of time is it possible to observe an effect on p53 levels in the total cell population.. In fact, cells arrested in mitosis with taxol treatment have been shown to accumulate p53.21,22 After mitotic arrest by tubulin poisons, cells either undergo mitotic slippage (leading to the appearance of cells that are tetraploid and multinucleated) or die. Here we investigated the levels of p53 in multinucleated cells obtained after JJ78:12 or nocodazole treatment and in both instances observed different levels of p53 staining in different micronuclei (Fig. 5). Hence, as a whole, it is possible to conclude that the effects of JJ78:12 in cells are the same as those observed with tubulin poisons.
Figure 5.
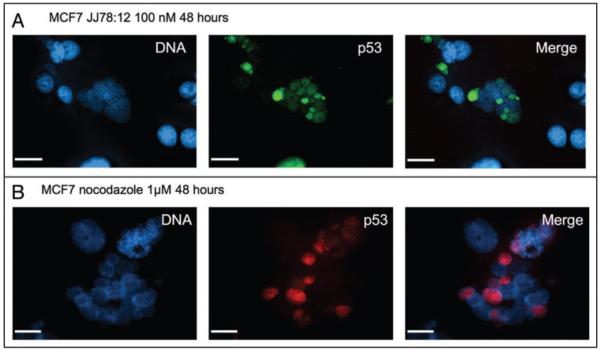
p53 accumulates in micronuclei of JJ78:12 treated cells. (A) MCF7 cells were treated with JJ78:12 (100 nM) for 24 hours and stained for p53 with DO1 antibody and DNA with Hoescht dye. (B) MCF7 cells were treated with nocodazole (50 nM) for 24 hours and stained for p53 with CM1 serum27 and DNA with Hoescht dye. As with JJ78:12, note that not all nuclei in the multinucleated cells are positive for p53.
JJ78:12 shows anti-tumor activity in vivo
Further evidence of the increased potency of JJ78:12 over the initial hit compound JJ78:1 was demonstrated by cell growth assays. As shown in Figure 6A using the melanoma derived cell line ARN8 the JJ78:1 GI50 value equals 223 nM, whereas for JJ78:12 GI50 equals 63 nM. Given the ability of JJ78:12 to reduce tumor cell growth at two digit nanomolar concentrations we tested its ability to reduce tumor growth in vivo. In spite of the fact that JJ78:12 showed a relatively short half life in vivo (Fig. 6B), mice receiving JJ78:12 daily at 30 mg/kg as a single agent showed a reduction in ARN8 tumor growth (Fig. 6C) without noticeable toxic effects on the animals over 15 days.
Figure 6.
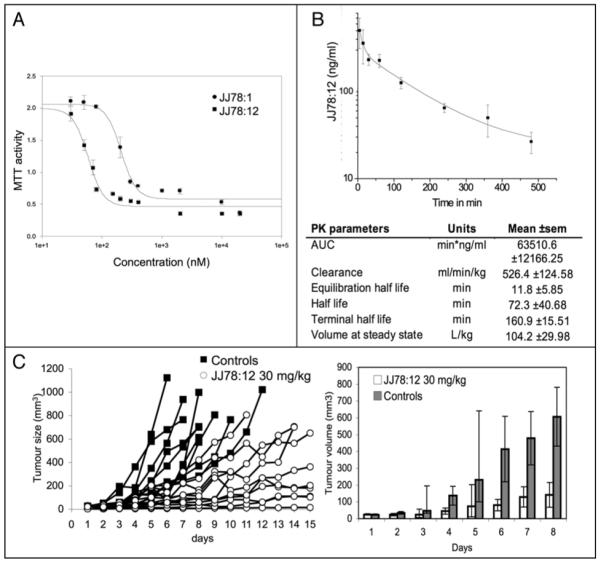
Antitumor activity of JJ78:12. (A) ARN8 human melanoma cells (p53 wild type) were incubated with the indicated concentrations of compound for 48 hours. Viable cells were detected by MTT assay. (B) Pharmacokinetic parameters for JJ78:12. Curve shows the elimination of JJ78:12 in mouse (n = 3). Extrapolation of the PK parameters was carried out using a two-compartmental model. (C) ARN8 melanoma cells were injected in the flank of SCID mice and allowed to develop into tumours. JJ78:12 (in 20% cyclodextrin) was administered once daily (a total of 14 times) by intraperitoneal injection at 30 mg/kg (n = 10) and tumour growth was measured over a period of 15 days. Control animals (n = 10) were treated with 20% cyclodextrin. Tumour size was measured with callipers. Left panel shows the measurements obtained from each individual tumour. Note that untreated tumours would reach sizes far beyond 1,000 mm3 by day 15. Bearing this in mind, but in order to provide a statistical analysis, measurements performed on days 1 through 8 were averaged between groups and plotted (right). Error bars correspond to 95% confidence intervals. Mice receiving JJ78:12 had significantly reduced tumour growth as analysed by appropriate choice of 2-sample t-test (normal distributions) and Mann Whitney U-test (non-normal distributions). Difference between the JJ78:12 and control distributions were statistically significant on days 3 to 8 (p = 0.014, 0.013, 0.004, 0.009, 0.002 and 0.001, respectively). JJ78:12 did not induce signs of toxicity at the concentration tested.
Discussion
We have used a p53-based phenotypic assay to identify novel bioactive small molecules. Unexpectedly, we found that the majority of the hit compounds from this screen arrest cells in mitosis. Furthermore, and like nocodazole (which was one of the hit compounds in the primary screen), most of these compounds impair polymerization of tubulin. Hence, we can conclude that screening for activators of p53 in a cell based assay is an effective way to identify inhibitors of mitosis progression and that a significant proportion of these, but not all, are likely to be direct tubulin depolymerising agents. A spread of bioactivity levels as assessed by cell-based assays can be influenced by such variables as solubility, cell permeability, compartmental localization and compound stability in addition to genuine drug-receptor interactions. One of the novel compounds with inhibitory activity on tubulin polymerization, JJ78:1, was subjected to SAR analysis leading to the identification of JJ78:12, which is significantly more potent than JJ78:1 and shows clear indications of antitumor activity in a mouse xenograft model.
The clinical success of tubulin targeting drugs such as the taxanes and vinca alkaloids has led to the drive to discover novel compounds that act in a similar manner but with improvements in the areas of potency, toxicity, pharmacology, synthetic tractability and formulation. Several classes of new chemotherapeutics that prevent tubulin polymerization include combretastatin and semi-synthetic vinca alkaloid derivatives such as vinflunine.23 It is important to note that it is possible to administer a therapeutically active amount of JJ78:12 on a daily basis and that the synthesis of this compound is relatively simple. These properties of JJ78:12 suggest that this compound or its derivatives could be interesting alternatives to current tubulin poisons.
In order to support that JJ78:12 activates p53 mainly through its ability to inhibit of tubulin polymerization, we have compared its effects on p53 levels with those of nocodazole, a well established tubulin poison. An increase in p53 levels is apparent only after prolonged treatment with JJ78:12, a feature that is also characteristic for nocodazole and other tubulin poisons.21 Another feature shared between JJ78:12 and nocodazole is the appearance of different levels of p53 staining amongst the micronuclei of cells that have undergone mitotic slippage. Studying the underlying causes for the differences in p53 detection observed amongst micronuclei may be of interest in understanding aspects of p53 regulation. One possibility is that different micronuclei contain different amounts of factors that determine the rate of p53 degradation or export to the cytoplasm. Alternatively or additionally, it is possible that the efficiency of p53 import from the cytoplasm varies amongst micronuclei.
In summary, we have identified a series of bioactive small molecules from a cell-based screen and through a series of simple secondary assays, we have been able to quickly identify the mechanism of action for twelve of them as inhibitors of tubulin polymerization. An optimized analogue of one of these compounds, JJ78:12 operates at two digit nanomolar concentrations in cells and clearly reduces tumor growth in vivo. Together with our previous report,6 these results stress that using a simple and inexpensive p53 activation cell based assay can lead to the identification of compounds with potential therapeutic activity and identifiable mechanisms of action.
Materials and Methods
Cell lines
ARN8 cells derive from the A375 melanoma cells and have been previously described.24 MCF-7 and HeLa cells were obtained from the American Type Culture Collection and grown in Dulbecco’s modified Eagle medium supplemented with 10% FCS and 50 μg/ml gentamycin. SKNSH cell lines (CMVNeo and DNp53) and T22- RGC-ΔFos-LacZ cells were also cultured in this manner but the media was supplemented with 1 mg/ml G418 (Gibco). p53 transcriptional activity in the T22- RGC-ΔFos-LacZ was measured as described (Lain et al., Cancer Cell). Two dimensional FACS analysis of SKNSH cell lines (CMVNeo and DNp53) has been described before.14
Immunofluorescence staining and imaging of cells
Cells were seeded into 2 well glass slide chambers (Nunc) at 5 × 104 cells per chamber, incubated for 24 hours and treated as specified. Cells were washed in DPBS (Gibco) before being submerged in ice-cold 1:1 v/v mix of methanol and acetone for 8 minutes. Fixed cells were then washed in DPBS supplemented with 0.1% v/v Tween-20 (DPBST) 4 times for 5 minutes each. Cells were then blocked using 5% w/v non-fat milk in DPBST. Primary and secondary antibodies were diluted in this solution and all washes were carried out in DPBST. After blocking, cells were incubated with primary antibodies for 1 hour at room temperature before being washed 4 times for 5 minutes. Cells were then incubated with the corresponding species type secondary fluorescently labelled antibodies (Alexa Fluor 488 and 594 from Molecular Probes, Invitrogen) in a zero light environment for 45 minutes at room temperature. Cells were again washed twice for 5 minutes before being submerged in Hoescht 33258 DNA stain (Sigma) for 1 minute at 40 μg/ml. Once removed from Hoescht stain glass coverslips were mounted onto the cells using Hydromount (National Diagnostic) supplemented with 2.5% Dabco (1,4-Diazabicyclo-[2.2.2.]octane) supplied by Sigma. Samples were visualized using an Axiovert 200 M microscope from Zeiss powered by the Volocity software from Improvision. Images were captured using a Hamamatsu Orca ER camera and the light source was provided by the Exfo X-cite 120 light source.
Fluorescence based in vitro tubulin polymerization assays
In vitro tubulin polymerization assays were carried out following manufacturer’s instructions (Cytoskelton). This involved firstly reconstituting purified bovine neuronal tubulin to a final concentration of 10 mg/ml in ‘Buffer 1’ (80 mM piperazine-N,N’-bis[2-ethanesulfonic acid] sequisodium salt; 2.0 mM magnesium chloride; 0.5 mM ethylene glycol-bis(b-aminuteso-ethyl ether) N,N,N’,N’-tetra-acetic acid, pH6.9 10 μM fluorescent reporter) supplemented with GTP (10 mM). This tubulin was then further diluted to 2 mg/ml in a reaction buffer containing Buffer 1 and a Tubulin Glycerol Buffer (same as Buffer 1 with 60% glycerol v/v and no fluorescent reporter) to produce a 20% glycerol concentration. GTP was added to this mix to a final concentration of 1 mM. Compounds were then added (the final concentration of DMSO was 0.5% v/v in all samples). Polymerization was monitored by measuring fluorescence (Ex. 355 nm Em. 460 nm) every minute for 60 minutes using a plate reader heated to 37°C (Molecular Devices SPECTRA MAX GEMINI XS driven by SoftMax Pro 4.6). Data was exported to Microsoft Excel for analysis where the data was normalized by subtracting the fluorescence reading at zero minutes from each point.
Chemical synthesis
Ethyl 4-(2′,5′-difluorobenzoyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (JJ78:1)
Aluminium chloride (0.88 g, 6.6 mmol) and 2,5-difluorobenzoyl chloride (1.13 g, 6.4 mmol) were stirred together at ambient temperature in chlorobenzene (10 mL) for 5 minutes before cooling to 0°C. A solution of ethyl 3,5-dimethylpyrrole-2-carboxylate (3.0 mmol, 0.50 g) in chlorobenzene (5 mL) was then added and the mixture was stirred at 0°C for 90 minutes, then for 2.5 hours at ambient temperature. The reaction mixture was heated to 80°C for 2 hours then cooled back to ambient temperature. The resulting solution was poured onto ice-water (50 mL) and was extracted with dichloromethane. The organic phase was washed with 1 M aqueous NaOH solution (1 × 30 mL), saturated brine (1 × 30 mL) and dried over MgSO4. Concentration in vacuo provided a residue that was further purified by flash silica chromatography eluting with dichloromethane to give the desired compound JJ78:1 as a white solid (0.51 g, 55%): m.p. 137-138°C; 1H NMR (300 MHz, CDCl3) δ ppm 1.38 (3 H, t, J = 7.2 Hz), 2.28 (3 H, s), 2.31 (3 H, s), 4.34 (2 H, q, J = 7.0 Hz), 7.01-7.23 (3 H, m), 9.15 (1 H, br. s.); MS (ES+) m/z 330 [M + Na]+; HRMS calc’d for C16H15F2NO3Na 330.0918, found 330.0912; CHN analysis (C16H15NO3F2·0.1H2O) requires: C, 62.17; H, 4.96; N, 4.53; Found: C, 62.02; H, 4.82; N, 4.46. A sample of JJ78:1 was recrystallized from chloroform to give needle-like crystals that were suitable for X-ray crystallographic analysis.
General procedure for the parallel synthesis of JJ78:1 analogues
Aluminium chloride (147 mg, 1.1 mmol) was added to chlorobenzene (2 mL) in each of the parallel reactor tubes. The appropriate aroyl chloride was then added with stirring under argon atmosphere for 5 minutes. The parallel reactor was then cooled to −3°C in an ice-brine bath. To each reaction tube was added a solution of the pyrrole 1 (0.5 mmol) in chlorobenzene (1 mL) and the reaction stirred for 90 minutes at −3°C. The reaction was subsequently stirred at ambient temperature for 1 hour followed by heating to 80°C for 2 hours. Each reaction was then submitted to an aqueous work up as described in the synthesis of JJ78:1. Desired products JJ78:7-12 and JJ78:14-15 were isolated (24-82% yield) following purification by flash silica chromatography and subsequent re-crystallization.
Ethyl 4-(3′,4′-dichlorobenzoyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (JJ78:7)
Synthesized according to general procedure from 1 and 3,4-dichlorobenzoyl chloride. Yellow crystals, 41 mg (24%): 1H NMR (300 MHz, CDCl3) δ ppm 1.38 (3 H, t, J = 7.1 Hz), 2.22 (3 H, s), 2.28 (3 H, s), 4.35 (2 H, q, J = 7.1 Hz), 7.54 (2 H, m), 7.80 (1 H, s), 9.52 (1 H, br. s); 13C NMR (75 MHz, CDCl3) δ ppm 12.46, 13.60, 14.47, 60.59, 118.81, 122.42, 128.27, 128.87, 130.58, 131.14, 132.95, 136.57, 137.37, 139.93, 161.78, 191.18.
Ethyl 4-(4′-chlorobenzoyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (JJ78:8)
Synthesized according to general procedure from 1 and p-chlorobenzoyl chloride. 80 mg (52%): 1H NMR (300 MHz, CDCl3) δ ppm 1.39 (3 H, t, J = 7.1 Hz), 2.24 (3 H, s), 2.28 (3 H, s), 4.36 (2 H, q, J = 7.1 Hz), 7.57 (4 H, dd, J = 8.4, 75.8 Hz), 9.60 (1 H, br. s); 13C NMR (75 MHz, CDCl3) δ ppm 12.40, 13.52, 14.47, 60.50, 118.63, 122.86, 128.72, 128.98, 130.65, 137.06, 138.53, 138.61, 161.85, 192.58.
Ethyl 4-(4′-methylbenzoyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (JJ78:9)
Synthesized according to general procedure from 1 and p-methylbenzoyl chloride. Beige crystals, 73 mg (51%): 1H NMR (300 MHz, CDCl3) δ ppm 1.36 (3 H, t, J = 7.1 Hz), 2.23 (3 H, s), 2.25 (3 H, s), 2.41 (3 H, s), 4.33 (2 H, q, J = 7.1 Hz), 7.44 (4 H, dd, J = 8.0, 121.2 Hz), 9.38 (1 H, br. s); 13C NMR (75 MHz, CDCl3) δ ppm 12.33, 13.46, 14.51, 21.68, 30.99, 60.37, 118.38, 123.44, 129.08, 129.49, 135.53, 137.54, 142.97, 161.87, 193.69.
Ethyl 4-(4′-methoxybenzoyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (JJ78:10)
Synthesized according to general procedure from 1 and p-methoxybenzoyl chloride. Off white solid, 88 mg (58%): 1H NMR (300 MHz, CDCl3) δ ppm 1.37 (3 H, t, J = 7.1 Hz), 2.24 (3 H, s), 2.25 (3 H, s), 3.87 (3 H, s), 4.33 (2 H, q, J = 7.1 Hz), 7.33 (4 H, dd, J = 7.8, 243.7 Hz), 9.30 (1 H, br. s).
Ethyl 4-benzoyl-3,5-dimethyl-1H-pyrrole-2-carboxylate (JJ78:11)
Synthesized according to general procedure from 1 and benzoyl chloride. White needles, 95 mg (70%): 1H NMR (300 MHz, CDCl3) δ ppm 1.36 (3 H, t, J = 7.1 Hz), 2.23 (3 H, s), 2.24 (3 H, s), 4.33 (2 H, q, J = 7.1 Hz), 7.51 (3 H, m), 7.72 (2 H, m), 9.52 (1 H, br. s); 13C NMR (75 MHz, CDCl3) δ ppm 12.29, 13.46, 14.43, 60.37, 118.43, 123.14, 128.33, 129.14, 132.12, 136.98, 140.27, 161.88, 193.94.
Ethyl 4-(3′-chlorobenzoyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (JJ78:12)
Synthesized according to general procedure from 1 and m-chlorobenzoyl chloride. White solid, 64 mg (42%): m.p. 137-138°C; 1H NMR (300 MHz, CDCl3) δ ppm 1.39 (3 H, t, J = 7.2 Hz), 2.23 (3 H, s), 2.27 (3 H, s), 4.35 (2 H, q, J = 7.2 Hz), 7.35-7.45 (1 H, m), 7.48-7.56 (1 H, m), 7.60 (1 H, d, J = 7.7 Hz), 7.66-7.74 (1 H, m), 9.13 (1 H, br. s); MS (ES+) m/z 328 [M + Na]+, (ES−) m/z 304 [M - 1]-; HRMS calc’d for C16H16ClNO3Na 328.0716, found 328.0714; CHN analysis (C16H16ClNO3) requires: C, 62.85; H, 5.27; N, 5.48; Found: C, 62.78; H, 5.16; N, 4.46.
Ethyl 4-(2′,5′-difluorobenzyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (JJ78:13)
JJ78:1 (0.5 mmol, 0.15 g) was treated with triethylsilane (0.32 mL, 2 mmol) in trifluoroacetic acid (0.35 mL) in a sealed reaction tube heated to 70°C for 6 hours. The reaction mixture was concentrated in vacuo, redissolved in Et2O (50 mL) and washed with 1 M aqueous NaOH (1 × 30 mL) and saturated brine (1 × 30 mL). The organic layer was then concentrated in vacuo to give a residue that was purified by flash silica chromatography eluting with dichloromethane. The resulting solid was recrystallization from methanol to give the desired product JJ78:13 as an off-white solid (64 mg, 44%): 1H NMR (300 MHz, CDCl3) δ ppm 1.37 (3 H, t, J = 7.1 Hz), 2.20 (3 H, s), 2.21 (3 H, s), 3.74 (2 H, s), 4.32 (2 H, q, J = 7.0 Hz), 6.59 (1 H, ddd, J = 3.2, 6.0, 9.0, Hz), 6.76-6.88 (1 H, m), 6.97 (1 H, td, J = 4.6, 9.0 Hz), 9.00 (1 H, br. s); MS (CI+) m/z 294 [M + 1]+; HRMS calc’d for C16H17F2NO2 294.1306, found 294.1302.
Ethyl 4-(2′-methoxybenzoyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (JJ78:14) and ethyl 4-(2′-hydroxybenzoyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (JJ78:15)
Synthesized according to general procedure from 1 and o-methoxybenzoyl chloride. Beige solid, 123 mg (82%). JJ78:14: 1H NMR (300 MHz, CDCl3) δ ppm 1.35 (3 H, t, J = 7.1 Hz), 2.21 (3 H, s), 2.25 (3 H, s), 3.78 (3 H, s), 4.31 (2 H, q, J = 7.1 Hz), 6.99 (2 H, m), 7.26 (1 H, dd, J = 1.8, 7.4 Hz), 7.40 (1 H, ddd, J = 1.8, 7.4, 8.3 Hz), 8.99 (1 H, br. s); 13C NMR (75 MHz, CDCl3) δ ppm 11.76, 13.94, 14.41, 55.70, 60.31, 111.34, 118.10, 120.66, 123.49, 128.61, 130.35, 131.28, 131.98, 139.01, 156.61, 161.86, 192.46; MS (ES+) m/z 324 [M + Na]+; HRMS calc’d for C17H19NO4Na 324.1212, found 324.1208.
JJ78:15 was isolated as a by-product. Off-white solid, 5 mg. JJ78:15: 1H NMR (75 MHz, CDCl3) δ ppm 1.38 (3 H, t, J = 7.1 Hz), 2.27 (3 H, s), 2.29 (3 H, s), 4.35 (2 H, q, J = 7.1 Hz), 6.86 (1 H, m), 7.03 (1 H, dd, J = 0.7, 8.3 Hz), 7.48 (2 H, m), 9.13 (1 H, br. s), 12.21 (1 H, s); MS (ES+) m/z 310 [M + Na]+; HRMS calc’d for C16H17NO4Na 310.1055, found 310.1052.
Ethyl 4-(5′-fluoro-2′-morpholin-4-yl-benzoyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (JJ78:16)
A mixture of JJ78:1 (100 mg, 0.33 mmol), morpholine (4 mL) and water (0.2 mL) was heated to 150°C in a sealed tube under microwave irradiation for 2 hours. The reaction mixture was then concentrated in vacuo and the resulting residue partitioned between EtOAc (30 mL) and water (20 mL). The aqueous layer was acidified to pH 2 with dilute HCl and extracted with EtOAc (3 × 30 mL). The combined organic layers were then washed with saturated brine (1 × 50 mL), dried (MgSO4) and concentrated in vacuo. The resulting solid was purified by recrystallization from ethanol to give the desired compound JJ78:16 as an off-white solid (85 mg, 69%): 1H NMR (300 MHz, CDCl3) δ ppm 1.37 (3 H, t, J = 7.2 Hz), 2.17 (3 H, s), 2.21 (3 H, s), 2.83-2.96 (4 H, m), 3.39-3.51 (4 H, m), 4.33 (2 H, q, J = 7.0 Hz), 6.98 (1 H, d, J = 8.7 Hz), 7.04-7.18 (2 H, m), 9.37 (1 H, br. s); 13C NMR (75 MHz, CDCl3) δ ppm 11.58, 13.69, 53.05, 60.47, 66.84, 116.04 (d, J = 23.4 Hz), 117.64 (d, J = 22.1 Hz), 118.15, 119.84 (d, J = 7.7 Hz), 122.68, 130.08, 137.69, 138.56, 146.35, 158.66 (d, J = 243.7 Hz), 161.72, 192.77; MS (ES+) m/z 397 [M + Na]+; HRMS calc’d for C20H23N2O4NaF 397.1540, found 397.1522.
Pharmacokinetic studies of JJ78:12 in mice
Three mice were dosed by i.p. injection at 30 mg/kg with a 10 mM JJ78:12 solution in 5% DMSO and 20% cyclodextrin. 10 μl of blood were collected directly from the tail vein at 5, 15, 30, 60, 120, 240, 360 and 480 minutes, mixed with 10 μl of heparin solution (15 IU/ml) in a 1.5 ml tube and kept on ice until storage at −80°C. To each blood sample, after thawing at room temperature, 20 μl of internal standard (gemcitabine 10 μg/ml in methanol) and 80 μl of methanol were added, before being vortexed for 1 minute. Subsequently, 80 μl of sulfosalicylic acid (10%) were added and samples vortexed for 1 minute, followed by a 5 minute sonication. The samples were then centrifuged at 2500 g for 10 min. Supernatants were transferred to vials and analysed by LC Q-TOF. In parallel, a calibration curve of JJ78:16 was carried out by spiking a fixed amount in blood (0-2500 ng/ml), followed by the extraction procedure outlined above. The percentage of extraction of JJ78:12 from the blood sample was equal to 62.5%. LC Q-TOF analysis was carried out using a Waters 2695 HPLC coupled to a Q-TOF Micro mass (MicroMass, UK) using the positive electrospray ionization (ESI) mode. Samples were analysed on a Luna Phenyl-Hexyl (150 mm × 3 mm, 5μm particle size, Phenomenex). The injection volume was 100 μl. The following elution program was used: a temperature of 20°C, a flow rate of 0.2 ml/min, and an isocratic mobile phase composed of 5% water, 90% methanol containing 0.1% formic acid and 5% acetonitrile containing 0.1% formic acid. The capillary voltage was adjusted to 4000 V, sample cone to 31 V, the cone to 50 L/h, the desolvation temperature to 300°C with a nitrogen flow to 400 L/h, and the collision energy to 28.0 V. The transitions monitored were 306.17 > 139.03, 264.10 > 112.08 for JJ78:12 and gemcitabine respectively. MS-MS data were processed using the QuanLynx quantify option of MassLynx software, version 4.1, from Micromass. Pharmacokinetic parameters were calculated using the WinNonLin software, version 4.1.
In vivo efficacy of compounds on the growth of xenograft tumors
Female SCID mice (Harlan) were injected subcutaneously with 1 × 106 ARN8 cells suspended in 1:1 v/v solution of DMEM containing resuspended cells: matrigel (BD Biosciences). ARN8 cells had been harvested when approximately 60-70% confluent. Tumors were allowed to establish and reach a size of approximately 20 mm3. Mice were treated with 30 mg/kg JJ78:12 in 20% cyclodextrin everyday by intraperitoneal injection. Control animals were treated with the vehicle solution containing cyclodextrin 20% w/v and DMSO 10% v/v. Tumor diameters were measured using calipers and volumes were calculated using the equation V = π4/3[(d1 + d2)/4]3. The appropriate measure of centrality (mean or median) of tumor sizes was calculated for each time point, as well as the corresponding 95% confidence intervals. Comparison of control and drug treated tumor size distributions were made by statistical tests. A Two Sample t-test or a Mann-Whitney U-test was appropriately chosen depending on whether the data was normally or non-normally distributed. Normality of data was determined by an Anderson-Darling test. An alpha-level of 0.05 was considered appropriate for determination of statistical significance. All animal experiments were in compliance with the United Kingdom Coordinating Committee on Cancer Research.
Supplementary Material
Acknowledgements
The authors thank Tenovus Scotland, Breast Cancer Research (Scotland), Medical Research Council, The Royal Society (NJW University Research Fellowship) and CRUK for financial support.
Footnotes
References
- 1.Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10:1321–8. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 2.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 3.Peterson JR, Mitchison TJ. Small molecules, big impact: a history of chemical inhibitors and the cytoskeleton. Chem Biol. 2002;9:1275–85. doi: 10.1016/s1074-5521(02)00284-3. [DOI] [PubMed] [Google Scholar]
- 4.Schreiber SL. The small-molecule approach to biology. Chemical and Engineering News. 2003;81:51–61. [Google Scholar]
- 5.Zheng XS, Chan TF, Zhou HH. Genetic and genomic approaches to identify and study the targets of bioactive small molecules. Chem Biol. 2004;11:609–18. doi: 10.1016/j.chembiol.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 6.Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, Thompson A, Mathers J, Holland SJ, Stark MJ, Pass G, Woods J, Lane DP, Westwood NJ. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454–63. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casenghi M, Mangiacasale R, Tuynder M, Caillet-Fauquet P, Elhajouji A, Lavia P, Mousset S, Kirsch-Volders M, Cundari E. p53-independent apoptosis and p53-dependent block of DNA rereplication following mitotic spindle inhibition in human cells. Exp Cell Res. 1999;250:339–50. doi: 10.1006/excr.1999.4554. [DOI] [PubMed] [Google Scholar]
- 8.Liou JP, Chang CW, Song JS, Yang YN, Yeh CF, Tseng HY, Lo YK, Chang YL, Chang CM, Hsieh HP. Synthesis and structure-activity relationship of 2-aminobenzophenone derivatives as antimitotic agents. J Med Chem. 2002;45:2556–62. doi: 10.1021/jm010365+. [DOI] [PubMed] [Google Scholar]
- 9.Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006;209:13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- 10.Gurley LR, D’Anna JA, Barham SS, Deaven LL, Tobey RA. Histone phosphorylation and chromatin structure during mitosis in Chinese hamster cells. Eur J Biochem. 1978;84:1–15. doi: 10.1111/j.1432-1033.1978.tb12135.x. [DOI] [PubMed] [Google Scholar]
- 11.Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, Grushcow JM, Brame CJ, Caldwell JA, Hunt DF, Lin R, Smith MM, Allis CD. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000;102:279–91. doi: 10.1016/s0092-8674(00)00034-9. [DOI] [PubMed] [Google Scholar]
- 12.Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–7. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 13.Finzer P, Aguilar-Lemarroy A, Rosl F. The role of human papillomavirus oncoproteins E6 and E7 in apoptosis. Cancer Lett. 2002;188:15–24. doi: 10.1016/s0304-3835(02)00431-7. [DOI] [PubMed] [Google Scholar]
- 14.Smart P, Lane EB, Lane DP, Midgley C, Vojtesek B, Lain S. Effects on normal fibroblasts and neuroblastoma cells of the activation of the p53 response by the nuclear export inhibitor leptomycin B. Oncogene. 1999;18:7378–86. doi: 10.1038/sj.onc.1203260. [DOI] [PubMed] [Google Scholar]
- 15.Haggarty SJ, Mayer TU, Miyamoto DT, Fathi R, King RW, Mitchison TJ, Schreiber SL. Dissecting cellular processes using small molecules: identification of colchicine-like, taxol-like and other small molecules that perturb mitosis. Chem Biol. 2000;7:275–86. doi: 10.1016/s1074-5521(00)00101-0. [DOI] [PubMed] [Google Scholar]
- 16.Yamakawi I, Matsushita Y, Asaka N, Ohmori K, Nomura N, Ogawa K. Synthesis and aldose reductase inhibitory activity of acetic acid derivatives of pyrrolo[1,2-c]imidazole. Eur J Med Chem. 1993;28:481–90. [Google Scholar]
- 17.Mizen L, Burton G. The use of esters as prodrugs for oral delivery of β-lactam antibiotics. Pharmaceutical Biotechnology. 1998;11:345–65. doi: 10.1007/0-306-47384-4_15. [DOI] [PubMed] [Google Scholar]
- 18.Tripathi A, Fornabaio M, Kellogg GE, Gupton JT, Gewirtz DA, Yeudall WA, Vega NE, Mooberry SL. Docking and hydropathic scoring of polysubstituted pyrrole compounds with antitubulin activity. Bioorganic and Medicinal Chemistry. 2008;16:2235–42. doi: 10.1016/j.bmc.2007.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tani M, Ariyasu T, Nishiyama C, Hagiwara H, Watanabe T, Yokoyama Y, Murakami Y. β-Acylation of ethyl pyrrole-2-carboxylate by Friedel-Crafts acylation; scope and limitations (synthetic studies on indoles and related compounds. XXXVIII) Chemical and Pharmaceutical Bulletin. 1996;44:48–54. [Google Scholar]
- 20.Yam CH, Fung TK, Poon RY. Cyclin A in cell cycle control and cancer. Cell Mol Life Sci. 2002;59:1317–26. doi: 10.1007/s00018-002-8510-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blagosklonny MV. Prolonged mitosis versus tetraploid checkpoint: how p53 measures the duration of mitosis. Cell Cycle. 2006;5:971–5. doi: 10.4161/cc.5.9.2711. [DOI] [PubMed] [Google Scholar]
- 22.Demidenko ZN, Kalurupalle S, Hanko C, Lim CU, Broude E, Blagosklonny MV. Mechanism of G1-like arrest by low concentrations of paclitaxel: next cell cycle p53-dependent arrest with sub G1 DNA content mediated by prolonged mitosis. Oncogene. 2008;27:4402–10. doi: 10.1038/onc.2008.82. [DOI] [PubMed] [Google Scholar]
- 23.Attard G, Greystoke A, Kaye S, De Bono J. Update on tubulin-binding agents. Pathol Biol (Paris) 2006;54:72–84. doi: 10.1016/j.patbio.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 24.Blaydes JP, Hupp TR. DNA damage triggers DRB-resistant phosphorylation of human p53 at the CK2 site. Oncogene. 1998;17:1045–52. doi: 10.1038/sj.onc.1202014. [DOI] [PubMed] [Google Scholar]
- 25.Bartkova J, Bartek J, Vojtesek B, Lukas J, Rejthar A, Kovarik J, Millis RR, Lane DP, Barnes DM. Immunochemical analysis of the p53 oncoprotein in matched primary and metastatic human tumours. Eur J Cancer. 1993;29:881–6. doi: 10.1016/s0959-8049(05)80431-9. [DOI] [PubMed] [Google Scholar]
- 26.Woods AL, Hall PA, Shepherd NA, Hanby AM, Waseem NH, Lane DP, Levison DA. The assessment of proliferating cell nuclear antigen (PCNA) immunostaining in primary gastro-intestinal lymphomas and its relationship to histological grade, S + G2 + M phase fraction (flow cytometric analysis) and prognosis. Histopathology. 1991;19:21–7. doi: 10.1111/j.1365-2559.1991.tb00890.x. [DOI] [PubMed] [Google Scholar]
- 27.Midgley CA, Fisher CJ, Bartek J, Vojtesek B, Lane D, Barnes DM. Analysis of p53 expression in human tumours: an antibody raised against human p53 expressed in Escherichia coli. J Cell Sci. 1992;101:183–9. doi: 10.1242/jcs.101.1.183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.