

Abstract
We present a large portion of the transcriptome of Zea mays, including ESTs representing 484,032 cDNA clones from 53 libraries and 36,565 fully sequenced cDNA clones, out of which 31,552 clones are non-redundant. These and other previously sequenced transcripts have been aligned with available genome sequences and have provided new insights into the characteristics of gene structures and promoters within this major crop species. We found that although the average number of introns per gene is about the same in corn and Arabidopsis, corn genes have more alternatively spliced isoforms. Examination of the nucleotide composition of coding regions reveals that corn genes, as well as genes of other Poaceae (Grass family), can be divided into two classes according to the GC content at the third position in the amino acid encoding codons. Many of the transcripts that have lower GC content at the third position have dicot homologs but the high GC content transcripts tend to be more specific to the grasses. The high GC content class is also enriched with intronless genes. Together this suggests that an identifiable class of genes in plants is associated with the Poaceae divergence. Furthermore, because many of these genes appear to be derived from ancestral genes that do not contain introns, this evolutionary divergence may be the result of horizontal gene transfer from species not only with different codon usage but possibly that did not have introns, perhaps outside of the plant kingdom. By comparing the cDNAs described herein with the non-redundant set of corn mRNAs in GenBank, we estimate that there are about 50,000 different protein coding genes in Zea. All of the sequence data from this study have been submitted to DDBJ/GenBank/EMBL under accession numbers EU940701–EU977132 (FLI cDNA) and FK944382-FL482108 (EST).
Electronic supplementary material
The online version of this article (doi:10.1007/s11103-008-9415-4) contains supplementary material, which is available to authorized users.
Keywords: Full-length cDNA, Corn genome, Alternative splicing, Promoters, Gene prediction, GC content
Introduction
Corn (Zea mays) is an important worldwide crop that is relied upon for human food, animal feed and for starch ethanol production. In 2007, 93.6 million acres were planted in the US and over 13.1 billion bushels of corn were harvested. Over the last 50 years, there has been a steady increase in corn grain yield averaging an increase of 1.6 bushels per acre per year. In addition, the number of acres planted with corn has gone from 68 million in 1966 to 93.6 million in 2007 (National Agricultural Statistics Service, http://www.nass.usda.gov/). The increasing use of corn for the production of ethanol has further added to the need for increased production.
Due to the commercial importance of the crop, there is significant interest in understanding the underlying DNA sequence. The corn genome is consequently being sequenced, producing on average over 800 BAC clone sequences every month during 2007, and a draft sequence has been announced (Wilson 2008). The sequencing of the genome is a large undertaking not only because the size of the genome is nearly as large as mammalian genomes (2,800 million base pairs (Mbp) in contrast to Arabidopsis (130 Mbp) and rice (430 Mbp)) but also due to the abundance of mobile elements within the genome. Nevertheless, the genome sequence will be an invaluable asset to accelerate further enhancements in the improvements of this crop.
Because corn has been an important plant cultivated for centuries, there is a wealth of information available from phenotypic descriptions, disease effects and resistance, marker data and quantitative trait loci. As has been done for rice, this information can be synergistically integrated with the genome sequence and linked with well-defined genes.
The identification of genes that are important in yield and yield preservation, which can serve as markers for polymorphisms and their use in improved breeding, offers a huge advantage toward that goal. While the genome sequence will be completed shortly, a quicker and complimentary approach to identifying a large number of corn genes is EST and full-length cDNA sequencing. These resources will prove invaluable for annotating the genomes of corn and other monocots and as substrates for transgenic improvement of crops. As in Arabidopsis and rice, these tools will prove to be critical in speeding up the genetic improvement of corn.
The complete genome sequences of several plant species are known and the rate at which whole genomes are being sequenced is increasing. Correct annotation of these genomes remains problematic in spite of gene prediction algorithms becoming ever more sophisticated. ESTs, full-length cDNAs and tiling arrays are extremely helpful toward annotating genomes correctly. As more full-length cDNAs become available from different plant species, the accuracy of annotations improves not only for the newly sequenced genomes but also for evolutionarily related species, including those previously annotated.
In the last several years two research groups submitted greater than 5,000 full-length corn cDNAs to GenBank. Lai et al (2004) sequenced 5′ and 3′ ends of ~13,000 cDNAs from three endosperm specific libraries. 2,168 unique sequences were overlapped with just the 5′ and 3′ reads and 992 additional clones were overlapped by primer walking on the 3,400 clones that had paired reads but where the reads did not overlap. They assembled a unique set of 5,326 non-redundant clones when the transposon-related sequences were eliminated. Interestingly, 22% of these did not match rice sequences, suggesting that they were lost from rice or gained in maize over the last 50 million years. Jia et al. (2006) sequenced 20,000 cDNA clones from PEG-treated corn tissues and obtained 2,073 clones that were scored as full-length. Of the 84 clones that they could align to annotated maize BAC sequences, only 51% were annotated correctly. In addition to these full-length sequences, and others that have been submitted to GenBank by individual researchers, there are 1,923,065 maize Genome Survey Sequences and 1,014,105 ESTs.
Here we present sequences from our corn sequencing program based on 484,032 cDNA clones made from a diversity of libraries. The 5′ ESTs fall into 63,476 clusters. 36,432 cluster representatives, deemed to be full-length and novel at the time of being selected, have been fully sequenced. Within these, we identified 9,951 that are non-redundant and where we have high confidence they are full-length and lacking any errors. All sequences are available in GenBank. We have added to this set 133 cDNA sequences created from publicly available ESTs from GenBank. Analysis of these 10,084 highest quality clones indicates that they can be divided into two groups based solely on their GC content. We found this bimodal distribution in gene catalogs from other grasses, but genes from dicots and other monocots have a unimodal distribution of GC content. High GC content genes in grasses are less homologous to dicot genes than the low GC content genes suggesting a large accumulation of novel gene sequences was associated with the divergence of grasses from other plants over 60 million years ago. In addition, the genes with high GC content have a much smaller number of introns, further suggesting the novel genes may have originated in a non-plant species or from reverse transcription of RNAs.
Methods
Full-length cDNA library construction and sequencing
The tissues used to generate cDNA libraries were derived from a number of hybrids that were available to us at different parts of the growing season. The libraries generated from the Mixed male/female infl., root tissues all came from Pioneer Hi-Bred International, Inc Hybrid 35A19. The rest of the libraries were generated from tissues obtained from several other hybrids. As our goal was to generate unique full-length cDNAs, the tissues and RNAs from various libraries could have been mixed. As such, these ESTs are useful to look at polymorphisms between ESTs but not between various parents except for those in the Mixed male/female infl., root libraries.
For each library, 130 μg of mRNA was ligated with 5′ end oligonucleotide linkers (5′-rGrCrArCrGrArGrArCrCrAUUrArCrCUrArGrArArCrAUrCrCUrArAUrCrGrArArArA-3′, or 5′-rCrGUrCUrCrArCrCrCrCUrArGrArArArArArA-3′ for mRNA isolated from various stress treatments). After ligation, excess free oligonucleotide was removed by column chromatography. Approximately 10 μg of oligonucleotide ligated mRNA was obtained and used for the cDNA synthesis using SuperscriptTM II reverse transcriptase following the instructions of the vendor (Invitrogen, Carlsbad, CA, USA). The oligo-dT primer (5′-GTACGTCTCGAGTTTTTTTTTTTTTTTTTTVN-3′) was annealed to mRNA. After removal of RNA by alkaline hydrolysis, first-strand cDNA was precipitated using isopropanol to eliminate the excess free primer. Second strand cDNA was synthesized with Klenow using the 5′-end oligonucleotide linkers 5′-ATCAAGAATTCGCACGAGACCATTACCTAGAACATCCTAATC-3′, or 5′-GATCGTAGAATTCGTCTCACCCCTAGAAA-3′. The quantity of double stranded DNA was estimated using a picogreen. ds cDNA was digested with EcoRI and XhoI and ligated into pBluescript SK+ (Stratagene, CA). Ligated cDNA was transformed to DH10B cell (Invitrogen, Carlsbad, CA).
In addition to the traditional approach for cDNA normalization (Soares et al. 1994; Carninci et al. 2003), RNA/DNA hybridization and double stranded deoxynuclease were employed to remove cDNAs that were already sequenced from existing libraries. The probe, or driver, was either mRNA or cRNA that was in vitro transcribed using T3 RNA polymerase from the plasmid DNA pool. 10 μg of the probe was hybridized to FL first-strand cDNA (Zhulidov et al. 2004) for 4 h. Kamchatka crab duplex-specific nuclease was added to break the DNA strand of the RNA/DNA duplex. The unhybridized intact single-stranded cDNA was used to complete library construction.
The development of a transposon full length sequencing methodology was considered for the purpose of producing a high throughput alternative to primer walking. The main consideration was pooling a large number of cDNAs together in one transposon reaction and then deconvoluting these through sequencing. A 5′ tag was used as an anchor to facilitate the deconvolution. The method was developed based on the GPS-1 Genome Priming System from New England Biolabs. High quality plasmid DNA from 16 to 32 cDNA plasmids were normalized for concentration and subjected to the TnsABC transposase reaction. During this reaction, a single transposon was inserted into each plasmid. Post-reaction cleanup consisted of ethanol precipitation and elution in water. Each pooled sample was electroporated and grown in SOC media for 1 h at 37°C. Using large square Petri plates containing SOC agar, the cells were plated and incubated for 16–18 h. After colonies were of appropriate size they were picked and moved to 384 well plates for processing. Since transposons insert randomly within a plasmid, all colonies needed to be screened to determine whether the transposon had inserted into the vector backbone or into the cDNA insert. To facilitate this PCR, oligos were designed to amplify the 3 kb vector backbone and a simple size scan was done on agarose gels. If the band size was larger than expected, indicating a transposon within the vector, the sample was discarded. The remainder of the colonies, in which the transposon inserted into the cDNA, went through a sequencing process where the DNA sequencing reaction was primed off the inserted transposon. For each pool of 16–32 cDNAs, a 384 well plate was sequenced and the resulting reads were clustered using by the 5′ tag of the cDNAs that went into the pool.
MegaBACE sequencers (Amersham Pharmacia/Molecular Dynamics) were used at Genset to generate the 5′ ESTs while 377 sequencers (Applied Biosystems) were used for full-length cDNA sequencing. Ceres used 3700 and 3730xl sequencers (Applied Biosystems) for both 5′ ESTs and full-length cDNA sequencing. Quality scores for the Genset sequences are not available making it difficult to identify reliable polymorphisms in the clusters. The type of sequencer used for each sequence is noted in each GenBank entry.
EST clustering
Clustering of 5′-tags was done using the Washington University Blastn program (Gish 1996–2004). Two sequences were clustered together if there were no more than six mismatches in any 30-nucleotide window of their blast alignment and their alignment covered the entire overlapping region. Information about the relationship between selected clones and other 5′-tags, including their relative start positions and other relevant information was stored in an Oracle database (Alexandrov et al. 2006).
Gene models
All ESTs, Ceres cDNA, and public mRNA were aligned against the corn genomic sequences available from GenBank using the spliced alignment method (Alexandrov et al. 2006). Alignments with lower than 98% overall identity (i.e., the ratio of matching nucleotides on the transcript to the overall length of the transcript) were discarded. If a transcript matched to more than one location on the genome, only the annotations with the best overall identity were considered for further analysis. The annotations were then checked for inversion. If an overwhelming majority of an annotation’s splice sites were non-canonical, they were compared to canonical splice sites of annotations on the opposing strand. If each non-canonical splice site had a matching canonical splice site on the opposing strand (within three nucleotides), the offending annotation was marked as inverted, and removed from further analysis. Mutually overlapping annotations were grouped into loci such that each annotation inside a locus overlaps with at least one other annotation in the locus.
Transcription start site prediction (TSSer algorithm)
We have developed a novel algorithm, called TSSer, to reliably predict positions of transcription start sites. We aligned EST and mRNA sequences against the corresponding genomic sequence using Washington University Blast. Alignments are further refined using the spliced alignment algorithm (Alexandrov et al. 2006). If there are two or more loci matching a transcript sequence, we select the best one based on the identity of the match. Positions of the 5′ ends of sequence alignments are clustered and the most frequent position that does not contradict the ORF prediction for a locus is designated as the best TSS for this locus. TSSer allows more accurate determination of the transcription start site, as compared to the traditional approach of using the longest cDNA for prediction.
Functional annotation
To obtain the GO annotations of corn proteins, we downloaded the GO annotations of Arabidopsis proteins from the TAIR website as the reference annotations, performed a BLAST similarity search of the protein sequences of our corn clones against the Arabidopsis protein sequences and propagated the GO annotations of the Arabidopsis proteins to the corn clones.
Since the Arabidopsis proteins from TAIR include clone sequences and genome predictions, to make a fair comparison between the two species, we used the expressed Arabidopsis protein sequences. We performed a similar BLAST search procedure to generate the GO annotations. The GO terms used in this annotation procedure are represented as GO slim terms, which are clusters of GO terms with similar functions in a broad sense.
Pfam annotation was performed for the two sets of proteins by blasting the protein sequences against the latest Pfam BLAST database downloaded from the Pfam website (ftp://selab.janelia.org/pub/Pfam/).
Results and discussion
cDNA libraries
Full-length cDNA libraries were prepared from a mixture of floral, root, stem and leaf tissues obtained from Pioneer Hi-Bred International, Inc. Hybrid 35A19, as well as from separate collections of embryo, callus, root, female flower and abiotic stress-induced tissues obtained from various other hybrids (Table 1). Size fractions were generated for some of the cDNA preparations, and normalization and depletion strategies were employed to enrich for novel clones (see Methods). Our depletion strategy involved hybridizing the most abundant clones (generally 5,000 clones) to a batch of cDNAs to minimize their presence and sequencing additional clones from the depleted library. This strategy proved effective at depleting the most frequent clones: histones, thioredoxins and ribosomal proteins from mixed libraries, and seed storage proteins from embryo libraries (data not shown). The number of clones, clusters and good full-length clones sequenced from libraries made from the various tissues and size fractions are summarized in Table 1. For the mixed tissue libraries it is not possible to gain insight into the gene distribution and frequency for the separate tissue types.
Table 1.
cDNA libraries generated from corn
| Library ID | Size fraction | Tissue | Number of clones | Number of clusters | Number of high quality selected clones |
|---|---|---|---|---|---|
| 176, 196, 213, 225, CL11 | Short | Mixed male/female infl., roota | 57,317 | 13,623 | 1,601 |
| 177, 191, 197, 214, 226 | Medium | Mixed male/female infl., roota | 111,196 | 22,519 | 4,102 |
| 199, 222, 227, CL12 | Long | Mixed male/female infl., roota | 89,052 | 18,269 | 1,263 |
| 259, 260, 264, CN2 | None | Mixed male/female infl., roota | 8,656 | 5,616 | 93 |
| 298, 301, C298, CN, CN10, CN101, CN22, CN23, CN24, CL13, CN31, CN32 | None | Embryo (20 DAP)a | 77,855 | 16,507 | 1,067 |
| 501 | 0.0–0.5 | Treated tissues | 6,400 | 4,396 | 34 |
| 502 | 0.5–1.0 | Treated tissues | 19,318 | 6,700 | 294 |
| 503, 507 | 1.0–2.0 | Treated tissues | 24,850 | 10,050 | 629 |
| 504, 504N, 505, 506 | >2 | Treated tissues | 49,074 | 14,225 | 672 |
| 600, 601, 602, 604 | None | Female flower | 3,423 | 1,894 | 19 |
| 605 | >1 kb | Female flower | 700 | 549 | 5 |
| 606 | 0.5–1.0 | Female flower | 9,382 | 3,626 | 61 |
| 607 | <0.5 | Female flower | 588 | 535 | 2 |
| CB5, CB6 | >2 | Immature M/F flower, kernel, embryo | 22,728 | 3,265 | 31 |
| CC1 | <1 kb | Callus tissue | 1,135 | 790 | 32 |
| CC2 | >1 kb | Callus tissue | 275 | 190 | 4 |
| CR1 | None | Corn roots (different treatments) | 1,915 | 1,544 | 42 |
| Other | 35 | 35 | |||
| GenBankb | 133 | 133 | 133 | ||
| Total | 484,032 | 10,084 |
Source of the libraries, number of clones and number of clusters in each sample are listed. Treated tissues were collected from seedlings that were subjected to a variety of stress treatments including heat, cold, drought, salt and N-deficiency
aLibraries were derived from Pioneer Hi-Bred International, Inc Hybrid 35A19. All other libraries were derived from various other hybrids
bThese sequences were made full-length by assembling GenBank ESTs per clone
5′ sequencing and EST clustering
We sequenced the 5′ end of >600,000 randomly selected clones from a diverse set of corn cDNA libraries. Usually, the 5′ ends of 1,536 random clones from each library were sequenced initially and the sequences were used to assess the proportion of full-length and novel clones. If the library was of sufficient quality (high percentage of full-length and novel clones), 10–20,000 additional clones were sequenced from the 5′ end. Sequences that were of low quality, very short (<50 nucleotides) or suspected to be from a different organism were discarded leaving 484,032 that were further analyzed and submitted to GenBank. Sequences that are derived from species other than the intended organism are quite common in EST sequencing projects. For example, of the 1,187 non-redundant sequences identified by Lai et al. (2004) as having no match to rice sequences, 61 (>5%) were closely related to E. coli sequences.
The 5′ reads were clustered into 63,476 clusters (any two clones that have >6 nt differences in any 30 nt window are clustered apart). The number of clones in each cluster ranged from 1 to 1,350 with an average of 7.64 clones per cluster. The 20 proteins with the largest number of 5′ ESTs are shown in Table 2. Histones and seed storage proteins are the most abundant in the top 20. The latter obviously reflects the abundance of seed storage protein mRNAs in seed storage tissues, in spite of attempts to minimize seed storage tissue in our libraries.
Table 2.
The 20 proteins with the largest number of 5′ ESTs
| SEL_CLONE_ID | Number of 5′ ESTs | NR_FUNCTION |
|---|---|---|
| 1372232 | 1,350 | Histone H2A |
| 220147 | 1,139 | Histone H2A-like protein |
| 999034 | 1,078 | Oleosin Zm-II |
| 218272 | 1,053 | Thioredoxin |
| 1286395 | 1,016 | 15 kD Beta zein |
| 1279712 | 1,015 | Histone H2A |
| 865719 | 1,000 | Glutelin-2 precursor (Zein-gamma) (27 kDa zein) |
| 1289363 | 907 | 17 kDa Oleosin |
| 207756 | 746 | Histone H4 |
| 207750 | 712 | 60S Ribosomal protein L6 |
| 1387617 | 695 | MFS18 Protein precursor |
| 1283579 | 667 | Zein protein |
| 207824 | 649 | Translationally controlled tumor protein homolog |
| 1470151 | 648 | Histone H1 |
| 218432 | 583 | Polyubiquitin |
| 263948 | 547 | Chalcone synthase |
| 220162 | 539 | Anther-specific protein MZm3-3 precursor |
| 1000184 | 524 | Tonoplast water channel |
| 280720 | 491 | Histone H1 |
| 1272660 | 472 | Metallothionein |
As these libraries were made from hybrids, reliable polymorphisms were observed in about half of the clusters: about one third of these polymorphisms are 1–6 nt insertion/deletions (indels) and the other two thirds are base substitutions (data not shown). If the indels are seven or more nucleotides, we would not identify them in our analysis because those ESTs would form separate clusters. Interestingly, nearly 10% of the clusters with 40 or more ESTs contain one or a few ESTs with a 5′ end that is 60 or more nucleotides longer than the other members of the cluster indicating a possible alternative transcription start site.
Clone sequencing
From each EST cluster, the clone that had the longest 5′ end at the time of selection was used as a cluster representative (Selected Cluster Representative, SCR). Out of 63,476 SCRs, 39,769 clones possessed an ATG translation start site and were not redundant. 15,308 SCRs were overlapped after sequencing from the 3′ end with an average length of 689 nts. We used primer walking in an attempt to overlap the remaining SCRs. After the clones were overlapped they were reanalyzed and the acceptable clones were sequenced on the second strand. We tried to eliminate ambiguities by generating another primer and sequence to get a consensus sequence. At each step, if a clone was determined to be (1) identical to another already sequenced clone in nucleotide or protein sequence, (2) truncated, (3) wrong organism, (4) chimeric, or (5) the clone could not be overlapped, sequencing of the clone was stopped. Using this approach, 35,497 clones were overlapped. We also experimented with transposon sequencing (Strathmann et al. 1991) and overlapped 935 clones with this approach. From these 36,432 clones, we have carefully selected the highest quality non-redundant clones (9,951) which, to the best of our knowledge are free of common errors, such as redundancy, contamination, truncations, frame-shifts and chimerism. Using our EST clustering system we have also overlapped 5′ and 3′ reads of corn cDNA clones from the GenBank EST database and have added 133 full-length clones to our set of high quality corn transcripts. In most of the analyses in this paper we use this set of 10,084 clones. We estimate that there are several thousand additional good full-length corn cDNA clones in our GenBank submission which we did not use in most of the analyses in this paper, because we have less confidence that these clones contain a complete CDS.
Statistical properties of corn cDNAs
The distribution of the number of 5′ ESTs in clusters corresponding to this set of 10,084 clones is shown in Fig. 1 and follows Zipf’s law stating that a frequency of occurrence of some event, is a power-law function of the rank of this event where rank is determined by the frequency (Zipf 1949). Zipf’s law for cDNA clusters may imply that the rate of evolutionary changes in gene expression (assessed by the number of 5′ ESTs in a cluster) is proportional to the gene expression level (Gibrat 1931).
Fig. 1.

Distribution of the number of clones (5′ ESTs) among clusters represented by a set of 10,084 full-length corn cDNA clones. Relatively few genes have many ESTs whereas many have only one or few ESTs. The distribution can be approximated by a power function (inset; linear function in log scale)
Length distributions of CDS, 5′ and 3′ UTRs, and comparisons with other full-length corn transcripts from GenBank, revealed that Ceres cDNA clones have longer 5′ UTRs, shorter coding regions and similar size 3′ UTRs (Table 3). A comparison of our set of corn cDNAs to the annotations of the rice and Arabidopsis genomes also showed that Ceres clones are overall shorter. This is likely to be due to our approaches taken in cloning, selecting and sequencing and probably does not reflect a biological difference in the libraries.
Table 3.
Median lengths of 5′ UTRs, CDSs, and 3′ UTRs in corn (from Ceres and GenBank), Arabidopsis and rice
| 5′ UTR | CDS | 3′ UTR | Number of sequences | Description | |
|---|---|---|---|---|---|
| Corn (Ceres) | 124 | 741 | 228 | 10,084 | Ceres full-length cDNA sequences |
| Corn (GenBank) | 80 | 1044 | 223 | 931 | Non-redundant set of corn full length mRNA sequences from GenBank |
| Rice | 123 | 1017 | 279 | 24,368 | Rice gene predictions having full-length mRNA support from TIGR genome annotation, release 5 (Ouyang et al. 2007) |
| Arabidopsis | 88 | 1097 | 184 | 18,468 | Arabidopsis gene predictions having full-length mRNA support from TAIR genome annotation, release 6 (Swarbreck et al. 2007) |
Corn transcripts, especially coding regions, are more GC-rich (Table 4) than the overall genome (58% GC in CDS vs. 46% in the genome) which is consistent with previous observations (Haberer et al. 2005). Equivalent percentages for Arabidopsis are 45% and 36% (Alexandrov et al. 2006); 5′ UTRs are C-rich whereas 3′ UTRs are T-rich, similar to Arabidopsis. As in Arabidopsis, most transcripts start with A (Table 4). The consensus sequence around the initiating ATG (Fig. 2) is similar to Arabidopsis in the coding region, in that there is a strong preference for codons GCN that specify alanine as the 2nd amino acid, but different at the 5′ end in that corn is C-rich while Arabidopsis is A-rich (Alexandrov, Troukhan et al. 2006). The most frequently used stop codon is TGA (occurs in 51% of all transcripts) followed by TAG (30%) and TAA (19%). TGA is the most frequently used stop codon in Arabidopsis (44%) and rice (43%), but in Arabidopsis TAA (36%) is more frequent than TAG (20%), whereas similar frequencies of TAG (30%) and TAA (27%) are used in rice.
Table 4.
Nucleotide composition of 10,084 corn transcripts
| A | C | G | T | |
|---|---|---|---|---|
| Genome | 0.269 | 0.232 | 0.231 | 0.268 |
| cDNA | 0.222 | 0.275 | 0.277 | 0.226 |
| 5′ UTR | 0.206 | 0.342 | 0.249 | 0.203 |
| CDS | 0.218 | 0.284 | 0.296 | 0.202 |
| 3′ UTR | 0.246 | 0.199 | 0.233 | 0.322 |
| TSS | 0.679 | 0.112 | 0.172 | 0.037 |
The most unexpected frequencies are shown in bold
Fig. 2.

Sequence logo for the ATG consensus in corn. The logo is based on 9,920 sequences. The figure was produced using WebLogo tool (Crooks et al. 2004)
Nucleotide distribution in coding regions
Corn coding regions have elevated GC contents compared to the coding regions of Arabidopsis genes as well as compared to non-coding regions in the corn genome. More intriguingly, the GC distribution in the coding region has two peaks (Fig. 3) indicating that there are two major classes of genes in corn. Genes in the first peak have a mean G+C of about 0.5, i.e. there is no preference for G+C. Genes in the second peak have an unusually broad GC frequency distribution in the third position in the codons (GC3) with a peak at about 0.9. In contrast, the genes of Arabidopsis and other dicot species have a unimodal and narrow GC distribution. Analysis of rice genes reveals a broad GC distribution similar to corn (Campbell and Gowri 1990; Wang et al. 2004; Wang and Hickey 2007). GC content for the first two positions in codons (GC12) has a unimodal distribution for all three species, emphasizing that the main difference in GC content is due to the third nucleotide in the codons (Fig. 3). Analysis of other plant genes from GenBank revealed that the bimodal distribution is a characteristic feature of all grasses (Poaceae) for which a sufficient number of genes have been sequenced (Fig. 4).
Fig. 3.

Distribution of GC in the coding region of corn, Arabidopsis and rice. The GC content in the coding region of corn cDNAs is bimodal and the high GC content can be explained by the abundance of GC in the third position of the codons. A similar result is observed for rice but Arabidopsis is unimodal. GC indicates the ratio of GC versus AT. GC12 represent the ratio of GC versus AT in the 1st and 2nd positions of the codons. GC3 represents the ratio of GC versus AT in the 3rd position of the codons
Fig. 4.

Distribution of the GC content in the third codon position of CDSs of different plant species. All grasses have a broad distribution with two peaks whereas the dicots have a unimodal distribution. All CDS sequences except corn (we used sequences described in this paper) and Arabidopsis (we used TAIR annotation) were downloaded from the J. Craig Venter Institute (JCVI, formerly known as TIGR) ftp site ftp://ftp.tigr.org/pub/data/plantta/. The number of unique transcripts for each species is: switchgrass 7,638, Arabidopsis 27,983, poplar 12,687, canola 10,709, Medicago 20,414, cotton 24,797, corn 10,084, rice 49,870, sorghum 20,714 and wheat 62,121
We have analyzed genes that contribute to the high and low GC peaks. We hypothesized that due to the effects of cytosine methylation and cytosine deamination, “old” genes would tend to be more AT rich in the third position as in Drosophila and mammalian genomes (Petrov and Hartl 1999). Relatively new genes then would be GC-enriched in the third position. Indeed, genes encoding homologous proteins in corn and Arabidopsis as well as in rice and Arabidopsis tend to be in the lower GC peak (Fig. 5). Genes in the higher GC peak are “newer” genes, not present in Arabidopsis. Also, intronless genes are highly enriched in the higher GC peak (Fig. 5). This latter observation is consistent with a massive synthesis of existing variant genes by reverse transcription of existing gene transcripts that became stabilized into the evolving genome since mRNA transcripts lack introns. However, it is clear that genome evolution in plants is driven most often by genome duplication followed by gene loss and/or modification (Cronk 2001). Often genome duplication is achieved by polyploidization, but more rarely it may involve wider hybridizations. Given the large number of new genes with different GC structures in grasses, perhaps the lineage was initiated by a wide hybridization event with another species that had genes with a high GC content, followed by selective gene retention and loss to create today’s Poaceae. The wide hybridization, while most likely to have involved a plant species, could have been prokaryotic or algal and, a prokaryotic origin could explain the higher proportion of intronless Poaceae-specific genes. We tried to find prokaryotes responsible for such an invasion based on GC profiles of gene sequences but could not find clear candidates due to either lack of sequence data in the relevant species or significant divergence of the genes over the more than 100 million years since the dicot/monocot separation. However, we found that the corn genes with high GC content are similar to Myxobacteria genes (Fig. 5). Myxobacteria are soil dwelling gram-negative bacteria producing a wide range of secondary metabolites and can inhibit plant pathogenic fungi (Bull et al. 2002). Some algae, e.g. Chlamydomonas, have genomes with high GC codon usage (Merchant et al. 2007) and these could be a source of the novel genes.
Fig. 5.

Distribution of the GC content in the third codon position among different groups of rice genes. Only those genes which have mRNA evidence in TIGR version 5 annotation (total 23,721 genes) were considered. GC3 distribution of these genes has two peaks at about 0.5 and 0.9. Genes without introns (4,452) are more prevalent in the high GC3 peak. Genes sharing similarity with Arabidopsis (blast P-value < 1.e-50, best reciprocal hit, 7,924 genes) are mostly in the lower GC3 peak whereas genes (1,664) similar to Myxobacteria (blast P-value < 1.e-3 and not matching Arabidopsis) are mostly in the high GC3 peak. 17,100 known protein sequences of the order Myxococcales from GenBank were used for comparison
The results in Fig. 3 suggest that all Poaceae have high GC gene fractions and there is a wide distribution of gene frequencies with respect to the presence of GC in the third position. Also, some dicots such as canola, cotton and Medicago have a higher proportion of genes with high GC contents than Arabidopsis and poplar (Fig. 5). This shows that during evolution gene variants with different GC contents are stabilized in genomes to differing degrees. Thus, perhaps during evolution hybridization between organisms with different GC contents in coding sequences occurs relatively frequently and that the resulting processes in selective loss and retention produces genomes with genes having different codon usage and GC contents. The Poaceae would be examples of where such processes have occurred to generate a more extreme form. It is not possible at present to choose between various hypotheses to explain the divergent codon usage but the different ideas are not mutually exclusive. Of especial importance is to discover what forces could result in selection of plants with variant codon usage on such a massive scale. The answers may lie in RNA biology, chromatin control processes, epigenetics and heterosis.
Statistical features of gene structures
We determined gene structure by spliced alignment of full-length cDNA clones and EST sequences with available genomic DNA. This gives us additional information on parts of genes not present in transcripts, i.e. introns and promoters. 2,793 of the 10,084 cDNA sequences were aligned with 2,714 corn genomic sequences from GenBank. Only genomic sequences longer than 20,000 nucleotides were used to avoid additional bias towards genes with a smaller number of introns. 497 (18%) of 2,793 clones consist of a single exon. Distribution of the number of exons in corn genes is similar to the distribution for Arabidopsis genes having full-length cDNAs sequenced using the same technology (Fig. 6). As these sets of genes are enriched for genes with shorter cDNA transcripts (Table 3) we expect some change of this distribution when all corn transcripts are known (as illustrated for the distributions of all Arabidopsis and rice genes in Fig. 6).
Fig. 6.

Distribution of the exon number in corn, Arabidopsis and rice genes. 2793 of 10,084 full-length corn clones were mapped to corn genomic sequences of >20,000 bps to ascertain the number of exons. This subset is biased towards shorter genes which may overestimate frequencies of genes with a smaller number of exons and underestimate frequencies of genes with a larger number of exons. This effect can be seen in the distributions for Arabidopsis genes: one was obtained using Arabidopsis cDNA clones produced by a similar technology (Arabidopsis cDNA) and the other derived from all genes in the TAIR genome annotation having mRNA support (Arabidopsis all). Distribution of exons in rice genes were obtained from 23,721 genes with mRNA support from TIGR rice genome annotation, release 6 and are shown for comparison
As in rice and Arabidopsis, exon length distribution depends on the type of exons. The longest exons are single exons, followed by the terminal and initial exons. The shortest exons are internal (Table 5). Internal exons are of about the same size in corn, rice and Arabidopsis, but single exons are much shorter in corn most likely because of the biased subset of shorter Ceres cDNA clones used in this comparison. Initial and terminal exons may also be affected by this bias. It has been noted previously that the median intron length is greater in corn and rice genes compared to Arabidopsis genes (Haberer et al. 2005). However, the mode of intron length distribution is the same in all three species (Fig. 7). It also has been reported that first introns are longer than other introns in Arabidopsis (Seoighe et al. 2005) and in other species (Kriventseva et al. 1999). Using our much larger set of genes, we have shown that this also holds for corn (Table 5).
Table 5.
Range and median exon and intron lengths in nucleotides
| Corn | Rice | Arabidopsis | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Min | Max | Median | Count | Min | Max | Median | Count | Min | Max | Median | Count | |
| Single exons | 231 | 3,045 | 886 | 497 | 197 | 7,798 | 1292 | 4,814 | 120 | 5,238 | 1123 | 3,063 |
| Initial exons | 17 | 2,448 | 268 | 2,296 | 3 | 5,991 | 311 | 30,088 | 2 | 6,532 | 252 | 19,327 |
| Terminal exons | 19 | 2,673 | 401 | 2,296 | 3 | 5,538 | 554 | 30,088 | 1 | 5,467 | 423 | 19,327 |
| Internal exons | 4 | 2,054 | 107 | 7,996 | 1 | 7,835 | 114 | 144,070 | 1 | 6,040 | 113 | 96,485 |
| First Introns | 64 | 9,884 | 267 | 2,296 | 21 | 18,270 | 334 | 30,088 | 22 | 7,385 | 176 | 19,327 |
| Other Introns | 61 | 13,048 | 142 | 7,996 | 34 | 18,328 | 142 | 144,070 | 22 | 5,001 | 99 | 96,485 |
The number of exons or introns used in the analysis is shown in the Count column
Median values (shown in bold) can be compared between species
Fig. 7.

Intron length distribution in corn, Arabidopsis and rice. Introns in both the coding and non-coding parts of the mRNA were used in this analysis. All three species have similar modes for intron length, although corn and rice genes have longer introns in average
We have previously shown that the average expression of Arabidopsis genes with introns is higher than the expression of intronless genes (Alexandrov et al. 2006). Using the number of 5′ tags as a measure of expression level, we see a similar trend in corn (Fig. 8). This might be explained by the presence of regulatory sequences in introns (Gidekel et al. 1996), by increased mRNA stability or by different epigenetic chromatin structures given the lower GC content of introns.
Fig. 8.

Average gene expression increases with the number of introns in genes. The number of 5′ ESTs in each cluster was used to estimate expression. These 5′ ESTs were derived from primary libraries and so reasonably estimate mRNA abundance in the libraries. The greater the number of exons in a gene the greater its expression, as measured by the number of 5′ ESTs
Alternative splicing
Alternative splicing is an important mechanism of gene regulation and provides a significant addition to the total number of different transcripts and proteins. It is important to understand frequency and common types of alternative splicing events. A commonly used measure of alternative splicing is the fraction of genes having alternative transcripts. However, this number depends on the available number of transcript sequences—the more sequences, the greater the chance to observe spliced variants. We proposed using a Gini index (Mirkin 1996; Alexandrov et al. 2006) to compare the frequency of alternative splicing in corn and Arabidopsis. The Gini index changes from 0 to 1 and is equal to 0 when there are no alternative splice variants. Gini is also small when almost all transcripts support the major variant of splicing and only a few transcripts correspond to the other isoform. Gini is closer to 1 when the various isoforms are supported by about the same number of transcripts. While the majority of genes have a Gini index of 0 for both corn and Arabidopsis (meaning lack of alternative splicing), there is a higher frequency of corn genes, as compared to Arabidopsis, at every Gini score above 0 indicating that alternative splicing occurs more frequently in corn than in Arabidopsis (Fig. 9). The average Gini index for corn is 0.10, whereas, for Arabidopsis, the Gini index is 0.08.
Fig. 9.

Gini index for corn and Arabidopsis introns. 96% of Arabidopsis introns and 92% of corn introns have a Gini index equal to 0 meaning that there are no variants (the data point is not shown). A larger Gini index in corn means that corn transcripts are more variable
Typically, alternative splicing events are classified as one of four major types: intron retention, alternative acceptor, alternative donor or skipped exon (Wang and Brendel 2006). To compute relative frequencies of these events, we compared the transcripts within a cluster and classified all differences in splicing patterns to one of these types. Not all types of alternative splicing are symmetrical. For example, in the case of intron retention, we can see retention in transcript A when compared to transcript B, but when we compare B with A, retention is not observed, instead we see intron addition. The same is true for exon skipping. However, alternative donor and alternative acceptors show up in the two-way comparison: A to B and B to A. Thus comparing a pair of transcripts, we should count two alternative donors or acceptors, one case of intron retention (and intron addition) or one case of exon skipping (and exon addition). However, in our calculations, as in previous publications (Wang and Brendel 2006), we counted all events only once per transcript pair, combining intron retention with intron addition and exon skipping with exon addition. In our calculations we compared all possible pairs of relevant transcripts. We found the most common types of alternative splicing in corn, as in other plants, are different acceptor sites and intron retention. It is interesting that the relative frequency of these events is somewhat related to the number of transcripts in the cluster: with a lower number of transcripts, the most common event is intron retention, while for the genes with a larger number of transcripts, alternative acceptor site is more frequent (Fig. 10).
Fig. 10.

Relative frequencies of different types of alternative splicing events. The frequencies of different alternative splicing events were computed from the alignment of 563,251 transcripts with corn genomic sequences. 289,608 mapped transcripts are from Ceres libraries, the other 273,643 transcript sequences were downloaded from GenBank
Motifs in corn promoters
Promoter sequences are enriched with transcription factor binding sites. Detection of these motifs upstream of the 5′ ends of cDNA clones mapped to the corn genomic sequences confirms that our clones are indeed 5′-full-length clones. Not surprisingly, the most frequently occurring motifs appeared to be the TATA-box about 30 nucleotides upstream of the TSS and a three-letter motif exactly at the TSS. Nucleotide distribution around the TSS has a peak of A/T at position -30 and a peak of C/A just before the TSS (Fig. 11). We estimated the statistical significance of different motifs using approximation suggested by Waterman et al. (Galas et al. 1985). Significance level p of the word w is estimated as 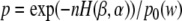 , where β is a fraction of sequences that contain the word w and α = p0 (w) is an expected fraction of sequences. Entropy of β relative to α is defined as
, where β is a fraction of sequences that contain the word w and α = p0 (w) is an expected fraction of sequences. Entropy of β relative to α is defined as 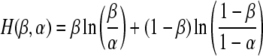 . Analyses of promoter regions of corn and Arabidopsis showed that they generally have two highly pronounced peaks of significance: at −30 bps, corresponding to the four-nucleotide pattern TATA, and the two-letter word CA at the TSS. Interestingly, significance of the CA motif in corn promoters appears to be much larger than that of the TATA box, whereas in Arabidopsis promoters the significance of these motifs is about the same (Fig. 12).
. Analyses of promoter regions of corn and Arabidopsis showed that they generally have two highly pronounced peaks of significance: at −30 bps, corresponding to the four-nucleotide pattern TATA, and the two-letter word CA at the TSS. Interestingly, significance of the CA motif in corn promoters appears to be much larger than that of the TATA box, whereas in Arabidopsis promoters the significance of these motifs is about the same (Fig. 12).
Fig. 11.

Distribution of nucleotides around the Transcription Start Site of corn based on 5,200 promoters that have TSSs predicted by at least four 5′ ESTs. There is a peak of A/T at position -30 and a peak of C/A just prior to the TSS
Fig. 12.

The most significant words in corn (a) and in Arabidopsis (b) promoters. The analysis is performed on a subset of 5,200 corn promoters and 5,050 Arabidopsis promoters that have TSS predicted by at least four 5′ ESTs. For Corn, there is a prominent CA peak at the TSS and a smaller TATA motif at position -30. This is in sharp contrast to Arabidopsis where TATA is more frequent than CA
The importance of the TATA box for variability of gene expression has been shown by several groups for various organisms (Tirosh et al. 2006). It is not clear, however, if the TATA box is important for strong gene expression. We have divided the promoters into five groups based on their expression level (measured by the number of ESTs in the cluster) and compared frequencies of TATA box containing promoters in each group. We found that the TATA box features in both strong and weak promoters but not as frequently in medium expressed genes. This observation is also true for Arabidopsis promoters, although the peak abundance of TATA boxes for corn is within the strongest promoters and for Arabidopsis is within the weakest promoters (Fig. 13).
Fig. 13.

Frequency of a TATA box in promoters of different strengths. Strong and weak promoters have a TATA box more often than genes with average expression. In corn, TATA boxes are more frequent in stronger genes whereas in Arabidopsis TATA boxes are more frequent in weaker promoters
We found that GC-rich corn genes have a TATA box more often than genes in the GC-poor peak, namely, only 17% of genes with GC3 < 60% contain a TATA-box as compared to 42% of genes with GC3 > 80%. This observation led us to consider transcriptional differences between GC3-rich and other genes in rice for which microarray chip data are available. We obtained a list of rice microarray experiments from NCBI GEO database (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE6893 (Jain et al. 2007) and GSE4438 (Walia et al. 2007)). For each probe on an Oryza sativa 50K Affymetrix GeneChip Rice Genome Array (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL2025) we computed standard deviation of the logarithm of intensities. Genes with GC3 > 80% have an average standard deviation of 0.6, while genes with GC3 < 60% have significantly smaller standard deviation (0.51). These observations imply that genes with a high GC content are on average expressed at more variable levels in different cell types or growth conditions. This more variable level of expression correlates statistically significantly with the presence of a TATA box.
CG skew around transcription start sites
A peak in the cumulative CG skew (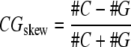 ) is associated with the transcription-coupled effects in the DNA template strand and location of the replication origin in bacteria (Beletskii and Bhagwat 1996; Grigoriev 1998). Previously, these ideas were extended to explain the CG skew peak near the TSS in Arabidopsis (Tatarinova et al. 2003). Analysis of the CG-skew at the TSS of several eukaryotic genomes was reported by Fujimori et al. (2005). Using our collection of 5′ EST for corn and genomic DNA from GenBank and TIGR, we predicted TSSs for corn with the TSSer algorithm (see Methods). We have computed the CG-skew plot for 5,200 promoters having at least four supporting 5′ ESTs as evidence. Figure 14 shows the skew present in corn promoters which is similar to the skew previously observed in Arabidopsis (Tatarinova et al. 2003).
) is associated with the transcription-coupled effects in the DNA template strand and location of the replication origin in bacteria (Beletskii and Bhagwat 1996; Grigoriev 1998). Previously, these ideas were extended to explain the CG skew peak near the TSS in Arabidopsis (Tatarinova et al. 2003). Analysis of the CG-skew at the TSS of several eukaryotic genomes was reported by Fujimori et al. (2005). Using our collection of 5′ EST for corn and genomic DNA from GenBank and TIGR, we predicted TSSs for corn with the TSSer algorithm (see Methods). We have computed the CG-skew plot for 5,200 promoters having at least four supporting 5′ ESTs as evidence. Figure 14 shows the skew present in corn promoters which is similar to the skew previously observed in Arabidopsis (Tatarinova et al. 2003).
Fig. 14.

CG skew plot for corn TSSs calculated as average CG skew in a sliding window of 40 nucleotides. The CG skew observed for corn is similar to what we have previously observed for Arabidopsis
Similarity of corn proteins to those of rice and Arabidopsis
We have compared 10,084 corn proteins with those of rice (Ouyang et al. 2007) and Arabidopsis (Swarbreck et al. 2007) derived from the genome annotations. As we expected, rice proteins are more similar to corn than Arabidopsis proteins (Fig. 15). 9514, or 94%, of corn clones have a match with 7,137 distinct rice genes (only the best rice match was counted for each corn protein) with blastp P-value ≤ 1.e-10 and 8,932 (88%) have a match with 5,853 distinct Arabidopsis genes (only the best Arabidopsis match was counted for each corn protein). Further examination of the 570 corn cDNAs not matching rice proteins, revealed that 192 cDNAs have a strong match with the rice genome indicating missed predictions in the rice genome annotation.
Fig. 15.

Corn proteins are more similar to rice than to Arabidopsis. The few exceptions are due to genes missed in the rice annotation, random fluctuations and possible contamination of corn cDNA clones by cDNAs from other organisms. 10,084 corn proteins, TAIR Arabidopsis genome annotation and TIGR rice annotation were used for comparison. Only matches with P-value ≤ 1.e-10, covering at least 70% of the protein length are shown in the plot
Protein functional characterization
Table 6 lists plant Gene Ontology (GO slim) terms (Berardini et al. 2004) associated with the 10,084 corn full length cDNA clones. We have compared frequencies of GO terms in this set of corn genes with the distribution of GO terms among all expressed proteins in the Arabidopsis genome. The corn set contains many more genes belonging to the “structural molecule activity” than one would expect from a comparison with the Arabidopsis genome, while genes in “transferase activity” are underrepresented. The top 10 Pfam families (Finn et al. 2007) are listed in Table 7. The results are consistent with the GO annotation: the most significant Pfam family is histone, which belongs to the overrepresented “nucleic acid binding” GO group.
Table 6.
Classification of the 10,084 full-length clones by GO slim categories
| GO classification | Number of corn proteins |
|---|---|
| Molecular function | |
| Structural molecule activity | 570 |
| Nucleic acid binding | 604 |
| Kinase activity | 667 |
| Transporter activity | 703 |
| Receptor binding or activity | 183 |
| DNA or RNA binding | 1,163 |
| Protein binding | 1,446 |
| Nucleotide binding | 898 |
| Transcription factor activity | 742 |
| Hydrolase activity | 1,301 |
| Transferase activity | 998 |
| Biological process | |
| Electron transport or energy pathways | 415 |
| Cell organization and biogenesis | 650 |
| Transport | 822 |
| Response to stress | 700 |
| Protein metabolism | 1,441 |
| DNA or RNA metabolism | 202 |
| Response to abiotic or biotic stimulus | 627 |
| Signal transduction | 257 |
| Transcription | 620 |
| Developmental processes | 647 |
| Cellular component | |
| Ribosome | 431 |
| Cytosol | 694 |
| Plastid | 952 |
| ER | 744 |
| Chloroplast | 3,765 |
| Extracellular | 352 |
| Plasma membrane | 969 |
| Cell wall | 875 |
| Nucleus | 2,270 |
| Mitochondria | 2,157 |
| Golgi apparatus | 437 |
Table 7.
Top ten Pfam families
| Pfam family | Description | Number of corn proteins |
|---|---|---|
| Pkinase | Protein kinase domain | 175 |
| zf-C3HC4 | Zinc finger, C3HC4 type (RING finger) | 140 |
| RRM_1 | RNA recognition motif. (a.k.a. RRM, RBD, or RNP domain) | 113 |
| Tryp_alpha_amyl | Protease inhibitor/seed storage/LTP family | 78 |
| Histone | Core histone H2A/H2B/H3/H4 | 76 |
| Myb_DNA-binding | Myb-like DNA-binding domain | 70 |
| WD40 | WD domain, G-beta repeat | 66 |
| p450 | Cytochrome P450 | 62 |
| F-box | F-box domain | 57 |
| efhand | EF hand | 56 |
Non-coding RNAs
In the course of looking at the complete set of 63,476 clusters (SCRs), we examined the abundance of potentially functional non-coding RNAs that might be contained within the cloned cDNAs by comparing the set to the RNA families maintained at RFAM (Griffiths-Jones et al. 2005). Table 8 summarizes the different types of RNAs that were found to be present. The most abundant species were spliceosomal followed by tRNA and small nuclear RNAs (sno RNAs) involved in RNA modifications—usually ribosomal RNAs. Self-splicing introns (both Groups I and II) were also identified in the set; these are usually found in mitochondrial or chloroplastic genes but not nuclear genes. Closer examination of the nine transcripts containing self-splicing introns indicates that six of these are encoded within chloroplastic DNA, two are encoded within mitochondrial DNA, and one can be found in both mitochondrial and chloroplastic DNA.
Table 8.
Non-coding RNAs in our collection of cDNA clones
| RNA type | Abundance |
|---|---|
| Spliceosomal RNAs | 35 |
| tRNA | 26 |
| RNA modification | 15 |
| Self splicing introns | 9 |
| Regulatory | 6 |
| Ribosomal RNA | 5 |
| Signal recognition | 1 |
Six cDNAs were identified that contained regulatory RNAs; two copies of the RNA-OUT were identified, which is the antisense to RNA-IN that inhibits transposition of the IS10 element (Kittle et al. 1989). Two copies of SnoRD14, also called U14, were identified; this RNA is involved in the processing of rRNA (Samarsky et al. 1996). Two mir elements (160 and 166) were also identified. Eighty five of the 97 non-coding RNAs identified are contained within a transcript that is at least 30% longer than non-coding RNA from RFAM suggesting that the non-coding RNA is contained within a larger gene although explanations based on chimeric clones are also possible.
Estimation of the number of protein-coding genes in corn
The total number of corn transcripts can be estimated from the number of matching sequences in two independent sets of transcripts. A similar approach was used to estimate the number of genes in the human genome (Ewing and Green 2000). If N is the total number of genes in the genome; the first set contains n1 randomly selected genes and the second set contains n2 independently selected genes, then the number m of the same genes in these two sets can be calculated as  ; hence N = n1n2/m. We compared our set of 31,552 (a part of 36,565 fully sequenced cDNA clones longer than 100 nucleotides) with a non-redundant set of 10,562 corn mRNAs longer than 100 nucleotides from GenBank. We identified 6,753 transcripts to be the same in these two sets. Thus the total number of transcripts in the corn genome can be estimated as 31,552 × 10,562/6,753 ≈ 50,000. This number is consistent with the previous estimate of the gene count in maize (from 42,000 to 56,000 genes) obtained by sampling BAC end sequences (Haberer et al. 2005).
; hence N = n1n2/m. We compared our set of 31,552 (a part of 36,565 fully sequenced cDNA clones longer than 100 nucleotides) with a non-redundant set of 10,562 corn mRNAs longer than 100 nucleotides from GenBank. We identified 6,753 transcripts to be the same in these two sets. Thus the total number of transcripts in the corn genome can be estimated as 31,552 × 10,562/6,753 ≈ 50,000. This number is consistent with the previous estimate of the gene count in maize (from 42,000 to 56,000 genes) obtained by sampling BAC end sequences (Haberer et al. 2005).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We are grateful to the library construction, clone curation and sequencing teams from Genset (lead by Jean-Baptiste Dumas) and from Ceres for generating cDNA libraries and sequencing of the cDNA clones. We acknowledge Monsanto for providing financial support for this project. We also are deeply indebted to our many other colleagues at Ceres for their contributions to this project
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s11103-008-9415-4) contains supplementary material, which is available to authorized users.
References
- Alexandrov NN, Troukhan ME et al (2006) Features of Arabidopsis genes and genome discovered using full-length cDNAs. Plant Mol Biol 60(1):69–85. doi:10.1007/s11103-005-2564-9 [DOI] [PubMed]
- Beletskii A, Bhagwat AS (1996) Transcription-induced mutations: increase in C to T mutations in the nontranscribed strand during transcription in Escherichia coli. Proc Natl Acad Sci USA 93(24):13919–13924. doi:10.1073/pnas.93.24.13919 [DOI] [PMC free article] [PubMed]
- Berardini TZ, Mundodi S et al (2004) Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiol 135(2):745–755. doi:10.1104/pp.104.040071 [DOI] [PMC free article] [PubMed]
- Bull CT, Shetty KG, Subbarao KV (2002) Interactions between Myxobacteria, plant pathogenic fungi, and biocontrol agents. Plant Dis 86:889–896. doi:10.1094/PDIS.2002.86.8.889 [DOI] [PubMed]
- Campbell WH, Gowri G (1990) Codon usage in higher plants, green algae, and cyanobacteria. Plant Physiol 92(1):1–11 [DOI] [PMC free article] [PubMed]
- Carninci P, Waki K et al (2003) Targeting a complex transcriptome: the construction of the mouse full-length cDNA encyclopedia. Genome Res 13(6B):1273–1289. doi:10.1101/gr.1119703 [DOI] [PMC free article] [PubMed]
- Cronk QC (2001) Plant evolution and development in a post-genomic context. Nat Rev Genet 2(8):607–619. doi:10.1038/35084556 [DOI] [PubMed]
- Crooks GE, Hon G et al (2004) WebLogo: a sequence logo generator. Genome Res 14(6):1188–1190. doi:10.1101/gr.849004 [DOI] [PMC free article] [PubMed]
- Ewing B, Green P (2000) Analysis of expressed sequence tags indicates 35, 000 human genes. Nat Genet 25(2):232–234. doi:10.1038/76115 [DOI] [PubMed]
- Finn RD, Tate J et al (2007) The Pfam protein families database. Nucleic Acids Res 36(Database issue):D281–288 [DOI] [PMC free article] [PubMed]
- Fujimori S, Washio T et al (2005) GC-compositional strand bias around transcription start sites in plants and fungi. BMC Genomics 6(1):26. doi:10.1186/1471-2164-6-26 [DOI] [PMC free article] [PubMed]
- Galas DJ, Eggert M et al (1985) Rigorous pattern-recognition methods for DNA sequences. Analysis of promoter sequences from Escherichia coli. J Mol Biol 186(1):117–128. doi:10.1016/0022-2836(85)90262-1 [DOI] [PubMed]
- Gibrat R (1931) Les Inégalités Économiques. Sirey, Paris
- Gidekel M, Jimenez B et al (1996) The first intron of the Arabidopsis thaliana gene coding for elongation factor 1 beta contains an enhancer-like element. Gene 170(2):201–206. doi:10.1016/0378-1119(95)00837-3 [DOI] [PubMed]
- Gish W (1996–2004) http://blast.wustl.edu
- Griffiths-Jones S, Moxon S et al (2005) Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33(Database issue):D121–D124. doi:10.1093/nar/gki081 [DOI] [PMC free article] [PubMed]
- Grigoriev A (1998) Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res 26(10):2286–2290. doi:10.1093/nar/26.10.2286 [DOI] [PMC free article] [PubMed]
- Haberer G, Young S et al (2005) Structure and architecture of the maize genome. Plant Physiol 139(4):1612–1624. doi:10.1104/pp.105.068718 [DOI] [PMC free article] [PubMed]
- Jain M, Nijhawan A et al (2007) F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol 143(4):1467–1483. doi:10.1104/pp.106.091900 [DOI] [PMC free article] [PubMed]
- Jia J, Fu J et al (2006) Annotation and expression profile analysis of 2073 full-length cDNAs from stress-induced maize (Zea mays L.) seedlings. Plant J 48(5):710–727 [DOI] [PubMed]
- Kittle JD, Simons RW et al (1989) Insertion sequence IS10 anti-sense pairing initiates by an interaction between the 5′ end of the target RNA and a loop in the anti-sense RNA. J Mol Biol 210(3):561–572. doi:10.1016/0022-2836(89)90132-0 [DOI] [PubMed]
- Kriventseva EV, Makeev V et al (1999) Statistical analysis of the exon-intron structure of higher eukaryote genes. Biofizika 44(4):595–600 [PubMed]
- Lai J, Dey N et al (2004) Characterization of the maize endosperm transcriptome and its comparison to the rice genome. Genome Res 14(10A):1932–1937. doi:10.1101/gr.2780504 [DOI] [PMC free article] [PubMed]
- Merchant SS, Prochnik SE et al (2007) The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318(5848):245–250. doi:10.1126/science.1143609 [DOI] [PMC free article] [PubMed]
- Mirkin B (1996). Mathematical classification and clustering. Kluwer Academic Publishers, Dordrecht
- Ouyang S, Zhu W et al (2007) The TIGR Rice Genome Annotation Resource: improvements and new features. Nucleic Acids Res 35(Database issue):D883–D887. doi:10.1093/nar/gkl976 [DOI] [PMC free article] [PubMed]
- Petrov DA, Hartl DL (1999) Patterns of nucleotide substitution in Drosophila and mammalian genomes. Proc Natl Acad Sci USA 96(4):1475–1479. doi:10.1073/pnas.96.4.1475 [DOI] [PMC free article] [PubMed]
- Samarsky DA, Schneider GS et al (1996) An essential domain in Saccharomyces cerevisiae U14 snoRNA is absent in vertebrates, but conserved in other yeasts. Nucleic Acids Res 24(11):2059–2066. doi:10.1093/nar/24.11.2059 [DOI] [PMC free article] [PubMed]
- Seoighe C, Gehring C et al (2005) Gametophytic selection in Arabidopsis thaliana supports the selective model of intron length reduction. PLoS Genet 1(2):e13. doi:10.1371/journal.pgen.0010013 [DOI] [PMC free article] [PubMed]
- Soares MB, Bonaldo MF et al (1994) Construction and characterization of a normalized cDNA library. Proc Natl Acad Sci USA 91(20):9228–9232. doi:10.1073/pnas.91.20.9228 [DOI] [PMC free article] [PubMed]
- Strathmann M, Hamilton BA et al (1991) Transposon-facilitated DNA sequencing. Proc Natl Acad Sci USA 88(4):1247–1250. doi:10.1073/pnas.88.4.1247 [DOI] [PMC free article] [PubMed]
- Swarbreck D, Wilks C et al (2007). The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res 36(Database issue):D1009–1014 [DOI] [PMC free article] [PubMed]
- Tatarinova T, Brover V et al (2003) Skew in CG content near the transcription start site in Arabidopsis thaliana. Bioinformatics 19(Suppl 1):i313–i314. doi:10.1093/bioinformatics/btg1043 [DOI] [PubMed]
- Tirosh I, Weinberger A et al (2006) A genetic signature of interspecies variations in gene expression. Nat Genet 38(7):830–834. doi:10.1038/ng1819 [DOI] [PubMed]
- Walia H, Wilson C et al (2007) Genome-wide transcriptional analysis of salinity stressed japonica and indica rice genotypes during panicle initiation stage. Plant Mol Biol 63(5):609–623. doi:10.1007/s11103-006-9112-0 [DOI] [PMC free article] [PubMed]
- Wang BB, Brendel V (2006) Genomewide comparative analysis of alternative splicing in plants. Proc Natl Acad Sci USA 103(18):7175–7180. doi:10.1073/pnas.0602039103 [DOI] [PMC free article] [PubMed]
- Wang HC, Hickey DA (2007) Rapid divergence of codon usage patterns within the rice genome. BMC Evol Biol 7(Suppl 1):S6. doi:10.1186/1471-2148-7-S1-S6 [DOI] [PMC free article] [PubMed]
- Wang HC, Singer GA et al (2004) Mutational bias affects protein evolution in flowering plants. Mol Biol Evol 21(1):90–96. doi:10.1093/molbev/msh003 [DOI] [PubMed]
- Wilson R (2008) Sequence and assembly of the maize B73 genome. In: 50th Annual Maize Genetics Conference, Washington, D.C.
- Zhulidov PA, Bogdanova EA et al (2004) Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucleic Acids Res 32(3):e37. doi:10.1093/nar/gnh031 [DOI] [PMC free article] [PubMed]
- Zipf G (1949). Human behavior and the principle of least effort. Addison-Wesley, Cambridge, USA
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.