

Abstract
Expression in E. coli of his-tagged human mevalonate diphosphate decarboxylase (hMDD) has expedited enzyme isolation, characterization, functional investigation of the mevalonate diphosphate binding site, and crystal structure determination (2.4 Å resolution). hMDD exhibits Vmax = 6.1 ± 0.5 U/mg; Km for ATP is 0.69 ± 0.07 mM and Km for (R,S) mevalonate diphosphate is 28.9 ± 3.3 uM. Conserved polar residues predicted to be in the hMDD active site were mutated to test functional importance. R161Q exhibits a ~1000-fold diminution in specific activity, while binding the fluorescent substrate analog, TNP-ATP, like wild-type enzyme. Diphosphoglycolyl proline (Ki = 2.3 ± 0.3 uM) and 6-fluoromevalonate 5-diphosphate (Ki = 62 ± 5 nM) are competitive inhibitors with respect to mevalonate diphosphate. N17A exhibits a Vmax = 0.25 ± 0.02 U/mg and a 15-fold inflation in Km for mevalonate diphosphate. N17A’s Ki values for diphosphoglycolyl proline and fluoromevalonate diphosphate are inflated (>70-fold and 40-fold, respectively) in comparison with wild-type enzyme. hMDD structure indicates the proximity (2.8 Å) between R161 and N17, which are located in an interior pocket of the active site cleft. The data suggest the functional importance of R161 and N17 in the binding and orientation of mevalonate diphosphate.
Keywords: human mevalonate diphosphate decarboxylase, isoprenoid biosynthesis, mevalonate pathway, competitive inhibition, active site function, protein structure
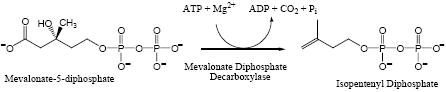
Mevalonate diphosphate decarboxyase (MDD1; EC 4.1.1.33) catalyzes the ATP dependent decarboxylation of mevalonate 5-diphosphate (MVAPP) to form isopentenyl 5-diphosphate [1]. The reaction is required for production of polyisoprenoids and sterols from acetyl-CoA. Inhibition of this enzyme effectively diminishes biosynthesis of cholesterol [2, 3] and low MDD activity correlates with decreased cholesterol levels in a hypertensive rat strain [4]. Genes encoding this enzyme have been detected in archaebacteria, some eubacteria, protozoa, plants, fungae, and animals. Substantial heterology is predicted between the various proteins encoded by these genes, suggesting that MDD could be targeted for development of antimicrobial agents [5].
MDD is a member of the GHMP kinase (galactokinase, homoserine kinase, mevalonate kinase, phosphomevalonate kinase) family of proteins. The significance of this enzyme has led to the elucidation of the X-ray structure of a recombinant form of the yeast protein [6]. More recently, proteins from Trypanosoma brucei and Staphyolococcus aureus were assigned as MDD on the basis of genetic complementation experiments and their structures were solved [7]. Since available MDD structures reflect unliganded protein, the strategy for initial mapping of active site amino acids [8] utilized the homology between MDD and mevalonate kinase (MK), an enzyme with a better characterized active site [9, 10, 11]. Thus, complementary approaches are required to demonstrate whether amino acids that have been implicated at the active site by structural results do, in fact, have important functions. Although MDD enzymatic activity has not been directly demonstrated for the S. aureus or T. brucei proteins, a model [7] for a ternary T. brucei protein-ATP-mevalonate diphosphate complex has been developed. This model utilized structural information available for ATP bound to the related enzyme, mevalonate kinase [11] and suggests the active site location of a variety of conserved amino acids. These include conserved aspartate [8] and serine [12] MDD residues that have been demonstrated to have major effects on MDD function.
Attempts at expression of recombinant human MDD in E. coli [13, 14] have not resulted in recovery of substantial amounts of highly purified enzyme, which would be useful in studies of enzyme function, structure, or inhibition. We now describe the isolation of a highly purified his-tagged form of recombinant human MDD, which has been utilized to carry out biochemical and structural work that tests the functional importance of active site residues predicted to interact with the substrate, mevalonate diphosphate. A preliminary account of parts of this study has appeared [15].
EXPERIMENTAL PROCEDURES
Materials
Deoxynucleotides and Pfu DNA polymerase used for mutagenesis were purchased from Stratagene. Primers used for mutagenesis were obtained from Integrated DNA Technologies. Plasmid DNA was propagated in E. coli JM109 cells (Promega). Reagents for plasmid DNA purification were purchased from Eppendorf (miniprep) and Qiagen (midiprep). DNA fragments were purified by agarose gel electrophoresis and isolated using a Qiaquick gel extraction kit (Qiagen). DNA sequencing was performed at the DNA Core Facility (University of Missouri - Columbia). For protein expression, E. coli BL21(DE3) cells were obtained from Novagen. Isopropyl-β-D-thiogalactopyranoside (IPTG) was purchased from Research Products International Corporation, Ni-Sepharose from GE Healthcare, and imidazole from Lancaster Synthesis Incorporated. 2’(3’)-O-(2,4,6-Trinitrophenyl)adenosine-5’-triphosphate (TNP-ATP) was obtained from Molecular Probes. Lactate dehydrogenase (rabbit muscle), pyruvate kinase (rabbit muscle), hexokinase (baker yeast), glucose 6-phosphate dehydrogenase (bakers yeast), 6-fluoromevalonate, β-NADH, β-NADP+, phosphoenolpyruvate, ATP, DEAE-Sephadex A-25 were purchased from Sigma. DTT was obtained from Acros Organics. Chemicals, buffers, media components and antibiotics were purchased from Fisher Scientific.
Syntheses of Mevalonate 5-Diphosphate
The synthesis of mevalonate 5-diphosphate has been previously reported [3] and is briefly summarized. Methyl 3-hydroxy-3-methyl-5-iodopentanoate was synthesized by reacting mevalonolactone with trimethylsilyl iodide, followed by diazomethane derivatization to form the methyl ester. The product was subsequently purified by silica gel chromatography. Methyl 5-diphosphomevalonate was synthesized by reacting the purified methyl 3-hydroxy-3-methyl-5-iodopentanoate with an excess of tetrabutylammonium diphosphate. The methyl 5-diphosphomevalonate was purified by anion exchange chromatography using a DEAE-Sephadex A25 (bicarbonate form) column. The chromatographically purified methyl 5-diphosphomevalonate was converted to the lithium salt by passage over a Dowex 50 column (lithium form). Deesterification was accomplished by alkaline hydrolysis in 0.5 N LiOH for 20 hours at 4°C. The pH was adjusted to ~ 8.0 with cold HCl. The product was then analyzed and the concentration of the physiologically active R isomer was determined by enzymatic end point assay [16].
Enzymatic Synthesis of 6-fluoromevalonate 5-diphosphate
Five milligrams (33.8 μmol) of (R,S) 6-fluoromevalonolactone was dissolved in 1 ml of 0.1 N KOH and delactonized by incubation for 1 hour at 37°C. The reaction was neutralized by the addition of ice-cold 6 N HCl to a pH of 7.5. The reaction mixture for the formation of 6-fluoromevalonate 5-diphosphate included: 30 mM Tris-Cl (7.5), 22.5 mM (R,S) 6-fluoromevalonate, 5 mM ATP, 6 mM MgCl2, 0.7 mM DTT, 50 mM phosphoenolpyruvate, 6 U pyruvate kinase and 2.7 U mevalonate kinase. The final volume of the reaction was 1.5 ml. The reaction was incubated at 30°C. 16.9 umol of (R) 6-fluoromevalonate were converted to the monophosphate in one hour as determined by end-point assay with phosphomevalonate kinase. At this point, 4 U of phosphomevalonate kinase (human) was added to the reaction and incubation was continued at 25°C for 12 hr. 10 umol (~60 %) of the (R) 6-fluoromevalonate 5-phosphate was converted to the diphosphate as determined by end-point assay with phosphomevalonate kinase (reverse reaction). The reaction was deproteinized by boiling (95°C) for 3 minutes, placing the reaction on ice for 5 minutes, and centrifugation for 15 minutes at 16,100 × g. The pH of the collected supernatant was adjusted to ~5.0 with ice-cold 6 N HCl. To precipitate most nucleotides from the sample, ice-cold absolute ethanol was added to a final concentration of 65 % and incubated on ice for 30 minutes. The solution was centrifuged (7700 × g, 20 minutes). The resulting pellet was washed with 1 ml of ice-cold absolute ethanol and centrifuged (7700 × g, 20 minutes). The supernatants were combined and ethanol removed by a speed vacuum concentrator. The pH of the remaining solution (containing 8.5 umol (R)-6-fluoromevalonate 5-diphosphate, 2 umol ATP) was adjusted to ~ 8.0 with cold KOH and diluted with ice-cold water to match the conductivity of 20 mM triethylammonium bicarbonate (TEA-HCO3). The above solution was applied to a DEAE-Sephadex A-25 column (1.5 × 25 cm) pre-equilibrated with 20 mM TEA-HCO3 (pH 8.0). The column was washed with 10 column volumes of 20 mM TEA-HCO3. The compounds were eluted with a linear TEA-HCO3 gradient (20 – 500 mM, 500 ml Vt). (R)-6-fluoromevalonate 5-diphosphate (7.8 umol; determined by end-point assay with phosphomevalonate kinase (reverse reaction)) eluted at ~ 450 mM TEA-HCO3 and contained only 1.1 % (0.088 umol) ATP. The eluate was concentrated by rotary evaporation. The overall yield of purified (R) 6-fluoromevalonate 5-diphosphate (7.8 umol) from (R) 6-fluoromevalonate (16.9 umol) was 46 %.
Synthesis of Diphosphoglycolylproline
The competitive inhibitor diphosphoglycolyproline ({[(Phosphonatooxy)phosphinato]oxy}acetyl)pyrrolidine-2-carboxylate) was synthesized using the strategy outlined by Vlattas et al. [17] using methodology described by Krepkiy and Miziorko [12].
Plasmid Construction and Protein Expression
The open reading frame encoding human MDD (generously supplied by Dr. K. M. Gibson as a glutathione-S-transferase fusion construct) was sub-cloned into the expression plasmid pET23d(+) using standard molecular biology techniques. Briefly, the gene was amplified by polymerase chain reaction using primers complementary to the 5’ and 3’ region of the gene. The 5’ primer (5’-TTCCCCATGGCCTCGGAGAAGC-3’) encoded an NcoI site (underscored) harboring the ATG (boldface) start codon; the 3’ primer (5’-GAGGCTCGAGGGCAGCTGGCTTCGGCAGGC-3’) encoded a XhoI site (underscored). The amplified cDNA and pET23d were digested with the appropriate restriction enzymes and gel purified. The purified DNA was ligated with NcoI/XhoI digested vector overnight at 4°C. The ligation product represents an expression construct that encodes human MDD containing a C-terminal His6 affinity tag. The integrity of the gene was verified by DNA sequence analysis.
To determine whether his-tag position could affect human MDD expression level, an expression plasmid encoding recombinant N-terminal His6 -tagged hMDD was also constructed. The gene was amplified using primers, which were designed to construct a 5’Nde1 site (underlined) harboring the start codon (bold), and the beginning of HMDD gene: 5’-GGAATACATATGGCCTCGGAGAAGCCGCTG-3’. The 3’ primer encoded a Bam H1 restriction site (underlined), a stop anticodon (bold) and the reversed complementary sequence of last nucleotides of the MDD gene : 5’-CATTGGATCCTCAGGCAGCTGGCTTTCGGCA-3. The PCR product was first subcloned into a pGEM vector and then into pET-15b vector, using Nde1 and BamH1 restriction sites. Expressed target protein contained a His6 affinity tag at the N-terminus.
Chemically competent E. coli BL21(DE3) cells were transformed with the appropriate expression vector. The transformed cells were plated onto LB agar containing ampicillin (amp; 100 ug/ml). These plates were incubated overnight at 37°C. 5 ml of LB-amp was inoculated with a single colony and allowed to grow to moderate turbidity (A600 ~0.3; 6-7 hrs). This culture was used to inoculate 20 LB-amp plates (130 μl/plate). The plates were incubated overnight at 37°C. The resulting lawns were used to inoculate 1 L LB-amp (100 ug/ml); optical density was generally between 0.9 - 1.2. The liquid culture was incubated at 18°C for 1 hr and induced with 0.75 mM IPTG. The culture was harvested 24 hours post induction by centrifugation. Approximately 5 to 6 grams of bacterial pellet are obtained per liter of induced culture. Similar conditions were used for mutant protein expression.
Mutagenesis
A full circle PCR method, which employed a Stratagene QuikChange site-directed mutagenesis protocol, was used to generate the desired mutations. Bases mutagenized are underlined. The presence of the mutation and the integrity of the remaining coding sequence were verified by DNA sequencing. Primer sequences used in the mutagenic reactions are:
N17A forward: 5’- GTACAGCGCCGGTCGCCATCGCGGTCATCAAG -3’,
N17A reverse: 5’- CTTGATGACCGCG ATGGCGACCGGCGCTGTAC -3’;
R161Qforward: 5’- GGCAGCGCCTGCCAGAGCCTGTATGGG -3’,
R161Q reverse: 5’- CCCATACAGGCTCTGGCAGGCGCTGCC -3’;
S162A forward: 5’- GGCAGCGCCTGCCGGGCCCTGTATGGG -3’,
S162A reverse: 5’- CCCATACAGGGCCCGGCAGGCGCTGCC -3’.
Enzyme Purification
Bacterial pellets from expression of wild type or mutant enzymes were resuspended in 100 ml lysis buffer containing 20 mM Tris-Cl (pH 7.8), 300 mM NaCl, 0.2 mM imidazole, 2 mM β-mercaptoethanol. DNAse and PMSF were added to the lysis buffer to a final concentration of 1 ug/ml and 0.5 mM, respectively. Lysis was accomplished by passage thru a microfluidizer at ~17 kpsi. The lysate was clarified by centrifugation at ~100,000 × g and the supernatant loaded onto ~1.0 – 1.5 ml of Ni-Sepharose Fast Flow resin. The column was washed with 20 mM Tris-Cl (pH 7.5), 1.0 M NaCl, 20 mM imidazole, and 2 mM β -mercaptoethanol until A280 < 0.005. This extensive wash was followed by washing the column with the original lysis buffer to lower the NaCl concentration to the original 300 mM (~10 column volumes). The protein was eluted with a linear gradient of imidazole (80 ml Vt; 20 – 250 mM) in 20 mM Tris-Cl, 300 mM NaCl, 2 mM β-mercaptoethanol at pH 7.8. The fractions containing MDD were pooled and the concentration determined by the method of Bradford [18] using bovine serum albumin as a standard.
Crystallization, data collection, structure solution, and refinement
Crystals of hMDD were grown at 19 °C in 4 μl drops, composed of 2 μl protein solution (10mg/ml with 1 mM MgATP) and 2 μl well solution of 0.1M MES buffer pH 6.5, 20% PEG5000 MME, 0.1M NaCl, 1mM DTT and 0.2M ammonium sulfate. The crystals were indexed in the space group P21 with unit cell dimensions of a=87.0 Å, b=53.2 Å, c=97.8 Å and β=107.3°. Two monomers were found in an asymmetry unit, corresponding to the Mathews coefficient Vm=2.5 Å3/Da and a solvent content of 49 %. The data for hMDD in the presence of MgATP were collected at 4 °C using our in-house R-AXIS IV++. DENZO and SCALEPACK [19] were used for data processing. Data collection and processing statistics are summarized in Table 1.
Table 1.
X-Ray Data Collection and Refinement Statistics
| Data collection | |
| Resolution, Å | 30.0-2.40 (2.44-2.40) |
| # of total reflections | 124510 |
| # of unique reflections | 33621 (1643) |
| Completeness (%) | 99.0 (97.9) |
| I/σ(I) | 18.4 (2.9) |
| Cell Dimensions (Å; °) a, b, c; β | 87.0, 53.2, 97.8; 107.3 |
| Space group | P21 |
| Rmerge | 0.063 (0.474) |
| Vm (Å3/Da), Solvent content (%) | 2.5, 49.2 |
| Monomers/asymmetric unit | 2 |
| Refinement | |
| No. of protein atoms | 5676 |
| No. of water molecules | 131 |
| No. of bound ligand atoms (SO4=) | 10 |
| B average of main chain atoms (Å2) | 49.2 |
| B average of side chain (Å2) | 50.0 |
| B average of ligand(SO4=) (Å2) | 39.9 |
| B average of water molecule (Å2) | 42.8 |
| Rcrystal/ Rfree | 0.222/0.274 |
| r.m.s.d.in bond lengths / bond angles | 0.007 Å /1.30 Å |
Numbers in parentheses are values for the highest resolution shell.
The structure of hMDD was solved by the molecular replacement with the structure of yeast MDD [6](PDB code 1FI4) as the search model, using MOLREP within the CCP4 program suite (Collaborative Computation Project Number 4, 1991). The initial solution gave the two monomers for the structure of hMDD with the correlation coefficient of 0.294 and R-factor of 0.497 in the resolution range of 20 - 4.0 Ǻ. After one full cycle of the CNS refinement (Rfree = 44.6 %), the residues that were not identical between the two species were replaced with the sequence of human MDD. The model was adjusted and the electron density maps were inspected with program TURBO-FRODO [20] working on a Silicon graphics workstation. ATP was not found in the crystal structure in the final difference map. The statistics of refinement for the structure are given in Table 1. Atomic coordinates have been deposited (accession code: 3D4J). Mevalonate diphosphate coordinates were obtained from RCSB file 2OI2.
Steady-State Kinetics
For measurement of enzyme activity, the standard spectrophotometric assay [16] was employed. Initial velocities were determined by coupling the production of ADP to the oxidation of NADH with pyruvate kinase/lactate dehydrogenase (4U/assay) and rate of decrease in absorbance at 340 nm monitored. All assays were done at 30°C in the presence of 100 mM Tris-Cl (pH 7.0), 100 mM KCl, 10 mM MgCl2, 0.2 mM NADH, 0.2 mM phosphoenolpyruvate, 8 mM ATP, and 0.4 mM mevalonate diphosphate. The catalytically impaired mutant (R161Q) was assayed fluorometrically using an excitation wavelength of 340 nm and monitoring the rate of change in NADH fluorescence (emission wavelength of 460 nm) using a Photon Technologies International spectrofluorometer. The activity of WT MDD was also determined fluorometrically and was in good agreement with activity measured spectrophotometrically. All assays were started with the addition of mevalonate 5-diphosphate. For estimates of maximum velocity (Vmax) and Michaelis constant (Km), the reaction velocities at various substrate concentrations were fit to the Michaelis-Menten equation using the Grafit program (Erithacus Software Ltd.). One unit of activity corresponds to one μmol of substrate converted to product in one minute.
TNP-ATP Binding to Wild-Type and Mutant PMK Proteins
The recombinant wild-type and mutant enzymes were tested for active site structural integrity using TNP-ATP, a fluorescent analogue of ATP. TNP-ATP was titrated into buffer alone or into buffer containing a fixed concentration of enzyme. The buffer used for these fluorescence measurements contained 50 mM Tris-Cl (pH 7.0), 0.5 mM MgCl2, and 1 mM DTT. Excitation wavelength used in these experiments was 409 nm. Emission spectra were scanned from 520 – 580 nm, using a 5 nm slit width. TNP-ATP concentration was determined by absorbance at 409 nm using the extinction coefficient of 26 400 M-1 cm-1 [21]. For data analysis, values measured at the fluorescent emission peak (530 to 538 nm) for bound probe were corrected for contributions from free TNP-ATP in buffer. Thus, these corrected fluorescence enhancement data were used to calculate free and bound TNP-ATP concentrations and the corrected data were analyzed by Scatchard analysis to yield an equilibrium dissociation constants (Kd) and binding stoichiometries (n) that reflect binding sites per 44 kDa subunit.
Competitive Inhibition of WT and Mutant MDDs
For inhibition by diphosphoglycolylproline or 6-fluoromevalonate 5-diphosphate, initial velocities were determined using the spectrophotometric assay and side fractions of the enzyme peak eluted from Ni-Sepharose using an imidazole gradient. For Ki determination of diphosphoglycolylproline, the concentration of the variable substrate, mevalonate 5-diphosphate, used in these assays ranged from 86 to 285 uM for WT and from 89 to 917 uM for N17A. The ATP concentration was kept constant at standard assay conditions (~ 8 mM). The concentration of the inhibitor, diphosphoglycolylproline, ranged from 5.5 to 11 uM for WT, and 0.31 to 1.22 mM for N17A. For Ki determination of 6-fluoromevalonate 5-diphosphate, the concentration of the variable substrate, mevalonate 5-diphosphate, used in these assays ranged from 33 to 657 uM for WT, and 0.21 to 1.25 mM for N17A. The ATP concentration was kept constant at 8 mM for WT and 19 mM for N17A. The concentration of the inhibitor, 6-fluoromevalonate 5-diphosphate, ranged from 32 to 484 nM for WT, and 3.4 to 9.1 uM for N17A. These data were fit to a competitive inhibition model using Sigmaplot 10.0/Enzyme Kinetics Module 1.3 (Systat Software, Inc.).
RESULTS
The expression of both C-terminal and N-terminal his-tagged proteins was tested in various E. coli strains and at various induction temperatures and IPTG concentrations. Since, in contrast to results with expression of human phosphomevalonate kinase [22], no advantage between the expression levels of N- and C-tagged proteins was observed, the C-tagged form was used (except as noted) in the experiments documented in this report. Induction of a 1 liter culture using 0.75 mM IPTG at 16-22°C for 20-24 hours results in recovery of homogenous protein (Figure 1) after chromatography on a nickel-Sepharose column at a yield of approximately 1.5-2.5 mg. The 44 kDa band observed on SDS-PAGE (Fig. 1, lanes 2-4) agrees well with the 44.4 kDa mass calculated for the C-tagged protein. Steady state kinetic characterization of wild type recombinant human MDD (Table 2) indicates Vmax = 6.1±0.5 U/mg, Km =0.69±0.07 mM for ATP, and Km = 28.9±3.3 uM for (R,S)-mevalonate diphosphate. Kinetic parameters for N-his tagged MDD are similar. Specific activity and yield of homogeneous enzyme represent a significant improvement in comparison with previous reports on E. coli expression of human MDD [13, 14].
Figure 1.
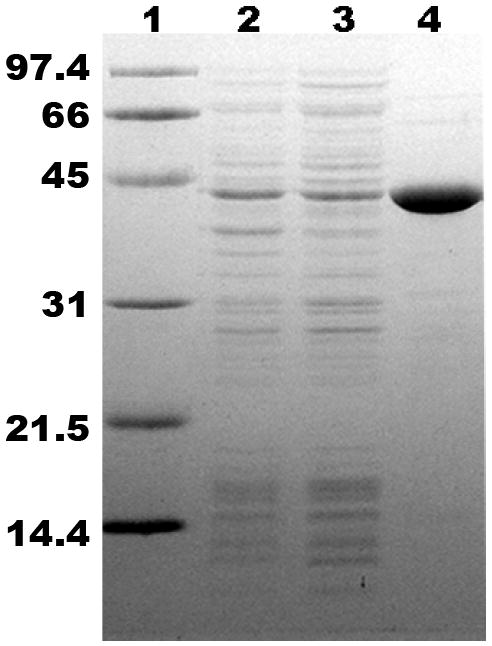
SDS-PAGE of C-his tagged human MDD, expressed in E. coli BL21(DE3). Lane 1, molecular mass standards (phosphorylase b, 97.4 kDa; bovine serum albumin, 66 kDa; ovalbumin, 45 kDa; carbonic anhydrase, 31 kDa; trypsin inhibitor, 21.5 kDa; and lysozyme, 14.4 kDa); lane 2, total lysate (15 μg) of transformed, IPTG-induced bacteria; lane 3, 100 000 × g supernatant (15 μg); lane 4, human MDD after Ni-Sepharose purification (5 μg). The observed 44 kDa band agrees with the subunit mass calculated for this recombinant hMDD protein.
Table 2.
Steady-State Kinetic Constants of Wild-Type and Mutant Human MDDsa
| Enzyme | kcat (s-1) | Vmax (U/mg) | Km(ATP) (mM) | Km(RS-MVPP) (μM) | kcat/Km,ATP (M-1s-1) | kcat/Km,RS-MVPP (M-1s-1) |
|---|---|---|---|---|---|---|
| WT | 4.5±0.4b 5.6±0.4c |
6.1±0.5b 7.6±0.6c |
0.69±0.07 | 28.9±3.3 | 6.52 × 103 8.12 × 103 |
1.56 × 105 |
| N17A | 0.2±0.01 | 0.25±0.02 | 7.7±1.2d | 425±74e | 26.0 | 4.71 × 102 |
| R161Q | 0.0022±0.0015f | 0.003±0.002f | nd | nd | nd | nd |
| S162A | 2.1±0.2 | 2.8±0.3 | 4.9±1.3 | 125.7±6.4 | 4.29 × 102 | 1.67 × 104 |
Spectrophotometric assays were performed at 30°C using the method of Cardemil and Jabalquinto (1985). Kinetic parameters were determined by fitting data to the Michaelis-Menten equation. Standard errors fo the fits are indicated.
In comparison with the C-his tagged human MDD, the N-his tagged enzyme exhibits a kcat = 4.6±0.1 s-1; Km(ATP) = 1.7±0.1 mM; Km(RS-MVPP) = 42.1±8.4 μM.
kcat estimate determined using a fluorometric assay with the same substrate concentrations used in the standard spectrophotometric assay.
Measured using elevated levels of RS-MVPP (8.8 mM).
Measured using elevated levels of ATP (20 mM) and MgCl2 (22mM).
Specific Activity determination by a fluorescence assay using the same reaction components employed in the spectrophotometric assay. Error represents the standard deviation of 6 measurements. nd indicates not determined.
Crystal structure of human MDD
The structure of human MDD is the first reported for an animal form of the protein and could greatly expedite any future development of inhibitors that requires discrimination between effects on the human enzyme versus MDD from human pathogens. The structure of the human enzyme is similar to structures of bacterial, protozoan, and yeast MDD [6,7]. The rmsd values for main chain atoms between the human structure and those of yeast, protozoan, and bacterial MDDs range from 1.1 to 1.4 Å. The structure is composed of two domains and belongs to the GHMP kinase superfamily. Ball and stick models depict conserved residues N17, R161, D305, and a tightly bound PO4=/SO4= anion (Figure 2A). There are two monomers of hMDD in the asymmetric unit, forming a head-to-head dimer (Figure 2B). The dimer interface area is about 2250 Å2 per dimer, which is only about 7 % of the total surface area. The rather small dimer interface appears to be common in several GHMK family members; the surface areas buried in the dimer of rat mevalonate kinase and M. jannaschii homoserine kinase [23] are 7.7 % and 10.6 %, respectively.
Figure 2.
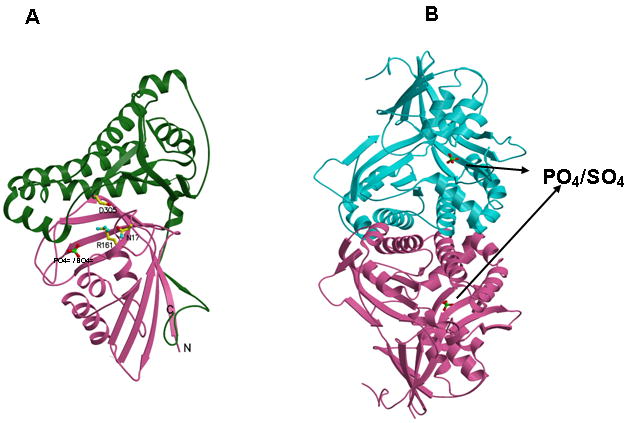
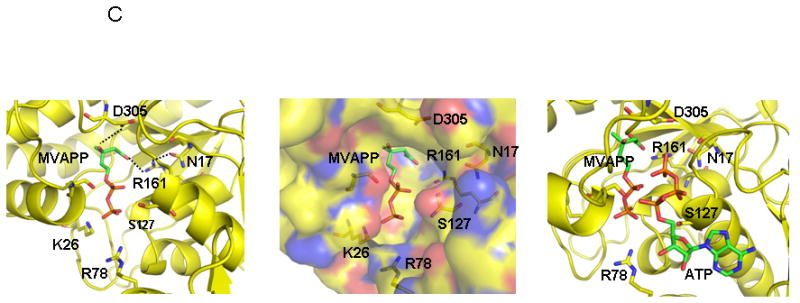
Human MDD structure and models of substrate complexes. A. The two domains of the monomer structure are shown with the N-terminal domain in pink and the C-terminal domain in green. R161, N17, D305, and the tightly bound phosphate/sulfate are shown in ball and stick models. N17 and R161 side chains are within hydrogen bonding distance (2.8 Å) of each other. B. The structure of a dimer of human MDD is shown in a view perpendicular to its 2-fold axis. Arrows indicate the position of bound phosphate/sulfate. The view of the pink monomer is rotated about 90° around the y-axis from the view shown in panel A. C. Structural models of human MDD-substrate complexes. Left panel depicts the active site of a MDD-MVAPP binary complex, with MVAPP docked into the protein using the Z-dock algorithm (http://zdock.bu.edu). Estimated distance between D305’s carboxyl and substrate’s C3 oxygen is ≤4.0 Å. For R161’s guanidinium and substrate’s C1 carboxyl oxygen, the estimate is ≤3.5 Å. Middle panel shows a surface representation of the active site region of the binary complex model, which suggests that the C1 end of MVAPP is situated in a pocket deep in the active site while the substrate phosphoryls may be more surface exposed. In the interior of the pocket are R161 and D305, which has been proposed to interact with MVAPP’s C3 hydroxyl [8]. Right panel represents a model of a ternary MDD-MVAPP-ATP complex. The position of ATP is based on an overlay of a structure of the mevalonate kinase-ATP complex [11] on the human MDD structure (Fig. 2A). The position of S127 agrees with the previous suggestion of its interaction with the phosphoryl chain of ATP [12]. The juxtaposition of ATP’s gamma phosphoryl group with respect to MVAPP’s C3 oxygen is in accord with production of a 3-phosphoMVAPP reaction intermediate [26] and supports the docking position of MVAPP in the binary complex model.
Although the crystallization medium contained 1 mM MgATP, the structure does not show bound ATP. Instead, there is a strong density that can be fit as an inorganic phosphate ion (Fig. 2A) in the final Fo-Fc map near K26 (2.7 Å), R78 (2.7 Å) and the main chain amide of G157 (3.3 Å). Since the medium included 0.2 M ammonium sulfate, the density could also be due to a sulfate ion. A similar observation was made for the structure of T. brucei protein [7]. In either case, it is reasonable to assume that the extra density is assignable to a bound phosphate;this phosphate could be attributed to either a phosphoryl group of ATP or to the alpha or beta phosphate of mevalonate diphosphate.
The Z-dock algorithm (http://zdock.bu.edu; [24]) was used to model a binary complex of hMDD with MVAPP (Fig. 2C; left panel). Similar results were obtained using the Glide docking program (Schrödinger, Inc, Portland, OR). An ordered sequential kinetic mechanism has been proposed for avian MDD by Jabalquinto and Cardemil [25] and MVAPP was assigned as the first substrate to bind, supporting the rationale for modeling a binary MDD-MVAPP complex. The beta phosphoryl of modeled MVAPP is within 1 Å of the observed electron density for a phosphate/sulfate ion, which is positioned (Fig. 2A) between the side chains of K26 and R78. This observation suggests that the location of MVAPP is reasonable. The model predicts interactions between R161’s guanidinium group and the C1 carboxyl of MVAPP (≤3.5 Å) as well as between D305’s carboxyl and the C3 oxygen of MVAPP (≤4.0 Å). N17, which is observed to form a hydrogen bond to R161 (Fig. 2A), is also located in this site. A surface representation of this region of the protein (Fig. 2C, middle panel) suggests that binding of the carboxyl end of MVAPP involves a pocket deep in the active site, while the phosphorylated end of this substrate is more surface exposed. Contributing to the surface of this pocket are residues K26 and R78, which are detected near the bound sulfate/phosphate anion (Fig. 2A). Situated in the intererior of this pocket is R161, as well as D305, which has been proposed to interact with the C3 hydroxyl of MVAPP [8].
In order to provide an additional test of the proposed MVAPP site, the empirically determined structure of ATP bound to mevalonate kinase [11], a protein exhibiting high structural homology with MDD, was overlayed on the binary MDD-MVAPP structural model. This overlay generates a model for the MDD-MVAPP-ATP ternary complex (Fig. 2C, right panel; [7]). No steric problems for the substrates are apparent, supporting the docking position of MVAPP in the human MDD binary complex. In this ternary model, the juxapositioning of ATP’s gamma phosphoryl to C3 of MVAPP is appropriate for phosphoryl transfer to form a transient 3-phosphoMVAPP intermediate [26]. In addition to the residues previously mentioned in the context of MVAPP interactions, it should be noted that S127’s position near ATP’s phosphoryl groups agrees with the previous active site assignment and functional predictions for this residue [12].
Strategy for mutagenesis of human MDD
The recombinant human enzyme was employed to test the functional influence of putative active site residues. An overlay of the human MDD structure on the homologous structure of the ATP-mevalonate kinase complex supported investigation of residues R161 and N17, which make a hydrogen bond to each other in the hMDD structure (Fig. 2A), These as well as S162, a previously uninvestigated conserved residue with side chain hydrogen bonding capability (to N17’s side chain amide N and to P15’s main chain carbonyl) were selected for mutation. All three residues are highly conserved in MDD proteins (Figure 3). Mutant proteins, in which functional side chains were conservatively substituted, were expressed and purified as described for wild type enzyme. In order to evaluate whether results of mutant enzyme characterization could be straightforwardly interpreted, a biophysical approach was tested for utility in determining whether R161Q, N17A, and S162A proteins retained structural integrity in comparison with wild-type MDD.
Figure 3.

Sequence alignment of eukoryotic and prokaryotic mevalonate diphosphate decarboxylases. Alignment was generated using the Bioedit program [36]. Residues selected for mutagenesis are denoted by an asterisk. Numbers above the sequence alignment corresponds to the residues number in human MDD. All sequences were obtained from public databases. Sequences correspond to the following organisms and accession numbers: Homo sapience (human), P53602; Sacchramyces cerevisiae (baker’s yeast), P32377; Rattus norvegicus (rat), Q642E5; Mus musculus (mouse), Q99JF5; Staphylococcus aureus, Q9FD84; Streptococcus pyogenes, Q9FD58; Brachydanio rerio (Zebrafish), 5U403; Trypanosoma brucei, Q388P2; Arabidopsis thaliana(thale cress), 023722; Ginko biloba, Q5UCT8.
Binding of TNP-ATP to Wild-Type and Mutant Human MDD Proteins
Precedent with various phosphotransferases [10,27] suggested that the fluorescent ATP analog trinitrophenyl-ATP (TNP-ATP) could function as a useful active site probe for human MDD. Accordingly, binding of this substrate analog to human MDD was tested and it was observed that fluorescence emission was enhanced upon binding to protein (Figure 4). A “blue shift” of the emission spectrum for enzyme bound TNP-ATP implicates a hydrophobic region of the protein in binding of this analog. Titrations of TNP-ATP solutions with wild-type or mutant proteins were performed and, after correction for any contributions from free probe, the fluorescence enhancement was used to estimate enzyme bound TNP-ATP. The binding data were analyzed by Scatchard plots (Figure 5) to yield comparative information on binding stoichiometries (n) and equilibrium dissociation constants (KD) for these proteins (Table 3). For wild-type human MDD, n=0.77 per 44 kDa subunit and Kd=0.55 uM. The reasonable agreement between values of binding stoichiometries (0.77-1.03) and binding constants (0.55-0.90 uM) for wild-type human MDD, N17A, and R161Q indicates that these mutant MDD proteins retain binding properties similar to wild-type enzyme, i.e., they contain a functional substrate binding site, which requires substantial overall structural integrity. On the basis of the ability to these mutant proteins to form binary complexes with the substrate analog, it seems reasonable that any substantial contrast between wild-type and mutant MDD proteins observed in other mutant characterization studies can be interpreted without reservations about major structural changes.
Figure 4.
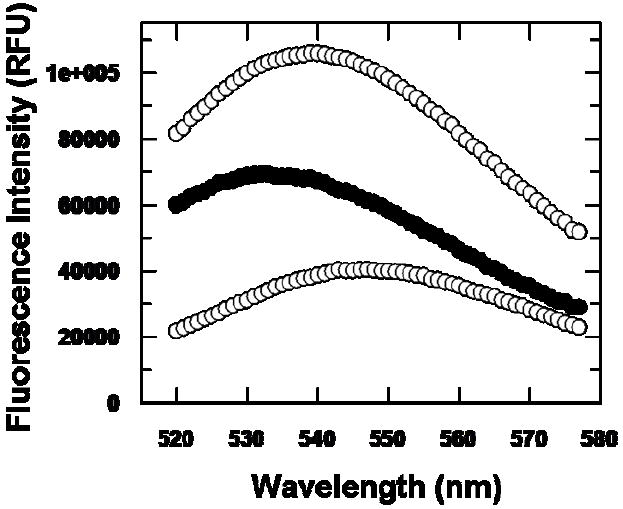
Fluorescence enhancement of TNP-ATP upon binding to wild type HMDD. The lower (control) spectrum represents 5 μM TNP-ATP in buffer (containing 50 mM Tris-Cl (pH 7.0), 0.5 mM MgCl2, and 1 mM DTT). The upper spectrum represents fluorescence emission of a sample containing 5 μM TNP-ATP and 2.5 μM wild type human MDD in buffer. The middle spectrum indicates fluorescence enhancement of enzyme bound TNP-ATP (λmax~532 nm) as reflected by subtraction of the buffer control spectrum (TNP-ATP in buffer; lower trace) from the uncorrected spectrum (enzyme + TNP-ATP; upper trace). Excitation wavelength is 409 nm.
Figure 5.
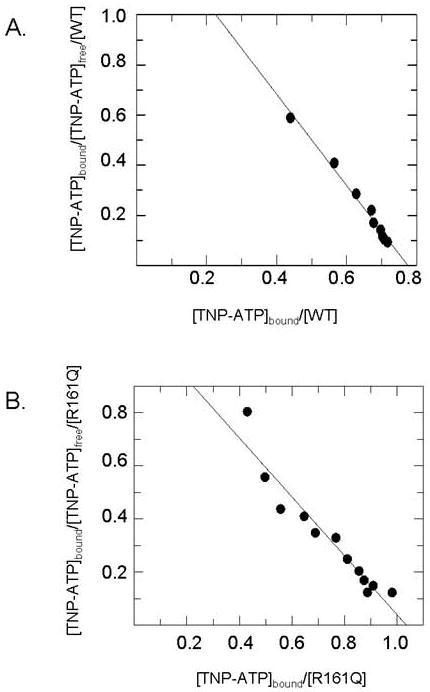
Scatchard analysis of TNP-ATP binding to wild-type and mutant human MDD proteins. Panel (A) displays the Scatchard plot for wild-type human MDD. Panel (B) displays the Scatchard plot for R161Q. Binding measurements were performed under the conditions described in the Methods and in Table 2.
Table 3.
Comparison of TNP-ATP Binding to Wild-Type and Mutant Human MDD Proteinsa
Fluorescence titrations were performed at 25°C in 50 mM Tris-Cl (pH 7.0), containing 0.5 mM MgCl2, and 1 mM DTT. Fluorescence measurements were performed using a PTI fluorometer with an excitation wavelength of 409 nm. Emission was measured over the wavelength range of 520-575 nm. Maximum emission wavelengths are 532 nm (WT); 530 nm (N17A); 532 nm (R161Q).
Equilibrium dissociation constants were calculated using the slopes of Scatchard plots of the binding data.
The n value (binding stoichiometry) was determined from the x-axis intercept of Scatchard plots of the binding data and reflects binding sites per 44 kDa subunit.
Kinetic Characterization of Mutant Human MDD Proteins and Contrasts in Efficacy of Competitive Inhibitors
Upon comparison of kinetic parameters (Table 2) for wild-type human MDD and S162A, only modest differences are apparent, with a 2-fold decrease in Vm and 4-7 fold increases (inflations) in substrate Km values. In contrast, the activity of R161Q is decreased by about 3 orders of magnitude. Specific activity of R161Q was measured using a spectrofluorometric assay. This assay affords more sensitivity than the standard spectrophotometric assay and provides Vmax results equivalent to standard assay results when used with dilute samples of wild-type enzyme (Table 2). Due to the low level of R161Q activity, estimates of substrate Km are not available. The Vmax effect is compatible with a role for R161 in MDD reaction chemistry. Mixed effects are observed for N17A. The decease in Vmax (~20-fold) is more modest than for R161Q. For N17A, the Km value for ATP is inflated by11-fold, but Kd for TNP-ATP (Table 3) is similar to wild-type MDD. A significant inflation in Km (15-fold) is measured for mevalonate diphosphate. Km comparisons between wild-type and mutant enzymes may be complicated by contributions from both catalytic and binding terms that contribute to Km. For this reason, another approach seemed desirable in order to further test whether N17’s observed interaction with R161 (Figure 2A) has functional importance.
Comparison of Ki values (instead of Km values) for competitive inhibitors of wild-type and mutant enzymes offers the advantage that Ki differences should reflect only changes in binding affinity. For this reason, we investigated inhibition of the N17A mutant enzyme (which exhibits an inflated Km for mevalonate diphosphate) by diphosphoglycolyl proline (DPGP) and 6-fluoromevalonate diphosphate (FMVAPP). Both compounds have been characterized [3, 17] as mevalonate diphosphate analogs. As demonstrated in Figure 6 and Table 4, both of these compounds inhibit recombinant human MDD, competing with the substrate, mevalonate diphosphate (Ki DPGP=2.3 uM; Ki FMVAPP=62 nM). For (R,S)-diphosphoglycolyl proline (DPGP), the ratio of Ki values for N17A/wild-type MDD is 160-fold (70-fold under elevated ATP levels in assays). For 6-fluoromevalonate diphosphate, the ratio of Ki values (N17A/wild-type MDD) is 42-fold. Inflation of the Ki values for N17A in the case of both inhibitors (Table 4) clearly demonstrates weaker binding of these analogs at the mutant’s mevalonate diphosphate substrate site and indicates a functional role for N17 in the active site of human MDD.
Figure 6.
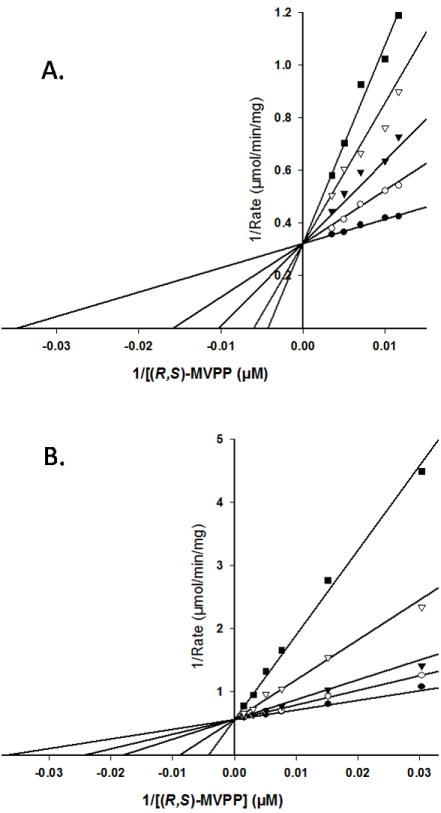
Competition for the mevalonate diphosphate site of wild-type human MDD. Double-reciprocal plots of the reaction velocity as a function of mevalonate 5-diphosphate concentration, measured at different levels of the inhibitors diphosphoglycolyl proline (A) or 6-fluoromevalonate 5-diphosphate (B). Data were fit to a competitive inhibition model using SigmaPlot 10.0/Enzyme Kinetics 1.3. (Systat Software, Inc.). Panel (A) displays the inhibition by diphosphoglycolyl proline measured at the following concentrations: (●) 0.0 mM, (○) 2.7μM, (▼) 5.5 μM, (▽) 11.0 μM, (■) 16.5 μM. Panel (B) displays the inhibition by 6-fluoromevalonate 5-diphosphate measured at the following concentrations: (●) 0.0 nM, (○) 32 nM, (▼) 65 nM, (▽) 194 nM, (■) 484 nM.
Table 4.
Comparison of Km and Ki Estimates for Wild-Type and Mutant MDDs.a
| Enzyme | Km(R,S-MVAPP)a (μM) | Fold Inflation | Ki(R,S-DPGP)b (μM) | Fold Inflation | Ki(R)-FMAVPP)c (μM) | Fold Inflation |
|---|---|---|---|---|---|---|
| WT | 28.9±3.3 | - | 2.3±0.3 | - | 0.062±0.005 | - |
| N17A | 425±74 | 14.7 | 368±27d | 160 | 2.6±0.4 | 42 |
Inhibition experiments were performed as described in Methods. Spectrophotometric assays (30°C) employed the method of Cardemil and Jabalquinto [16]. Ki values were estimated by fitting the data (Sigmaplot/Enzyme Kinetics module) to a competitive inhibition model using the equation velocity = Vmax[S]/(Km(1+[I]/Ki) + [S]). Standard errors of the fits are indicated.
(R,S)-diphosphoglycolyl proline (DPGP) concentrations ranged from 2.4 – 4.8 times the Ki value for WT and 0.8 – 3.3 times Ki value for N17A.
(R)-6-fluoromevalonate diphosphate (FMVAPP) concentrations ranged from 0.5 – 8 times the Ki value for WT and from 1.3 – 3.5 times the Ki value for N17A.
This inhibitor constant was determined using standard assay conditions (8 mM ATP). Measurements performed at elevated (20 mM) ATP indicated a Ki value of 161 μM.
DISCUSSION
The significance of mevalonate diphosphate decarboxylase (MDD) to polyisoprenoid/ sterol biosynthesis and ultimately to cell viability is underscored by a variety of observations. The gene is essential for yeast viability [28]. Disruption of the MDD gene in Streptococcus pneumonia blocks growth of the pathogenic bacteria [29]. Inhibition of the enzyme suppresses proliferation of Ras-transformed animal cells [30]. Fluoromevalonate inhibition of sterol biosynthesis in animal tissue extracts [2, 3] is explained by formation of fluoromevalonate diphosphate and inhibition of MDD. Despite the important role of this enzyme in metabolism, a convenient recombinant form of human MDD has been elusive. The report of Toth and Huwyler [13] indicated that E. coli expression of human MDD was very low, prompting an attempt to improve yield by expression in baculovirus infected cells. Recovery of partially purified enzyme validated assignment of the cDNA that encodes human MDD. Hinson et al. [14] employed E. coli expression to produce in low yield a GST-human MDD fusion protein useful in studies that identified the site of feedback inhibition of sterol biosynthesis by isoprenoid diphosphates. We isolated cDNA from that GST-MDD fusion construct and inserted the coding sequence into pET vectors to produce expression plasmids. Our results with E. coli expression of C-terminal his-tagged human MDD indicate an improvement in yield of homogeneous protein over previous published attempts. The specific activity (>6 U/mg) is comparable to the estimate for MDD purified from yeast [26] or chicken liver [16]; no activity estimates have been published for T. brucei or S. aureus proteins that have been assigned as MDD enzymes [7]. Human MDD activity is adequate to allow contrasting measurements with mutants in which catalytic efficiency is substantially decreased. The enzyme is stable upon extended storage in 20% glycerol at -80 °C and has proven useful for biophysical experiments, structural work, and functional tests by mutagenesis.
Based on the initial kinetic characterization (Table 2) of mutated residues in the putative phosphoryl acceptor binding site, N17A and R161Q were selected for functional tests. In testing the function of the invariant residue N17, characterization of N17A mutant enzyme indicated significant perturbation of Vmax and substrate Km values. The Vmax effect is much less prominent than observed upon mutation of R161 or the aspartate and serine residues (corresponding to hMDD D305 and S127) that are depicted in Figure 7. The effects of D305 [8] and S127 [12] on catalytic function have been attributed to interactions with MVAPP and the phosphoryl chain of ATP, respectively (Fig. 7). Binding of the ATP substrate analog TNP-ATP did not indicate much difference between N17A (or R161Q) and wild-type MDD, so further study of N17A focused on substrate/analog binding at the mevalonate diphosphate site. Compounds previously implicated as mevalonate diphosphate analogs and MDD inhibitors, diphosphoglycolyl proline [17] and 6-fluoromevalonate diphosphate [2, 3], were synthesized and demonstrated to very effectively inhibit human MDD competitively with respect to the phosphoryl acceptor substrate. For N17A, inhibition constants of both analogs were significantly inflated (40-160 fold) suggesting that N17’s amide substituent has considerable influence on ligand binding at the mevalonate diphosphate site. These functional results represent a significant experimental test of proposed active site models (Fig. 2C; [7]). In these models, the substrate mevalonate diphosphate is positioned so that the terminal phosphate is located near the sulfate/phosphate ion which is observed in the structure (Fig. 2A) The proposed general base catalyst, D305, is believed to deprotonate MVAPP’s C3 hydroxyl, to facilitate attack on the gamma phosphoryl of ATP (Fig. 7). In the model of liganded hMDD, it maps (Fig. 2C) in close proximity to the substrate’s C3 hydroxyl, which is consistent with its proposed function. On the basis of functional characterization of N17A and R161Q mutants, human MDD N17 and R161 are predicted (Fig. 7) to be oriented toward the C1 carboxyl group of the modeled phosphoryl acceptor substrate. This prediction is consistent with models of the human enzyme-substrate complexes (Fig. 2C). Optimal orientation may be attributed to the observed formation of a hydrogen bond between N17’s amide carbonyl oxygen and the guanidinium side chain of R161.
Figure 7.

Proposed human MDD active site residues and MDD reaction chemistry. D305 is shown juxtaposed to MVAPP’s C3 hydroxyl, which is consistent with this residue’s large functional contribution [8] and its positional homology with the general base catalyst in the active site of mevalonate kinase [11]. Similar observations [10, 12] suggest that hMDD’s S127 interacts with ATP to orient the phosphoryl chain for productive gamma phosphoryl transfer to MVAPP. Observations of impaired binding of MVAPP analogues by N17A and a large diminution in catalytic efficiency for R161Q are interpreted in the context of structural models to implicate these residues in interaction with the C1 carboxyl of MVAPP. Four chemical species involved in the reaction pathway (from left to right) include: substrate MVAPP; a transient 3-phosphoMVAPP intermediate (in which the C3 alcohol is transformed to the improved leavind group needed to produce the next intermediate); a transient beta carboxy carbenium intermediate (providing an electon sink to drive the decarboxylation reaction); reaction product isopentenyl diphosphate. The participation of a carbocation intermediate is suggested by the potency of MDD inhibition by MVAPP analogs with positively charged atoms corresponding to MVAPP’s C3 (e.g. diphosphoglycolyl proline [17]; Fig. 6A).
R161’s importance to the enzymatic function of MDD seems quite clear based on the large decrease in specific activity observed for the R161Q mutant. With such a low Vmax value for R161Q, accurate Km values could not be determined. However R161Q effectively binds the substrate analog TNP-ATP so it appears that the nucleotide binding site is intact and that this mutant’s tertiary structure and active site are not grossly perturbed. For this reason, it seems reasonable to propose a catalytic function to the guanidinium moiety of R161. After the preliminary account of our results on human MDD [15] was submitted, a paper by Qiu et al. [31] appeared; it included a report of nondetectable enzyme activity upon alanine substitution of several rat MDD amino acids. Among these mutated amino acids are arginine and aspartate residues that correspond to human MDD R161 and also to human MDD D305, for which we previously demonstrated catalytic function (Fig. 7; [8]). The rat MDD work was limited in detection sensitivity due to reliance solely on a spectrophotometric MDD assay. Rat mutants with “nondetectable” activity were not tested for structural integrity nor were mutagenesis effects quantitated. Thus, no discrimination between structural perturbation or decreased catalytic function as the explanation for observations of diminished activity for those alanine mutants seems possible.
The three orders of magnitude effect on specific activity attributed to mutation of R161, as well as the close proximity of R161 to N17 (this study, Figure 2A; [7]), suggest that a functional role for these residues at the mevalonate diphosphate site is reasonable. Arginine and asparagine residues of malic enzyme [32] each interact with C1 carboxylate oxygens of the malate substrate. However, these residues participate in binding rather than catalysis since malate’s C4 carboxyl group is involved in the decarboxylation reaction to form pyruvate. In contrast, orientation and alignment of the C1 carboxyl group of mevalonate diphosphate by R161 could influence catalysis of substrate decarboxylation and would account for a large mutagenesis effect. The influence of the hydrogen bond to N17’s amide carbonyl oxygen in orientation of the R161 guanidinium group (Fig. 2A, Fig. 2C) could also be significant. The catalytic contribution of an interaction between arginine and a substrate carboxylate has solid precedent in decarboxylation reactions. In the case of glutaryl-CoA dehydrogenase, a substitution of glutamine for the arginine that orients the C5 carboxyl group of glutaryl-CoA results in a 300-fold diminution of the rate of the C5 decarboxylation partial reaction [33]. For PEP carboxykinase, substitution of glutamine for the arginine that interacts with the C4 carboxyl of oxaloacetate (lost upon decarboxylation during formation of PEP) results in a 3900-fold diminution in catalytic rate [34]. In a recent report of structural work on PEP carboxykinase, Sullivan and Holyoak [35] provide an explanation of this mutagenesis effect, noting that the active site arginine orients the substrate carboxyl so that an unfavorable electron orbital overlap between a nitrogen of the arginine side chain and the substrate carboxyl drives the decarboxylation process. The effect observed upon mutagenesis of human MDD R161 is similar in magnitude to the effects observed for glutaryl-CoA dehydrogenase and PEP carboxykinase. Thus, precedent supports the conclusion that proper orientation and interaction of R161 with the C1 carboxyl of mevalonate diphosphate (Figure 7) accounts for our experimental observation of a notable catalytic contribution for this active site residue.
Acknowledgments
Dr. K.M. Gibson generously provided the plasmid encoding a GST-human MDD fusion construct. Dr. Dmitriy Krepkiy provided preliminary evidence for the importance of an arginine corresponding to human MDD R161 in unpublished work on yeast MDD. We thank Dr. Todd Holyoak for suggestions and help with some of the moldecular modeling. This work was supported, in part, by NIH DK53766.
Footnotes
Abbreviations used are: MDD, mevalonate diphosphate decarboxylase; MK, mevalonate kinase; GHMP, galactokinase, homoserine kinase, mevalonate kinase, phosphomevalonate kinase; DPGP, diphosphoglycolyl proline; MVAPP, mevalonate 5-diphosphate, FMVAPP, 6-fluoromevalonate 5-diphosphate.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bloch K, Chaykin S, Phillips AH, deWaard A. J Biol Chem. 1959;234:2595–2607. [PubMed] [Google Scholar]
- 2.Nave JF, d’Orchymont H, Ducep JB, Piriou F, Jung MJ. Biochem J. 1985;227:247–254. doi: 10.1042/bj2270247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reardon JE, Abeles RH. Biochemistry. 1987;26:4717–4722. doi: 10.1021/bi00389a018. [DOI] [PubMed] [Google Scholar]
- 4.Sawamura M, Nara Y, Yamori Y. J Biol Chem. 1992;267:6051–6055. [PubMed] [Google Scholar]
- 5.Dassanayake RS, Cao L, Samaranayake LP, Berges T. Mol Genet Genomics. 2002;267:281–290. doi: 10.1007/s00438-002-0648-7. [DOI] [PubMed] [Google Scholar]
- 6.Bonanno JB, Edo C, Eswar N, Pieper U, Romanowski MJ, Ilyin V, Gerchman SE, Kycia H, Studier FW, Sali A, Burley SK. Proc Natl Acad Sci U S A. 2001;98:12896–12901. doi: 10.1073/pnas.181466998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byres E, Alphey MS, Smith TK, Hunter WN. J Mol Biol. 2007;371:540–553. doi: 10.1016/j.jmb.2007.05.094. [DOI] [PubMed] [Google Scholar]
- 8.Krepkiy D, Miziorko HM. Protein Science. 2004;13:1875–1881. doi: 10.1110/ps.04725204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Potter D, Miziorko HM. J Biol Chem. 1997;272:25449–25454. doi: 10.1074/jbc.272.41.25449. [DOI] [PubMed] [Google Scholar]
- 10.Cho, Rios SE, Kim JJP, Miziorko HM. J Biol Chem. 2001;276:12573–12578. doi: 10.1074/jbc.M011478200. [DOI] [PubMed] [Google Scholar]
- 11.Fu Z, Wang M, Potter D, Miziorko HM, Kim JJP. J Biol Chem. 2002;277:18134–18142. doi: 10.1074/jbc.M200912200. [DOI] [PubMed] [Google Scholar]
- 12.Krepkiy D, Miziorko HM. Biochemistry. 2005;44:2671–2677. doi: 10.1021/bi0484217. [DOI] [PubMed] [Google Scholar]
- 13.Toth MJ, Huwyler L. J Biol Chem. 1996;271:7895–7898. doi: 10.1074/jbc.271.14.7895. [DOI] [PubMed] [Google Scholar]
- 14.Hinson DD, Chambliss KL, Toth MJ, Tanaka RD, Gibson KM. J Lipid Res. 1997;38:2216–2223. [PubMed] [Google Scholar]
- 15.Miziorko HM, Voynova NE, Fu Z, Battaile K, Kim JJP. FASEB J. 2008;22:611.4. [Google Scholar]
- 16.Cardemil E, Jabalquinto AM. Methods Enzymol. 1985;110:86–92. doi: 10.1016/s0076-6879(85)10063-7. [DOI] [PubMed] [Google Scholar]
- 17.Vlattas I, Dellureficio J, Ku E, Bohacek R, Zhang X. Bioorg & Med Chem Lett. 1996;6:2091–2096. [Google Scholar]
- 18.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 19.Otwinowski Z, Minor W. In: Processing of X-ray Diffraction Data Collected in Oscillation Mode. Carter C, Sweet RM, editors. Academic Press; Boston: 1996. pp. 307–325. [DOI] [PubMed] [Google Scholar]
- 20.Roussel A, Inisan AG, et al. Turbo-Frodo, Version OpenGL:1. Marseille, France: CNRS/Universitite – Marseille; 1999. [Google Scholar]
- 21.Hiratsuka T, Uchida K. Biochim Biophys Acta. 1973;320:635–647. doi: 10.1016/0304-4165(73)90143-8. [DOI] [PubMed] [Google Scholar]
- 22.Herdendorf TJ, Miziorko HM. Biochemistry. 2006;45:3235–3242. doi: 10.1021/bi052231u. [DOI] [PubMed] [Google Scholar]
- 23.Zhou T, Daugherty M, Grishin NV, Osterman AL, Zhang H. Structure. 2000;8:1247–1257. doi: 10.1016/s0969-2126(00)00533-5. [DOI] [PubMed] [Google Scholar]
- 24.Wiehe K, Peterson MW, Pierce B, Mintseris J, Weng Z. Methods Mol Biol. 2007;413:283–314. doi: 10.1007/978-1-59745-574-9_11. [DOI] [PubMed] [Google Scholar]
- 25.Jabalquinto AM, Cardemil E. Biochim Biophys Acta. 1989;996:257–259. doi: 10.1016/0167-4838(89)90256-2. [DOI] [PubMed] [Google Scholar]
- 26.Dhe-Paganon S, Magrath J, Abeles RH. Biochemistry. 1994;33:13355–13362. doi: 10.1021/bi00249a023. [DOI] [PubMed] [Google Scholar]
- 27.Runquist JA, Narasimhan C, Wolff CE, Koteiche H, Miziorko HM. Biochemistry. 1996;35:15049–15056. doi: 10.1021/bi9619334. [DOI] [PubMed] [Google Scholar]
- 28.Berges T, Guyonett D, Karst F. J Bacteriol. 1997;179:4664–4670. doi: 10.1128/jb.179.15.4664-4670.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilding EI, Brown JR, Bryant AP, Chalker AF, Holmes DJ, Ingraham KA, Iordanescu S, So CY, Rosenberg M, Gwynn MN. J Bacteriol. 2000;182:4319–4327. doi: 10.1128/jb.182.15.4319-4327.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuthbert JA, Lipsky PE. Cancer Res. 1995;55:1732–1740. [PubMed] [Google Scholar]
- 31.Qiu Y, Gao J, Guo F, Qiao Y, Li D. Biorg Med Chem Lett. 2007;17:6164–6168. doi: 10.1016/j.bmcl.2007.09.033. [DOI] [PubMed] [Google Scholar]
- 32.Tao X, Yang Z, Tong L. Structure. 2003;11:1141–1150. doi: 10.1016/s0969-2126(03)00168-0. [DOI] [PubMed] [Google Scholar]
- 33.Dwyer TM, Rao KS, Westover JB, Kim JJP, Frerman FE. J Biol Chem. 2001;276:133–138. doi: 10.1074/jbc.M007672200. [DOI] [PubMed] [Google Scholar]
- 34.Cristina Ravanal M, Flores M, Perez E, Aroca F, Cardemil E. Biochimie. 2004;86:357–362. doi: 10.1016/j.biochi.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 35.Sullivan SM, Holyoak T. Biochemistry. 2007;46:10078–10088. doi: 10.1021/bi701038x. [DOI] [PubMed] [Google Scholar]
- 36.Hall TA. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]