

Abstract
DNA topoisomerase I (top1) is the target of potent anticancer agents, including camptothecins and DNA intercalators, which reversibly stabilize (trap) top1 catalytic intermediates (cleavage complexes). The aim of the present study was to define the structural relationship between the site(s) of covalently bound intercalating agents, whose solution conformations in DNA are known, and the site(s) of top1 cleavage. Two diastereomeric pairs of oligonucleotide 22-mers, derived from a sequence used to determine the crystal structure of top1–DNA complexes, were synthesized. One pair contained either a trans-opened 10R- or 10S-benzo[a]pyrene 7,8-diol 9,10-epoxide adduct at the N6-amino group of a central 2′-deoxyadenosine residue in the scissile strand, and the other pair contained the same two adducts in the nonscissile strand. These adducts were derived from the (+)-(7R,8S,9S,10R)- and (−)-(7S,8R,9R,10S)-7,8-diol 9,10-epoxides in which the benzylic 7-hydroxyl group and the epoxide oxygen are trans. On the basis of analogy with known solution conformations of duplex oligonucleotides containing these adducts, we conclude that top1 cleavage complexes are trapped when the hydrocarbon adduct is intercalated between the base pairs flanking a preexisting top1 cleavage site, or between the base pairs immediately downstream (3′ relative to the scissile strand) from this site. We propose a model with the +1 base rotated out of the duplex, and in which the intercalated adduct prevents religation of the corresponding nucleotide at the 5′ end of the cleaved DNA. These results suggest mechanisms whereby intercalating agents interfere with the normal function of human top1.
DNA topoisomerase I (top1) is an essential enzyme in higher eukaryotes (1, 2). It can relax DNA supercoiling and relieve torsional strain during DNA processing, including replication, transcription, and repair. It can also perform intermolecular religation leading to DNA recombinations (3, 4). The catalytic intermediate of top1 is a cleavage complex in which a tyrosine (Tyr-723 for human top1) in the enzyme attacks a DNA phosphodiester and forms a covalent bond to the phosphorus at the 3′ side of the cleavage site while a 5′-hydroxyl is generated at the other side (1, 2). The reaction occurs with short duplex oligodeoxynucleotides (5, 6), as evidenced by recent crystal structures of top1–DNA complexes (7, 8).
top1 is the cellular target of several anticancer drugs, including the camptothecins, which have recently been approved by the Food and Drug Administration for the treatment of colon and ovarian carcinomas. The anticancer activity of these drugs results from the trapping of top1 cleavage complexes such that the enzyme is reversibly inactivated as it cleaves DNA (9–11). These agents are often referred to as “top1 poisons.” They represent an exemplary case of pharmacological interference whereby the drug stabilizes the interactions of two macromolecules (i.e., top1 and DNA) by formation of a ternary complex. Because of the clinical efficacy of camptothecins, novel top1 inhibitors are being actively pursued. Among them, some DNA intercalators are potent top1 poisons, and two of them, intoplicine and the indolocarbazole derivative NB-506, have been introduced in clinical trials (for a review see ref. 2).
The aim of the present study was to define the structural relationship between the site(s) of intercalated adducts and the site(s) of top1 cleavage. We synthesized oligonucleotides similar in sequence to those used to determine the crystal structures of top1–DNA complexes (7, 8). This sequence is derived from Tetrahymena R-chromatin at a previously identified and well-characterized top1 cleavage site (12–14). To obtain DNA oligonucleotides with intercalated adducts at selected positions relative to the normal top1 cleavage site, single trans-opened benzo[a]pyrene (BaP) diol epoxide (DE-2, benzylic 7-hydroxyl group and epoxide oxygen are trans) adducts at N6 of dA (Fig. 1) were incorporated at the +1 position of the upper, scissile, strand, replacing a dG residue in the normal substrate (see Fig. 2A). In a second set of experiments using the normal sequence (15), the trans-opened BaP DE-2 dA adducts were introduced at the −1 position (lower, nonscissile, strand) relative to the normal top1 cleavage site (Fig. 5A). trans-opened BaP DE-2 adducts at N6 of dA were selected because several NMR studies have shown that such adducts intercalate into the DNA without significant disruption of the DNA structure, whereas the previously studied (15) trans-opened BaP DE-2 adducts at N2 of dG lie in the minor groove (16). trans-R dA adducts are intercalated toward the 5′ side of the modified base (17–21). Although the evidence is less compelling for trans-10S-BaP DE-2 dA adducts, such adducts are thought to intercalate toward the 3′ side of their adducted base (22, 23). Thus, four unique structural motifs with intercalated adducts (compare Figs. 2A and 5A) were available for study. We assume that binding of top1 to these duplexes does not markedly alter their solution conformation. The present results demonstrate that intercalation between the base pairs at either the normal top1 cleavage site or immediately 3′ from this site efficiently traps top1 cleavage complexes.
Figure 1.

Structures of the trans-opened 10R- and 10S-N6-BaP DE-2 dA adducts.
Figure 2.

(A) Sequence of the 22-mer oligonucleotide, with the top1 cleavage site in the control oligonucleotide shown by a caret between the central A and T bases in the sequence at the top, and schematic representations of the oligonucleotides with intercalated hydrocarbon (shaded rectangles) based on the solution conformations of the trans-10R- and 10S-N6-BaP-dA adducts from DE-2. The trans-R and trans-S adducts are intercalated toward the 5′ and 3′ ends of the adducted strand, respectively. Labeling of the upper (scissile) strand with [α-32P]cordycepin is schematically represented as 32P-A. Note that the 3′-end label (cordycepin) introduces an additional A residue into the sequence. (B) Accumulation of top1-mediated DNA cleavage in the presence of the BaP DE-2 dA adducts. Oligonucleotide substrates are indicated at the top. Lanes 1, DNA alone; lanes 2, + top1; lanes 3, + top1 + 10 μM camptothecin (CPT). Reactions were for 30 min at 25°C. Size of the fragments (number of nucleotides) generated by top1 with the 3′-end-labeled oligonucleotide (including the cordycepin label) is indicated to the left.
Figure 5.

Position-specific effects of the trans-10R- and 10S-N6-BaP DE-2 dA adducts at position −1 in the lower strand. (A) Sequence of the oligonucleotides used and schematic representation of the intercalated BaP dA adducts (shaded rectangles). Labeling of the upper (scissile) strand with [α-32P]cordycepin is schematically represented as 32P-A. (B) top1-mediated DNA cleavage of the unadducted (control) oligonucleotide, and of the two oligonucleotides containing the BaP dA adduct at position −1 in the lower strand. Lanes 1, 4, and 6, unreacted oligonucleotides; lanes 2, 5, and 7, reactions performed in the presence of top1; lane 3, reactions performed in the presence of both top1 and camptothecin (10 μM). Reactions were for 30 min at 25°C.
Materials and Methods
Enzymes and Chemicals.
Human recombinant top1 was purified from baculovirus-infected insect cells as described previously (13). Terminal deoxynucleotidyltransferase and T4 polynucleotide kinase were purchased from GIBCO/BRL. [α-32P]Cordycepin 5′-triphosphate and [γ-32P]ATP were purchased from New England Nuclear; polyacrylamide, from Bio-Rad. Aliquots of 10 mM camptothecin in dimethyl sulfoxide (DMSO) were stored at −20°C, thawed, and diluted in water just before use.
Oligoncleotide Synthesis.
The modified oligonucleotides with an adduct (A*) (see Figs. 2A and 5A) formed by trans opening at C-10 of the epoxide ring of (+)-(7R,8S,9S,10R)- and (−)-(7S,8R,9R,10S)-BaP DE-2 by the 6-amino group of dA (Fig. 1) were prepared from diastereomeric mixtures of appropriately protected phosphoramidites (24, 25) derived from (±)-BaP DE-2 (trans-N6-BaP-dA phosphoramidites). Syntheses were carried out on a 2-μmol scale by using an automated DNA synthesizer to generate the sequence 3′ to the modified nucleotide followed by a manual step (24, 25) (8–10 μmol phosphoramidite, 50 μl of 0.5 M 4,5-dicyanoimidazole in CH3CN, 20 h at room temperature) for the coupling of the diastereomeric trans-N6-BaP-dA phosphoramidites. Efficiency for the manual coupling step ranged from 15% to 20% (determined by a spectrophotometric (495 nm) assay of the 4,4′-dimethoxytrityl cation released upon detritylation of the coupling product).
The upper- and lower-strand oligonucleotide 22-mers containing trans-N6-BaP-dA adducts were purified by reverse-phase HPLC on a Zorbax Eclipse XDB-C18 column or a Hamilton PRP-1 column, which separated the trans 10R- and 10S-adduct-containing oligonucleotides derived from the diastereomeric trans-N6-BaP-dA phosphoramidites (supplemental material at www.pnas.org). Isolated yields ranged from 5 to 6 A260 units of each oligonucleotide. The synthesized oligonucleotide 22-mers gave the expected mass spectra for the upper strand (C239H286N87O128P21, mass 7076) and lower strand (C237H290N71O137P21, (mass 6976). Assignment of the absolute configuration for the trans-N6-BaP-dA adducts in the oligonucleotides was based on the circular dichroism (CD) spectra of the oligonucleotides (supplemental material at www.pnas.org), which showed positive long-wavelength (300–350 nm) bands for 10R- and negative bands for 10S-adducted oligonucleotides (26). The long-wavelength band for the late-eluting lower-strand oligonucleotide was extremely weak. Thus, this oligonucleotide was digested to the nucleoside level with calf spleen phosphodiesterase followed by alkaline phosphatase (27), and its configurational assignment was confirmed to be 10R on the basis of comparison of its CD spectrum with the spectra of the previously characterized trans-10R- and 10S-N6-BaP-dA adducts from DE-2 (28, 29).
The three oligonucleotide size markers 5′-A*GA AAA ATT TTT-3′, 5′-T A*GA AAA ATT TTT-3′, and 5′-TT A*GA AAA ATT TTT-3′ used for identifying the site of cleavage in the upper strand were synthesized by the same method. Details of the chromatographic separations as well as CD spectra of the adducted 12-, 13-, and 14-base markers are given in the supplemental material at www.pnas.org.
top1 Reactions.
Single-stranded oligonucleotides were 3′-end-labeled with [32P]cordycepin (4, 13). Annealing to the complementary strand was performed in 1× annealing buffer (10 mM Tris⋅HCl, pH 7.8/100 mM NaCl/1 mM EDTA) by heating the reaction mixture to 95°C and overnight cooling to room temperature.
DNA substrates (approximately 50 fmol per reaction) were incubated with 5 ng of top1 with or without camptothecin for indicated times at 25°C in 10 μl of reaction buffer (10 mM Tris⋅HCl, pH 7.5/50 mM KCl/5 mM MgCl2/0.1 mM EDTA/15 μg/ml BSA, final concentrations). Reactions were stopped by adding sodium dodecyl sulfate (SDS; final concentration 0.5%). For the heat reversal experiments, the SDS stop was preceded by transferring the samples to a heat block at 65°C for the indicated times. Sequencing of DNA oligonucleotides was performed by using the Maxam–Gilbert purine sequencing protocol (30).
Before loading the sample for electrophoresis, 3.3 vol of Maxam–Gilbert loading buffer (98% formamide/0.01 M EDTA/10 mM NaOH/1 mg/ml xylene cyanol/1 mg/ml bromophenol blue) was added to reaction mixtures. Denaturing 16% polyacrylamide gels (7 M urea) were run at 40 V/cm at 50°C for 2–3 h and dried on 3MM Whatman paper sheets. Imaging was performed with a PhosphorImager (Molecular Dynamics).
Modeling the Trapping of a top1 Cleavage Complex by the Adducted DNA.
The model was generated by using Insight II Molecular Modeling System (Molecular Simulations, Waltham, MA). Two structures were aligned: the NMR structure of a DNA duplex that contains a trans-10R BaP DE-2 dA adduct (BaP-DNA) (20) and the x-ray crystal structure of human top1 covalently complexed with a double-stranded DNA substrate (7). The unadducted strand of the BaP-DNA was aligned with the scissile DNA strand in the top1 covalent complex. Then the display for the BaP-DNA unadducted strand was turned off, as was the display for the nonscissile strand DNA in the top1 crystal structure. The resulting DNA duplex is composed of the adducted strand from the BaP DNA, and the scissile strand from the top1 crystal structure. The base sequence of the adducted strand was modified to match the sequence of the top1 substrate used in this study. Modifications were made to the bond angles for the +1 and +2 bases to orient the bases in the two strands for hydrogen bonding.
Results
Accumulation of top1-Mediated DNA Cleavage Complexes by BaP Intercalation Either at, or Immediately 3′ from, the top1 Cleavage Site.
We first examined the effect of either a single trans-R or trans-S dA adduct at position +1 in the scissile (upper) strand (Fig. 2) on top1-mediated DNA cleavage. Study of an intercalated adduct at this position necessitated replacing G with A in the upper strand of the normal substrate (see Figs. 2A and 5) (15). The oligonucleotides were labeled at their 3′ terminus with [α-32P]cordycepin (see Fig. 2) to identify unambiguously the DNA cleavage fragments generated by top1. Analysis of the fragments derived from the 3′ end of the 22-mer substrate was the method of choice because these fragments are free oligonucleotides, whereas the fragments derived from the 5′ end of the substrate are linked covalently to the enzyme at their 3′ ends (1) (see supplemental material at www.pnas.org).
The unmodified 22-mer oligodeoxynucleotide (Fig. 2) was efficiently cleaved by human top1 at the expected site in the presence of camptothecin (7, 12). DNA cleavage was between the T and A bases (caret in Fig. 2A) (13, 14). The resulting 3′-cleavage product is a 13-mer fragment including the [α-32P]cordycepin (Fig. 2B, control, lane 3).
We prepared oligonucleotides in which the dA residue immediately 3′ from the top1 cleavage site (position +1) was replaced with either of the two isomeric BaP DE-2 dA adducts: the trans-S or trans-R adduct (Fig. 1). For the trans-R isomer, the aromatic portion of the BaP is intercalated at the cleavage site on the 5′ side of the adducted base (between positions −1 and +1). In contrast, the trans-S isomer is expected to have a reverse orientation (intercalation between positions +1 and +2; see Fig. 2A). Comparison of lanes 1–3 in Fig. 2B demonstrates that top1-mediated DNA cleavage products were observed in the scissile strand with both the trans-R- and trans-S-adducted oligonucleotides and, notably, were not significantly enhanced by the presence of camptothecin. Neither of the BaP dA adducts induced cleavage of the nonscissile DNA strand (Fig. 2B). Together these results demonstrate that both the trans-R and trans-S dA adducts efficiently increase top1 cleavage complexes on the scissile strand of the DNA independently of camptothecin.
The adduct-containing DNA fragments derived from cleavage of the modified scissile strand migrated more slowly than the 13-mer fragment observed with the control oligonucleotide in the presence of camptothecin. Thus, the top1-mediated DNA cleavage sites were unambiguously identified by comparison of these fragments with a series of short synthetic oligonucleotides containing either the trans-R or the trans-S dA adduct (Fig. 3A). For both adducts, top1-mediated DNA cleavage was at the same site as in the normal oligonucleotide—i.e., between T(−1) and A(+1) in the center of the sequence (caret in the sequence of the control oligonucleotide C in Fig. 3A). In addition, a weak 15-mer band was detectable. Cleavage products containing a trans-S adduct migrated slightly more slowly than did the corresponding fragments containing a trans-R adduct. Differences in electrophoretic migration for oligonucleotides containing R- versus S-BaP DE adducts have been noted previously (31, 32).
Figure 3.

Determination of the top1-mediated DNA cleavage site induced by trans-10R- and 10S-N6-BaP DE-2 dA adducts in the upper strand. (A) Sequences of the marker oligonucleotides used. Note that the 3′-end label (cordycepin) introduces an additional A residue (32P-A) into the sequence. The oligonucleotides are, from top to bottom: upper strand 13-mer, 14-mer, and 15-mer and control oligonucleotide (23-mer, C). The asterisks above the A bases indicate the position of the BaP DE-2 adducts (for each sequence, both the trans-R and trans-S adducted oligonucleotides were prepared). (B) (Left) Cleavage induced by the trans-R adduct and electrophoretic migration of the trans-R markers. (Right) Cleavage induced by the trans-S adduct and electrophoretic migration of the corresponding trans-S markers. Lanes C, substrate DNA alone; lanes T, with top1. Reactions were for 30 min at 25°C.
Together these results demonstrate the marked accumulation of top1 cleavage complexes by intercalation of BaP DE adducts at the normal top1 cleavage site (between positions −1 and +1 for the trans-R BaP dA adduct, see Fig. 2A), and between the +1 and +2 base pairs (for the corresponding trans-S BaP dA adduct at position +1).
Accumulation of top1-Mediated Cleavage Complexes by Intercalated BaP DE-2 dA Adducts Is Caused by Inhibition of Religation.
Anticancer drugs (intercalators and camptothecins) poison top1 by inhibiting religation of the top1 cleavage complexes (2). To determine whether the present intercalated trans-N6-BaP-dA adducts at position +1 on the scissile strand had a similar effect, the heat stability of the top1 cleavage complexes was examined. Heat treatment is commonly used to shift the equilibrium of the top1–DNA complexes from cleavage complexes to religation (33). Fig. 4 shows that the substrates containing BaP dA adducts produce top1 cleavage complexes that persist in the presence of heat treatment, and that these complexes are much more stable than the complexes trapped by camptothecin in unmodified DNA. The top1 cleavage complexes observed with the BaP dA adducts were also more stable, albeit partially reversible, to salt reversal than those trapped by camptothecin in unmodified DNA (see Fig. 10 in supplemental material at www.pnas.org). Furthermore, 5′-end labeling of the oligonucleotide substrate demonstrated that DNA cleavage in the presence of the BaP-dA adduct was caused by covalent binding to the protein (see Fig. 11 in supplemental material). Thus, the observed lack of reversibility did not result from enzyme-catalyzed hydrolysis of the DNA. These results demonstrate that the trans-N6-BaP-dA adducts, when intercalated either at the normal cleavage site or between the +1 and +2 positions, result in the accumulation of top1 cleavage complexes by inhibiting their religation.
Figure 4.

Heat stability of the top1 cleavage complexes trapped by the trans-10R- and 10S-N6-BaP DE-2 dA adducts in the upper strand. The oligonucleotides indicated at the top of the figure were reacted as follows: for the control oligonucleotide, lane C, no treatment (control); lane T, top1 without camptothecin; lanes 0, 1, 3, 10, and 20, top1 + camptothecin (10 μM) for 30 min at 25°C followed by the indicated times (min) at 65°C. For the trans-R and -S adducted oligonucleotides (without camptothecin): lanes C, no treatment (control); lanes 0, 1, 3, 10, and 20, top1 for 30 min at 25°C followed by the indicated times (min) at 65°C. Size of the fragments generated by top1 with the 3′-end labeled oligonucleotide (including the cordycepin label) is indicated to the left of the gel.
Trapping of top1 Cleavage Complexes by the Intercalated BaP DE-2 dA Adducts Is Dependent on the Intercalation Position.
To delineate further the influence of the intercalation position on top1 activity, we next introduced BaP DE adducts on the dA residue in the nonscissile strand at position −1. The hydrocarbon portion of the trans-R adduct at this position intercalates at the scissile bond between the −1 and +1 position (see Fig. 5A). This trans-R adduct resulted in the accumulation of top1 cleavage complexes at the normal cleavage site, thereby generating a 13-mer DNA cleavage product (compare lanes 3 and 5 in Fig. 5B). A minor 15-mer cleavage product was also observed. In contrast, the trans-S dA(−1) adduct on the nonscissile strand, which intercalates between the −2 and −1 positions (see Fig. 5A), had markedly different effects. Cleavage at the normal top1 site (−1/+1) was not detectable, whereas cleavage complexes accumulated at the −2/−3 site, thereby generating a 15-mer cleavage product. Thus, intercalation at the top1 cleavage site, between positions −1 and +1, traps the normal cleavage complex when the intercalating agent is covalently attached to either the scissile [trans-R dA(+1)] or nonscissile [trans-R dA(−1)] strand. In contrast, when intercalation is immediately upstream from the top1 cleavage site between positions −2 and −1 [trans-S dA(−1)], cleavage at the normal site is not detected. In no case (data not shown) was cleavage of the lower strand observed.
Discussion
The present study demonstrates that trans-N6-BaP DE-2 dA adducts can act as potent inhibitors of top1 cleavage and religation. It also suggests how DNA intercalators can trap top1 cleavage complexes, depending on the position of the intercalator relative to the top1 cleavage site. Because both the solution conformations (NMR structure) of certain adducts in DNA and the crystal structure of human top1 are known, our results have structural implications regarding top1–DNA interactions.
Stacking of topoisomerase poisons with the DNA base pairs immediately flanking the topoisomerase cleavage site was initially hypothesized as the mechanism of poisoning of topoisomerase II by several inhibitors of this enzyme (34–38). The base-stacking hypothesis has been extended to top1 poisons (camptothecins) (6, 39). However, binding of camptothecin to DNA is weak (40, 41). We recently proposed, on the basis of structure–activity studies and base-specific alkylation experiments with a camptothecin derivative (42), that camptothecins intercalate between the base pairs flanking the top1 cleavage site (−1/+1) in the enzyme–DNA complex (43). Redinbo et al. (7) proposed an alternative model in which camptothecin is also bound between the −1 and +1 bases, but is associated with the +1 guanine base (at the 5′ terminus of the cleaved DNA fragment), which is rotated and flipped out of the DNA duplex. The exact geometry of camptothecin binding in the top1–DNA complexes has not yet been determined. The present data, showing that BaP adducts that intercalate between the −1 and +1 base pairs trap top1 (see Figs. 2 and 5), are generally consistent with both models (7, 43) although detailed molecular interactions are different (see below). The finding that top1–DNA complexes trapped by the trans-N6-BaP DE-2 dA adducts are more stable than those observed with camptothecin suggests that it might be possible to determine the structure of such complexes by x-ray crystallography.
The site of hydrocarbon intercalation of our present BaP DE-2 dA adducts relative to the normal top1 cleavage site depends on both the configuration of the adduct and whether it is bound to the +1 (upper strand) or −1 (lower strand) position (Fig. 6). Intercalation at or immediately downstream from the normal top1 cleavage site (either between positions −1 and +1, or +1 and +2) stabilizes (traps) the cleavage complexes. These dA adducts are initially invisible to the enzyme, so that cleavage occurs at the normal site, but they subsequently act in a “stealth-like” fashion to trap the cleavage complexes. In contrast, an adduct intercalated immediately upstream from the normal cleavage site [i.e., trans-S dA(−1), Fig. 5] was visible to the enzyme and suppressed top1 cleavage at this site, as no cleavage was observed at this site even in the presence of camptothecin (data not shown). Instead, cleavage occurred at a different site to give a 15-mer product (Fig. 5B, lane 7). This adduct [trans-S dA(−1)] is intercalated immediately downstream from the new cleavage site [in the same position relative to the cleavage site that resulted in trapping of the 13-mer by the trans-S dA(+1) adduct]. Thus, it is not surprising that the 15-mer cleavage product does not readily religate and is observed in the absence of camptothecin. On the basis of our previous observations of multiple cleavage sites for this substrate containing BaP DE-2 dG adducts (15), we presume that the catalytic site of top1 can bind and interact at several different positions on the oligonucleotide. However, binding and/or reaction of the enzyme at these alternative sites is less favorable than at the normal position, and consequently becomes kinetically competitive only when the normal cleavage is prevented.
Figure 6.
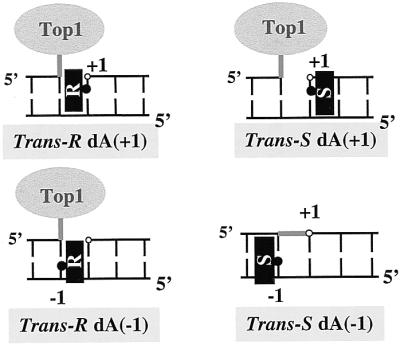
Schematic diagram of DNA substrates and orientation of the trans-10R- and 10S-N6-BaP DE-2 dA adducts. The black rectangles represent the BaP aromatic moiety intercalated in the DNA, and the black dots indicate the attachment to the alkylated base. Three of the adducts trap top1, whereas the trans-10S dA(−1) adduct on the nonscissile strand suppresses cleavage at the normal site.
The present results with intercalated dA adducts at or immediately downstream from the normal (+1/−1) cleavage site contrast with our previous observations (15) that a minor-groove trans-BaP DE-2 adduct at dG(+1) suppressed top1 cleavage at this site regardless of the orientation of the hydrocarbon [trans-S adduct toward the 5′ side of the adducted base, and trans-R adduct in the opposite orientation (16)]. This difference between intercalated and minor-groove-bound adducts is consistent with the importance of minor groove contacts at the +1 and +2 positions relative to the top1 cleavage site (7, 8). Preliminary modeling of these minor-groove N2-dG adducts in DNA indicates strong steric clashes between the adduct, in either orientation, and Arg-364 and Asp-533 of top1 (data not shown). Intercalated cis-N2-dG(+1) adducts at this same location also completely block top1-mediated cleavage at the normal site (15). Unlike the present trans dA adducts at the same position in the oligonucleotide, these cis-N2-dG(+1) adducts appear quite visible to the incoming top1, presumably because of their distortion of the duplex. Whereas little perturbation of the normal DNA structure occurs when trans dA adducts intercalate, the cis-N2-dG(+1) adducted base and its base pairing partner are flipped out of the helix and are replaced by the hydrocarbon, forming a bulge in the DNA structure (16).
Fig. 7 describes a hypothetical model to explain the inhibition of top1-mediated DNA religation when the DNA substrate contains the trans-R BaP DE-2 dA adduct in the lower, nonscissile strand at the normal cleavage site (see Figs. 5 and 6). Upon cleavage of the DNA containing the trans-R dA(−1) adduct, we speculate that the +1 guanine on the scissile upper strand rotates out of the duplex, and this appears to relieve the stress induced by the adduct on the +1 guanine, and consequently on the scissile strand. As a result, the “relaxed” +1 guanine is now lowered relative to its plane of exit (see Fig. 7). A similar mode of “flipping” the base out of the duplex is observed in several crystal structures of enzyme–DNA complexes (44, 45), as well as in a duplex DNA with a BaP adduct opposite to a mismatch (19). We propose that reentry of the +1 guanine into the DNA duplex is then hindered by the intercalated BaP DE adduct, and that the 5′-hydroxyl of the dG(+1) is prevented from attacking the enzyme tyrosyl-DNA phosphate bond.
Figure 7.

A proposed model for trapping of a top1 cleavage complex (at the normal cleavage site) by the intercalated trans-10R-N6-BaP DE-2 adduct (red) at dA(−1) [trans-10R dA(−1) in Fig. 6] on the lower, nonscissile, strand. The view is looking up at the major groove with the catalytic tyrosine, Tyr-723, in black. We speculate that when top1 makes a covalent complex with the BaP DNA at Tyr-723, the adduct will force the +1 guanine out of the duplex (see Discussion). We “flipped” the +1 guanine out of the duplex by rotating the nucleoside around the bond between the +2 base backbone P and +1 base O3′. The rotation was stopped when the +1 nucleoside O5′ came within hydrogen-bonding distance of Asp-533. This brings the guanine sugar O4′ within hydrogen-bonding distance of Arg-488, and the guanine N-7 within hydrogen-bonding distance of Arg-590. Potential hydrogen bonds are shown by black dashes.
The trans-S dA(+1) and trans-R dA(+1) substrates present a different case in that the adduct is attached to the +1 adenine on the scissile upper strand (Fig. 6). If the adducted adenine is rotated out of the duplex (as is the +1 guanine in Fig. 7), both DNA strands can then “relax.” When the adducted adenine then attempts to reenter the duplex there is likely a steric clash, which blocks entry into the now “relaxed” DNA duplex and again results in an irreversible cleavage complex (Fig. 4).
A model in which the +1 base on the scissile strand is rotated out of the duplex suggests several additional features for top1 catalysis. The +1 nucleotide residue could make three hydrogen bonds: with Asp-533, Arg-488, and Arg-590 (Fig. 7). Consequently, the free 5′ end of the DNA would not be entirely free to rotate, but instead would undergo controlled rotation (8) during relaxation of supercoiled DNA. Interestingly, mutation of Asp-533 to Gly results in an enzyme with enhanced religation activity relative to wild-type top1 (46), and which is resistant to camptothecin (47). This suggests that the hydrogen bond to Asp-533 is involved in keeping the 5′-hydroxyl of the +1 residue away from the Tyr-723/phosphate linkage.
Our present observations as well as our previous study of BaP DE-2 dG adducts (15) underline recent progress in the understanding of the molecular contacts between topoisomerases and their DNA substrates (7, 8, 15, 48–50), and the potential of such structures to elucidate the molecular interactions of various ligands, including carcinogenic adducts as well as antibacterial and anticancer drugs.
Supplementary Material
Abbreviations
- BaP
benzo[a]pyrene
- DE
diol epoxide
- DE-2
DE with the benzylic hydroxyl group trans to the epoxide oxygen
- top1
mammalian DNA topoisomerase I
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.190312697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.190312697
References
- 1.Wang J C. Annu Rev Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 2.Pommier Y, Pourquier P, Fan Y, Strumberg D. Biochim Biophys Acta. 1998;1400:83–105. doi: 10.1016/s0167-4781(98)00129-8. [DOI] [PubMed] [Google Scholar]
- 3.Christiansen K, Westergaard O. J Biol Chem. 1994;269:721–729. [PubMed] [Google Scholar]
- 4.Pommier Y, Jenkins J, Kohlhagen G, Leteurtre F. Mutat Res. 1995;337:135–145. doi: 10.1016/0921-8777(95)00019-g. [DOI] [PubMed] [Google Scholar]
- 5.Svejstrup J Q, Christiansen K, Andersen A H, Lund K, Westergaard O. J Biol Chem. 1990;265:12529–12535. [PubMed] [Google Scholar]
- 6.Jaxel C, Capranico G, Kerrigan D, Kohn K W, Pommier Y. J Biol Chem. 1991;266:20418–20423. [PubMed] [Google Scholar]
- 7.Redinbo M R, Stewart L, Kuhn P, Champoux J J, Hol W G J. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 8.Stewart L, Redinbo M R, Qiu X, Hol W G J, Champoux J J. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 9.Chen A Y, Liu L F. Annu Rev Pharmacol Toxicol. 1994;94:194–218. doi: 10.1146/annurev.pa.34.040194.001203. [DOI] [PubMed] [Google Scholar]
- 10.Bjornsti M-A, Benedetti P, Viglianti G A, Wang J C. Cancer Res. 1989;49:6318–6323. [PubMed] [Google Scholar]
- 11.Nitiss J L, Wang J C. Mol Pharmacol. 1996;50:1095–1102. [PubMed] [Google Scholar]
- 12.Bonven B J, Gocke E, Westergaard O. Cell. 1985;41:541–551. doi: 10.1016/s0092-8674(85)80027-1. [DOI] [PubMed] [Google Scholar]
- 13.Pourquier P, Ueng L-M, Fertala J, Wang D, Park H-J, Essigman J M, Bjornsti M-A, Pommier Y. J Biol Chem. 1999;274:8516–8523. doi: 10.1074/jbc.274.13.8516. [DOI] [PubMed] [Google Scholar]
- 14.Pourquier P, Ueng L-M, Kohlhagen G, Mazumder A, Gupta M, Kohn K W, Pommier Y. J Biol Chem. 1997;272:7792–7796. doi: 10.1074/jbc.272.12.7792. [DOI] [PubMed] [Google Scholar]
- 15.Pommier Y, Kohlhagen G, Pourquier P, Sayer J M, Kroth H, Jerina D M. Proc Natl Acad Sci USA. 2000;97:2040–2045. doi: 10.1073/pnas.040397497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geacintov N E, Cosman M, Hingerty B E, Amin S, Broyde S, Patel D J. Chem Res Toxicol. 1997;10:111–146. doi: 10.1021/tx9601418. [DOI] [PubMed] [Google Scholar]
- 17.Cosman M, Fiala R, Hingerty B E, Laryea A, Lee H, Harvey R G, Amin S, Geacintov N E, Broyde S, Patel D. Biochemistry. 1993;32:12488–12497. doi: 10.1021/bi00097a029. [DOI] [PubMed] [Google Scholar]
- 18.Schurter E J, Sayer J M, Oh-hara T, Yeh H J, Yagi H, Luxon B A, Jerina D M, Gorenstein D G. Biochemistry. 1995;34:9009–9020. doi: 10.1021/bi00028a009. [DOI] [PubMed] [Google Scholar]
- 19.Schurter E J, Yeh H J, Sayer J M, Lakshman M K, Yagi H, Jerina D M, Gorenstein D G. Biochemistry. 1995;34:1364–1375. doi: 10.1021/bi00004a031. [DOI] [PubMed] [Google Scholar]
- 20.Zegar I S, Kim S J, Johansen T N, Horton P J, Harris C M, Harris T M, Stone M P. Biochemistry. 1996;35:6212–6224. doi: 10.1021/bi9524732. [DOI] [PubMed] [Google Scholar]
- 21.Zegar I S, Chary P, Jabil R J, Tamura P J, Johansen T N, Lloyd R S, Harris C M, Harris T M, Stone M P. Biochemistry. 1998;37:16516–16528. doi: 10.1021/bi9817616. [DOI] [PubMed] [Google Scholar]
- 22.Yeh H J, Sayer J M, Liu X, Altieri A S, Byrd R A, Lakshman M K, Yagi H, Schurter E J, Gorenstein D G, Jerina D M. Biochemistry. 1995;34:13570–13581. doi: 10.1021/bi00041a037. [DOI] [PubMed] [Google Scholar]
- 23.Cosman M, Laryea A, Fiala R, Hingerty B E, Amin S, Geacintov N E, Broyde S, Patel D J. Biochemistry. 1995;34:1295–1307. doi: 10.1021/bi00004a024. [DOI] [PubMed] [Google Scholar]
- 24.Lakshman M K, Sayer J M, Jerina D M. J Am Chem Soc. 1991;113:6589–6594. [Google Scholar]
- 25.Lakshman M K, Sayer J M, Yagi H, Jerina D M. J Org Chem. 1992;57:4585–4590. [Google Scholar]
- 26.Page J E, Zajc B, Oh-hara T, Lakshman M K, Sayer J M, Jerina D M, Dipple A. Biochemistry. 1998;37:9127–9137. doi: 10.1021/bi980273v. [DOI] [PubMed] [Google Scholar]
- 27.Cheh A M, Yagi H, Jerina D M. Chem Res Toxicol. 1990;3:545–550. doi: 10.1021/tx00018a009. [DOI] [PubMed] [Google Scholar]
- 28.Cheng S C, Hilton B D, Roman J M, Dipple A. Chem Res Toxicol. 1989;2:334–340. doi: 10.1021/tx00011a011. [DOI] [PubMed] [Google Scholar]
- 29.Jerina D M, Chadha A, Cheh A M, Schudak M E, Wood A W, Sayer J M. In: Biological Reactive Intermediates IV. Molecular and Cellular Effects and Their Impact on Human Health, Advances in Experimental Medicine and Biology. Witmer C M, Snyder R, Jollow D J, Kalf G F, Kocsis J J, Sipes L G, editors. Vol. 283. New York: Plenum; 1991. pp. 533–553. [Google Scholar]
- 30.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 31.Shibutani S, Margulis L A, Geacintov N E, Grollman A P. Biochemistry. 1993;32:7531–7541. doi: 10.1021/bi00080a027. [DOI] [PubMed] [Google Scholar]
- 32.Chary P, Latham G J, Robberson D L, Kim S J, Han S, Harris C M, Harris T M, Lloyd R S. J Biol Chem. 1995;270:4990–5000. doi: 10.1074/jbc.270.10.4990. [DOI] [PubMed] [Google Scholar]
- 33.Hsiang Y H, Liu L F. Cancer Res. 1988;48:1722–1726. [PubMed] [Google Scholar]
- 34.Chen G L, Yang L, Rowe T C, Halligan B D, Tewey K M, Liu L F. J Biol Chem. 1984;259:13560–13566. [PubMed] [Google Scholar]
- 35.Capranico G, Kohn K W, Pommier Y. Nucleic Acids Res. 1990;18:6611–6619. doi: 10.1093/nar/18.22.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pommier Y, Capranico G, Orr A, Kohn K W. Nucleic Acids Res. 1991;19:5973–5980. doi: 10.1093/nar/19.21.5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pommier Y, Fesen M R, Goldwasser F. In: Cancer Chemotherapy and Biotherapy: Principles and Practice. Chabner B A, Longo D L, editors. Philadelphia: Lippincott–Raven; 1996. pp. 435–461. [Google Scholar]
- 38.Capranico G, Binaschi M. Biochim Biophys Acta. 1998;1400:185–194. doi: 10.1016/s0167-4781(98)00135-3. [DOI] [PubMed] [Google Scholar]
- 39.Tanizawa A, Kohn K W, Pommier Y. Nucleic Acids Res. 1993;21:5157–5166. doi: 10.1093/nar/21.22.5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leteurtre F, Fesen M, Kohlhagen G, Kohn K W, Pommier Y. Biochemistry. 1993;32:8955–8962. doi: 10.1021/bi00085a029. [DOI] [PubMed] [Google Scholar]
- 41.Yang D, Strode J T, Spielmann H P, Wang A H J, Burke T G. J Am Chem Soc. 1998;120:2979–2980. [Google Scholar]
- 42.Pommier Y, Kohlhagen G, Kohn F, Leteurtre F, Wani M C, Wall M E. Proc Natl Acad Sci USA. 1995;92:8861–8865. doi: 10.1073/pnas.92.19.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fan Y, Kohn K W, Shi L M, Weinstein J N, Pommier Y. J Med Chem. 1998;41:2216–2226. doi: 10.1021/jm9605445. [DOI] [PubMed] [Google Scholar]
- 44.Klimasauskas S, Kumar S, Roberts R J, Cheng X. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 45.Barrett T E, Schärer O D, Savva R, Brown T, Jiricny J, Pearl L H. EMBO J. 1999;23:6599–6609. doi: 10.1093/emboj/18.23.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gromova I I, Kjeldsen E, Svejstrup J Q, Alsner J, Christiansen K, Westergaard O. Nucleic Acids Res. 1993;21:593–600. doi: 10.1093/nar/21.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kjeldsen E, Bonven B J, Andoh T, Ishii K, Okada K, Bolund L, Westergaard O. J Biol Chem. 1988;263:3912–3916. [PubMed] [Google Scholar]
- 48.Cheng C, Kussie P, Pavletich N, Shuman S. Cell. 1998;92:841–850. doi: 10.1016/s0092-8674(00)81411-7. [DOI] [PubMed] [Google Scholar]
- 49.Berger J M. Biochim Biophys Acta. 1998;1400:3–18. doi: 10.1016/s0167-4781(98)00124-9. [DOI] [PubMed] [Google Scholar]
- 50.Cabral J H, Jackson A P, Smith C V, Shikotra N, Maxwell A, Liddington R C. Nature (London) 1997;388:903–906. doi: 10.1038/42294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}