

Abstract
Auditory hair cell function requires proper assembly and regulation of the nonmuscle gamma isoactin-rich cytoskeleton, and six point mutations in this isoactin cause a type of delayed onset autosomal dominant nonsyndromic progressive hearing loss, DFNA20/26. The molecular basis underlying this actin-dependent hearing loss is unknown. To address this problem, the mutations have been introduced into yeast actin, and their effects on actin function were assessed in vivo and in vitro. Because we previously showed that polymerization was unaffected in five of the six mutants, we have focused on proteins that regulate actin, in particular cofilin, which severs F-actin and sequesters actin monomers. The mutations do not affect the interaction of cofilin with G-actin. However, T89I and V370A mutant F-actins are much more susceptible to cofilin disassembly than WT filaments in vitro. Conversely, P332A filaments demonstrate enhanced resistance. Wild type actin solutions containing T89I, K118M, or P332A mutant actins at mole fractions similar to those found in the hair cell respond in vitro toward cofilin in a manner proportional to the level of the mutant present. Finally, depression of cofilin action in vivo by elimination of the cofilin-activating protein, Aip1p, rescues the inability to grow on glycerol caused by K118M, T278I, P332A, and V370A. These results suggest that a filament instability caused by these mutations can be balanced by decreasing a system in vivo that promotes increased filament turnover. Such mutant-dependent filament destabilization could easily result in hair cell malfunction leading to the late-onset hearing loss observed in these patients.
Our ability to perceive sounds hinges on the proper functioning of a highly specialized group of cells, called hair cells, which are housed in the cochlea of the inner ear. Their actin cytoskeleton plays a vital role in the ability of these cells to transduce mechanical stimuli into electrical responses (1). The columnar auditory hair cell consists of three actin-rich components (2). First are the three graded rows of finger-like projections from the apical surface of the cell referred to as stereocilia. These protrusions, comprised predominantly of bundles of actin filaments, are surrounded by the cellular membrane (3, 4). Sound-generated pressure waves in the cochlear fluid displace the basilar membrane, which in turn forces the stereocilia into the overlaying tectorial membrane, resulting in their mechanical deformation. Deflection of the stereocilia results in the opening of mechanoelectrical transduction ion channels residing on the stereocilia and ultimately triggers the initiation of a nerve impulse, which is relayed by the auditory nerve to the brain for interpretation (5).
Normal development, functioning, and maintenance of hair cell structures depend on tight regulation of the hair cell cytoskeleton as it controls the height and width of the stereociliary bundles. Each stereocilium inserts into the second actin-rich structure, the cuticular plate, a web-like arrangement of actin filaments and associated actin-binding proteins located just beneath the basal surface of the stereocilia (3, 6). This fibrous matrix produces a foundation into which the stereocilia are anchored, and it furnishes physical support for maintaining the stereociliary bundles in their upright positions. Finally, the zonula adherens contains a thick band of actin filaments which encircles each hair cell. Its function is to both unite the hair cells with the surrounding matrix of epithelial cells and provide tension across the cuticular plate in a manner analogous to the springs found on a trampoline. Based on actin involvement in cochlear cell function, it is not surprising that mutations in the components of these filamentous networks cause deafness.
In the majority of non-muscle cells in the body, the predominant actin isoform is β-nonmuscle actin with smaller amounts of γ-nonmuscle isoactin also present (7). These two isoforms differ at only 4 of the 375 amino acid residues in the protein. The N terminus of β-actin has three Asp residues, whereas that of γ-actin has three Glu residues. The other difference occurs at position 10; Val in β-actin is substituted by Ile in γ-actin. In the hair cell, however, γ-nonmuscle actin is the major form comprising about 75% of the total actin in the cell based on immunological studies with actin isoform-specific antibodies (8, 9).
Six point mutations in the coding region of the γ-nonmuscle actin gene (ACT1G) have been shown to cause a subtype of autosomal dominant nonsyndromic progressive sensorineural hearing loss designated DNFA20/26 (10–12). Patients carrying these mutations hear fine until their teens and twenties. Typically at this point the patients demonstrate marked decreases in their ability to hear high frequency sounds ranging from 4 to 8 kHz. As the patient ages the initial high frequency of hearing loss spreads over a broader range of frequencies (250 Hz to 8 kHz) until the overall loss becomes profound.
Until recently nothing was known about how the biochemical effects of these actin mutations might ultimately lead to hearing loss. To address this problem, we devised a model system in which we introduced these six point mutations into their corresponding positions in yeast actin whose sequence is 90% identical to that of γ-nonmuscle isoactin. Furthermore, each of the mutation sites in γ-nonmuscle actin is identical to the corresponding site in yeast actin. This system allows for the analysis of these mutation effects in vivo and for the purification of enough of the mutant proteins to assess their biochemical and biophysical properties. Although these mutations produced allele-specific effects in yeast cells harboring them, the purified mutant actins, with one exception, V370A, displayed polymerization characteristics virtually identical to those of WT yeast actin (13). The lack of apparent gross polymerization defects associated with these mutant actins suggested that, instead, the deafness associated with them might result from misregulation of cytoskeletal dynamics because of altered interactions with one or more of the battery of actin-binding proteins known to control cytoskeletal function and dynamics.
The actin filament is a polar structure (14, 15) with a barbed end, the preferred end for actin monomer addition, and a pointed end, which is the predominant end for monomer dissociation from the filament (16). This polarity is translated to each monomer, with the barbed end defined by subdomains 1 and 3 of the actin and the pointed end by subdomains 2 and 4 (Fig. 1). Four of the six mutations, K118M, T278I, P332A, and V370A, are in positions that could affect the barbed end surface, which interfaces with the pointed end of the next lower monomer in the filament strand (15). Moreover, three of the mutations, T89I and K118M and V370A, are predicted to be part of the actin surface comprising the tight and weak binding sites, respectively, for the actin-binding protein cofilin (17).
FIGURE 1.

Locations of the six γ-actin deafness mutations in yeast. A, front view of the crystal structure of the yeast actin monomer (63), modified from Protein Data Bank code 1YAG using Swiss-PdbViewer Version 4.01. The positions of the mutations are modeled in space-fill and color-highlighted as follows: T89I, orange; K118M, red; P264L, blue; T278I, green; P332A purple; cyan, V370A. ATP is modeled in ball-stick and colored in Corey-Pauling-Koltun. Numbers denote the actin subdomains, and N and C mark the respective termini. B, model illustrating the longitudinal contacts between two neighboring monomers within the same filament strand based on the Holmes filament model (15). Each monomer is labeled as in panel A with regard to the mutations, ADP, and subdomains. The red dashed circle denotes the general vicinity on the actin surface to which cofilin binds (21, 41, 42).
Cofilin is a small, 15–21-kDa actin-binding protein that, depending on its concentration, can sequester actin monomers (18, 19), sever/disassemble actin filaments (20–24), and promote filament nucleation by stabilizing spontaneously forming actin nuclei (25, 26). Cofilin plays an essential role in the regulation of actin cytoskeletal dynamics (18, 27–31) and is found in cochlear cells (neibank.nei.nih.gov). Cofilin-dependent filament scission/disassembly results from its disruption of both lateral (24, 32) and longitudinal (21) contacts between neighboring monomers within the filament. Thus, an actin mutation which results in destabilization of monomer-monomer interfaces or alters the cofilin binding site might make the filament more susceptible to cofilin severing. An alternative function of cofilin that might be affected by these mutations is its ability to bind to either ATP monomers being added to the barbed end of the filament or ADP-actin monomers being released from the pointed end of the filament (23, 33, 34). Altered binding could lead to altered degrees of monomer sequestration by the cofilin ultimately affecting filament length and polymer dynamics.
In this study we have assessed the ability of yeast actins carrying the deafness-causing actin mutations to interact with yeast cofilin in vitro. Based on our results in vitro, which demonstrated allele-specific altered actin-cofilin interactions, we then dampened cofilin function in vivo to determine whether altered regulation of the mutant actins by cofilin was responsible for the glycerol phenotypes associated with these mutations in vivo in yeast (13).
EXPERIMENTAL PROCEDURES
Materials
DNase I (grade D) was purchased from Worthington. Affi-Gel 10-activated resin and Micro Bio-Spin P-30 Tris gel filtration chromatography columns were obtained from Bio-Rad. DE52 DEAE-cellulose was acquired from Whatman. Platinum® TaqDNA Polymerase High Fidelity PCR kit was purchased from Invitrogen. DNA primers used for site-directed mutagenesis were obtained from Integrated DNA Technologies (Coralville, IA). N-(1-Pyrenyl)maleimide, ATP, ADP, hexokinase, and glucose were acquired from Sigma-Aldrich. Yeast cakes for WT2 actin preparations were purchased from a local bakery. All other chemicals were reagent-grade quality.
Purification of Yeast Actins and Cofilin
WT and mutant actins were purified from lysates of frozen cells via a combination of DNase I-agarose affinity chromatography, DEAE-cellulose chromatography, and polymerization/depolymerization cycling as described previously (35). The concentration of G-actin was determined from the absorbance at 290 nm using an extinction coefficient of 0.63 ml·mg−1·cm−1. Actin was stored as G-actin in G buffer (5 mm Tris-HCl, pH 7.5, 0.1 mm ATP, pH 7.5, 0.2 mm CaCl2, and 0.2 mm dithiothreitol). All actins were used within 4 days after completion of purification. Yeast cofilin was purified from Escherichia coli carrying a recombinant construct for the protein according to Lappalainen et al. (36), and concentration of the purified cofilin was determined by the Pierce® BCA Protein Assay Kit Reducing Agent Compatible.
Actin Polymerization
Polymerization of 4.8 μm G-actin in a total volume of 120 μl was induced by the addition of MgCl2 and KCl to final concentrations of 2 and 50 mm, respectively (F-salts). Polymerization was monitored at 25 °C by following the increase in light scattering of the sample in a FluoroMax-3 fluorescence spectrometer outfitted with a computer-controlled thermostatted four-position multi-sample exchanger (HORIBA Jobin Yvon Inc.). Both the excitation and emission wavelengths were set to 360 nm with the slit widths for both set at 1 nm. To determine the effects of cofilin on preformed actin filaments, the desired amount of cofilin was added to the polymerized actin sample, and the resulting change in light scattering was monitored. Alternatively, to assess the behavior of actin polymerization in the presence of cofilin, the cofilin was added to the G-actin solution before the induction of polymerization by the addition of F salts. For experiments examining the effects of different mole fractions of mutant actin on overall actin behavior, the appropriate amounts of 4.8 μm WT and mutant actins were mixed together before induction of polymerization to give a final actin concentration of 4.8 μm with the desired mole fraction of mutant actin. All polymerization experiments were performed at least three times with at least two different actin preparations.
Electron Microscopy
Two microliters of a sample containing 4.8 μm F-actin was deposited on carbon-coated Formvar grid and negatively stained with 1% uranyl acetate. Samples were then observed using a JOEL 1230 transmission electron microscope (University of Iowa Central Electron Microscopy Facility) equipped with a Gatan UltraScan 1000 2k × 2k CCD camera. Accelerating voltage of the transmission electron microscope was 100 kV. Image J was used to process the images.
Cofilin Binding Assay
ATP-G-Actin
Pyrene-labeled G-actin was made according to Feng et al. (37). An increasing amount of cofilin was added to a 1.5-ml sample containing 1 μm 100% pyrene-labeled G-actin, and the cofilin-dependent increase in pyrene fluorescence was recorded on a FluoroLog3 fluorescence spectrometer outfitted with a computer-controlled thermostatted sample exchanger with continuous sample mixing (HORIBA Jobin Yvon Inc.). All experiments were performed in G-buffer containing 50 mm KCl. Note that this concentration of KCl will not induce polymerization of yeast actin as, unlike muscle actin, it requires Mg+2 (38). The excitation and emission wavelengths were 344 and 386 nm, respectively, with the corresponding slit widths of 1 and 2 nm. Using Microsoft Excel, experimental data were fit to the quadratic binding isotherm,
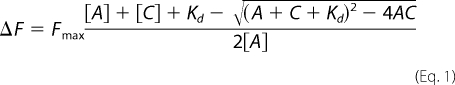 |
where ΔF is the observed fluorescence change of the actin-cofilin complex after the fluorescence of the G-actin alone has been subtracted. Fmax is the maximum fluorescence change at complete saturation of actin with cofilin. A and C are the concentrations of G-actin and cofilin, respectively, and Kd is the observed dissociation constant. The solver function was used to minimize the difference between the experimental data and the best fit to produce the Kd.
ADP-G-Actin
Unbound nucleotide was removed from the samples above using Micro Bio-Spin P-30 Tris gel filtration columns that had been equilibrated with nucleotide-free G-buffer (5 mm Tris-HCl, pH 7.5, 0.2 mm CaCl2) at 4 °C according to the manufacturer's specifications. After depletion, the 1 μm ATP-G-actin was converted to ADP-G-actin by the addition of 0.2 mm ADP, pH 7.5, 16 μm hexokinase, and 3 mm glucose. The samples were incubated on ice for at least 1 h before use, and ADP-G-actin experiments were executed within 4 h of ADP-actin preparation.
Co-sedimentation Assay
Samples containing 4.8 μm WT or mutant actin and the desired amounts of yeast cofilin were polymerized by the addition of F-salts and incubation at room temperature for about 1 h. Aliquots of 100 μl were removed and centrifuged at 75,000 rpm in a Beckman TLA100 rotor for 20 min at 25 °C. The supernatant fraction of each sample was removed, and the pellets were re-suspended in an equivalent amount of F-buffer. Then equal proportions of the supernatant and the pellet fractions were electrophoresed on a 12% SDS-polyacrylamide gel. The Coomassie-stained gels were optically scanned using a Hewlett Packard 2750 scanner, and the intensities of the actin bands were quantified by Image J.
ΔAip1p Deletion Strain Studies
Using an Δaip1:pCENWT strain as the host (39), pRS314 plasmids containing the promoter region, the TRP1 gene, and the coding sequence for WT or the various mutant yeast actins were transformed into this haploid yeast strain (40). Transformants were selected on tryptophan-deficient medium and then subjected to plasmid shuffling to eliminate the WT actin gene. Next, overnight cultures of the WT and mutant actin strains were diluted to an A600 of 0.2 in phosphate-buffered saline (140 mm NaCl, 2.7 mm KCl, 8 mm Na2HPO4, and 2 mm KH2PO4). Serial dilutions of 1×, 1/10×, 1/100×, and 1/1000× were made in phosphate-buffered saline and spotted onto YPG plates (2% yeast peptone, 2% glycerol, and 1% yeast extract), and their growth on the plate was observed at 24, 48, and 72 h.
RESULTS
Effects of Mutations on Filament Sensitivity to Cofilin
Cofilin regulation of actin filament dynamics derives in large part from its ability to sever F-actin. This role requires that cofilin first binds to a site residing between adjacent monomers within the same strand of the actin filament (Fig. 1B) (17, 21, 32, 41). Second, it requires that it be able to exploit the thermal motions between monomer-monomer interfaces to ultimately induce strand breakage. A number of the deafness-causing actin mutations lie either in cofilin binding sites (17, 41, 42) or near the barbed end of the monomer in a way that could affect the monomer-monomer interface between two monomers in the same strand. This latter possibility implies that if the mutations weakened the interface, the filament would be more susceptible to cofilin severing.
To assess this possibility, we examined the effects of yeast cofilin on the polymerization of these mutant actins for the following two reasons. First, we were using yeast actin, and previous work had shown that often pairs of proteins from the same organism evolved to work together more efficiently than proteins from heterologous sources (43, 44). Second, studies have demonstrated that stoichiometric amounts of yeast cofilin will actually decorate yeast F-actin rather than sever it (21), potentially affording us a larger range over which to assess filament stability. Increased breathing between monomers caused by the mutations might, therefore, decrease decoration by cofilin and increase severing.
We first used the change in light scattering to monitor the effect of these mutations on the ability of cofilin to interact with preformed F-actin. Increased light scattering would be suggestive of filament decoration as the thickness of the filament would increase. Large scale severing, on the other hand, would be expected to cause significant decreases in light scattering. We previously demonstrated that, by themselves, all of the mutant actins polymerized to the same extent as WT actin (13), so cofilin-dependent changes involving these actins could be easily compared. Consistent with a previous study (45), stoichiometric amounts of cofilin caused an average 42% increase in the light scattering shown by F-actin alone (Fig. 2) based on 21 different trials involving more than 5 different actin preparations. P332A mutant actin showed an average increased light scattering similar to that observed with WT actin, whereas smaller average increases of ∼15% were observed with the K118M, P264L, and T278I mutants (n = 9, 12, and 9, respectively). Student's t test analysis indicates that the cofilin-dependent change in light scattering for these three mutants is statistically different from that obtained with WT actin (p < 0.005). This slightly lower light scattering, consistent with our prediction, might be because of a small amount of filament disassembly coupled with filament decoration.
FIGURE 2.

Allele-specific effects of stoichiometric yeast cofilin on the light scattering capacity of preformed actin filaments. Samples containing 4.8 μm concentrations of either WT or mutant G-actins were polymerized by the addition of F-salts (see “Experimental Procedures”), denoted by the first set of arrows on the graphs, and the polymerization-dependent increase in light scattering was monitored as a function of time. Once the samples reached steady state, a stoichiometric amount of yeast cofilin was added to the samples (denoted by the second set of arrows), and the change in light scattering was recorded. Light scattering data were normalized so that the G-actin signal equaled 0 and the maximum no-cofilin F-actin signal equaled 1. Shown are representative data based on multiple trials with at least five different actin preparations. The number of trials for each actin follows: WT, n = 21; T89I, n = 15; K118M, n = 9; P264L, n = 12; T278I, n = 9; P332A, n = 8; V370A, n = 1. The high propensity of aggregation of V370A actin monomers makes it extremely difficult to purify in the amounts required.
The addition of stoichiometric cofilin to T89I filaments resulted in an average 77% decrease in light scattering. Stoichiometric cofilin also causes a marked decrease in light scattering of V370A, roughly 50%. This value was obtained from the titration curve shown in Fig. 4. However, we were unable to repeat the experiment multiple times because of the tendency of V370A monomers to aggregate during purification, preventing us from obtaining the material needed for the study. With both T89I and probably V370A, the profound decrease in light scattering caused by the addition of this amount of cofilin suggests significant filament disassembly. Control experiments in which buffer was added in place of the cofilin confirms that the changes in light scattering are not simply because of mixing of the samples.
FIGURE 4.

Effects of varying the cofilin concentration on the light scattering of WT and mutant F-actins. Experiments similar to those performed in Fig. 1 were carried out except we varied the amount of cofilin added to the samples once the steady state was reached. Data were normalized as in Fig. 1 so that the maximum light scattering (L.S.) values of the no-cofilin control samples were set to 100%. A, each point on the curve represents the average light scattering value and S.D. based on at least three independent trials using three different actin preparations. An exception are the data for V370A, which was from a single trial from the same actin preparation. A.U., arbitrary units. B, electron micrographs of 4.8 μm WT or P332A actin after the addition of 8.3 μm yeast cofilin. Bar = 250 nm.
Effects of Cofilin on Filament Morphology
To determine whether the changes in light scattering observed with these mutants resulted from filament decoration and/or mild to significant filament disassembly, negatively stained samples of the reaction solutions were visualized by electron microscopy. Fig. 3 shows a cofilin-dependent effect on filament morphology. WT and all of the mutants except T89I and V370A (data not shown) filaments subjected to stoichiometric amounts of cofilin appear to be more ragged and thicker relative to the filaments in the no-cofilin controls, consistent with filament decoration. These observations are consistent with a previous study (21). T89I (Fig. 3) and V370A (data not shown) actins both exhibited short filament fragments with irregular morphology, indicative of large changes in light scattering correlating with filament disassembly.
FIGURE 3.

Morphological effects of cofilin on F-actin. Electron micrographs of samples showing the differences between actin filaments alone versus those taken from Fig. 2 where stoichiometric amounts of cofilin were added. Bar = 50 nm.
Effects of Varying the Cofilin Concentration on F-actin Light Scattering
To determine the sensitivity of each of these mutant filaments to cofilin, 4.8 μm F-actin was subjected to concentrations of cofilin ranging from 0 to 11.7 μm (Fig. 4A). Under these conditions, T89I filaments disassemble at the lowest concentration tested. K118M filaments display a milder susceptibility to cofilin than WT when the cofilin concentrations were equal to or below 5.4 μm. Mild filament hyposensitivity to cofilin was observed for P264L and T278I filaments only when the cofilin concentration was 6.4 or 8.3 μm. Interestingly, across the entire cofilin concentration range tested, the P332A filaments displayed a generally higher resistance to cofilin than WT. This was especially apparent when the cofilin:actin ratios were higher than 1:1. As before, purification problems with V370A prevented multiple executions of the same experiment.
To verify that the WT filaments disassembled while the P332A filaments remained predominately intact at super-stoichiometric cofilin levels, samples at a cofilin:actin ratio of 1.7:1 were visualized by electron microscopy. Fig. 4B shows that the WT sample contains predominantly filament fragments, whereas P332A filaments appear intact.
We then performed a co-sedimentation experiment to determine whether the drop in light scattering actually represented decreased filamentous mass. If the amount of pellet-able WT actin in the absence of cofilin is taken as 100%, 85% of WT and 100% of P332A pelleted at a 1:1 cofilin/actin ratio. At a 1.7:1 ratio, 80% of P332A still pelleted compared with only 20% of the WT. This value of 20% is comparable with that obtained with the hypersensitive T89I mutant under the same conditions. These results are totally consistent with our light scattering and electron microscopy data.
Effects of Mutations on Cofilin-G-actin Binding
To determine whether the decrease in light scattering resulted completely or in part from simple sequestration of the T89I or K118M actin-ATP monomer by cofilin, the affinities for the cofilin/ATP-G-actin interaction were determined for all of the mutant except V370A. The apparent Kd value for WT actin (n = 3) was ∼0.6 μm, whereas the apparent Kd values for the mutants ranged between 0.4 and 0.8 μm (n = 2). These binding constants are comparable with those found from other cofilin studies (23, 33, 34, 46) and indicate that the mutations had no significant effect on the cofilin affinity for ATP monomers as all differed by less than a factor of 2. Likewise, the binding of cofilin to ADP-G-actin was assessed for samples of WT, T89I, and P332A. Previous experiments with muscle actin had yielded Kd ∼ 0.1 μm for the ADP-actin/cofilin interaction (33). We repeated the binding experiments with ADP G-actin for WT, T89I, and P332A actins. Fitting the data to the same quadratic binding equation used above, the Kd values for all of the species were ≤0.02 μm based on at least two determinations with different actin preparations for each species. A more accurate determination was precluded by the tightness of binding. Together our results suggest that the mutations do not materially affect the affinity of cofilin for either the ATP or ADP forms of G-actin.
Effects of Different Cofilin Concentrations on Actin Polymerization
Because of the different activities of cofilin, filament severing, monomer sequestration, and possibly filament nucleation, cofilin action during polymerization may differ from that in the presence of preformed actin filaments. We, thus, polymerized WT and mutant actins in the presence of three different cofilin concentrations using cofilin:actin ratios of 1:5, 1:1, and 1.7:1 to gain insight into which of these cofilin activities might be affected by the mutations. Sequestration in the absence of severing should result in a proportional increase in the nucleation time as the cofilin concentration increases, leading to either a noticeable lag in the onset of polymerization or inhibition of polymerization altogether. Conversely, filament severing or stabilization of nuclei in the absence of monomer sequestration should produce an increased rate of polymer formation, specifically during the elongation phase, as this would lead to the creation of more filaments ends. With simultaneous sequestration and severing, the net result would be determined by the predominant process.
Fig. 5 shows the allele-specific effects of increasing cofilin concentrations on the polymerization kinetics of these actins when it is added before polymerization. The lowest cofilin concentration, 0.92 μm, increases the rate of elongation of all of the actins compared with the no-cofilin controls, consistent with predominant filament severing and/or nucleation. Stoichiometric amounts of cofilin with either WT or K118M actins show lags consistent with predominant cofilin sequestering of monomers. Conversely, T89I, P264L, T278I, and P332A actins all result in faster elongation rates than their respective no-cofilin controls, consistent with predominant cofilin severing or nucleation. Interestingly, with stoichiometric cofilin, T89I exhibits an abrupt decrease in light scattering once the elongation phase reaches about 50% that of the total light scattering observed with the no-cofilin control sample. This decrease in light scattering to a value that is approximately one-fifth that attained in the no-cofilin control is consistent with filament disassembly as a result of severing and/or sequestering of monomers. With the exception of P332A actin, whose kinetics are unaffected, increasing the cofilin concentration to 8.3 μm (cofilin:actin ratio of 1.7:1) resulted in more pronounced lags (nucleation) in the polymerization curves of all the other actins, indicative of monomer sequestration. Based on the final extent of polymerization at this concentration of cofilin, P264L, T278I, and P332A actins are the least affected of the mutants studied.
FIGURE 5.

Effects of polymerizing deafness-causing mutant actins in the presence of different amounts of cofilin. Samples containing 4.8 μm actin (WT or mutant as indicated) were mixed in the presence of either 0 (black curves) 0.92 (red curves), 4.8 (green curves), or 8.3 μm cofilin (blue curves). Polymerization was induced by the addition of F-salts, and the change in light scattering (L.S.) was monitored as a function of time. Shown is a representative example of one of two different runs with essentially similar results from two different actin preparations. A.U., arbitrary units.
Correlation of Mutant Actin Mole Fraction with Sensitivity to Cofilin Severing
Our results thus far have shown that pure T89I, K118M, P264L, T278I, and P332A actins associated with deafness have a range of altered sensitivities to cofilin. However, based on immunological studies with actin isoform-specific antibodies (9), γ-actin comprises roughly 75% of the total actin in hair cells. Assuming that the γ-actin is equally produced from the normal and mutant gene present in the cell, about 35% of the total cellular actin would be the mutant actin. We, therefore, wished to determine the effect that different mole fractions of the more severely affected mutant actins would have on the susceptibility of WT actin to cofilin action. We first assessed the light scattering characteristics of 4.8 μm actin containing different mole fractions of either T89I (strongly hypersensitive), K118M (mildly hypersensitive), or P332A (hyposensitive). Next, yeast cofilin, in the amounts described in the Fig. 6 legend, were added to the F-actin hybrid samples, and the maximum level of light scattering was again recorded. The percent change in light scattering was plotted versus the mole fraction of mutant in the sample. For T89I (Fig. 6A) and K118M (Fig. 6B), increasing the mole fraction of the mutant actin produced a proportionate decrease in the extent of light scattering. However, in agreement with our results showing different relative sensitivities of the pure mutants, the slope of the line, indicative of the net effect, was much steeper for WT samples containing T89I versus K118M mutant actin. Conversely, increasing amounts of P332A actin, which by itself showed decreased cofilin sensitivity, actually protected the hybrid filaments against cofilin in proportion to the amount of mutant actin present. These results indicate that the mutant actins can affect the overall behavior of hybrid filaments when present in amounts likely to occur in the hair cell.
FIGURE 6.

Effects of increasing the mole fraction of mutant actin on F-actin sensitivity to cofilin. Mixtures of WT and mutant G-actins, 4.8 μm final concentration, containing different mole fractions of mutant actin were allowed to polymerize, and the final light scattering (L.S.) value was determined. A and B, stoichiometric amounts of yeast cofilin were added to each sample, and the final light scattering value was determined. Shown is the percent change in light scattering normalized to the final value of the WT sample. For C, 8.3 μm cofilin, almost twice the stoichiometric amount, was added. For the 100% WT sample, this amount of cofilin lowered the light scatting to about 20% that of the no-cofilin control. Increasing amounts of P332A afforded increasing protection against this drop. Data in C are presented relative to the light scattering value of the F-actin in the absence of cofilin. Data in all three panels show the averages and S.D. from three independent trials using two different actin preparations. A.U., arbitrary units.
Effect of Decreased Cofilin Action in Vivo on Cells Carrying the Deafness-causing Actin Mutations
Our in vitro results revealed that four of the six deafness mutations in actin produced altered cofilin sensitivity. However, there was a lack of agreement between the extents of the cofilin effects in vitro with the severity of the phenotypes associated with these mutations in vivo in yeast. One of the most hypersensitive mutations in vitro, T89I, produced only mild growth defects. Conversely, K118M, mildly hypersensitive, and P332A, hyposensitive to cofilin in vitro, produced severe growth defects including mitochondrial dysfunction (13).
This incongruity between in vivo and in vitro effects may result from the fact that our in vitro assays may not accurately reflect the more complex regulation of the actin cytoskeleton present in vivo. We, thus, wished to alter cofilin action in vivo and determine whether such a change would perhaps ameliorate the adverse phenotype associated with these mutations. Elimination of cofilin was not an option because in yeast cofilin is essential for viability (47). However, in yeast efficient cofilin action depends on the presence of an auxiliary protein, Aip1p (48, 49), which itself is nonessential (50). We, thus, determined how elimination of Aip1p would affect the in vivo phenotypes associated with these deafness-causing actin mutations. For this experiment we introduced a plasmid carrying each mutant actin into a cell deleted for Aip1 (ΔAip1) and then eliminated the plasmid encoding the WT actin. We then assessed the ability of these cells to grow on glycerol as a sole carbon source (Fig. 7). Elimination of Aip1p clearly rescues the mitochondrial defect associated with the K118M, T278I, P332A, and V370A mutations. This result suggests that the original mutations somehow resulted in a change in overall cytoskeletal stability which ultimately resulted in the associated phenotype in vivo.
FIGURE 7.

In vivo rescue of mitochondrial function in mutant actin cells after deletion of Aip1p. Serial dilutions showing rescue of the ability of mutant actin strains to grow on YPG plates (2% yeast peptone, 2% glycerol, and 1% yeast extract) after 72 h at 30 °C. Left panel, mutant actin strains. Right panel, same in A but in cells deleted for Aip1p.
DISCUSSION
Because our earlier work (51) demonstrated that the deafness-associated mutations in γ-actin did not significantly interfere with polymerization per se, our focus instead turned to the effects of these mutations on the ability of actin-binding proteins to regulate polymerization and filament dynamics. An interaction that might be affected by some or all of these mutations is that between actin and cofilin. A current model for cofilin-dependent severing of F-actin involves cofilin binding to the side of an actin filament in the groove separating two adjacent monomers in the same filament strand. This interaction would then impart or stabilize a twist to the filament weakening the monomer-monomer interface ultimately leading to filament scission (17, 21, 32, 41, 52).
Cofilin binds across subdomains 1 and 3 of one monomer and subdomain 2 of the monomer immediately below it in the same filament strand (Fig. 1B). Three of these deafness mutations fall within the predicted cofilin binding site (17); T89I within the strong cofilin binding site and K118M and V370A in a weak binding site. V370A lies near the C terminus of actin, which has been shown to be important for allosteric regulation of the top of subdomain 2 (38, 53–55), and P332A lies near a hinge region separating subdomains 1 and 3. which appears to be important in the scissor-like opening and closing of the nucleotide cleft (56). Moreover, the P332A substitution results in a residue that is less torsionally constrained. This change may substantially affect cleft movement leading to more flexible protomers and consequent changes in the actin monomer-monomer interface. This prediction is consistent with our previous in vitro data (51). In short, the mutations might interfere with proper cofilin regulation of F-actin by directly altering the binding site, altering the accessibility of cofilin to the binding site, or by altering the inherent topology of the monomer-monomer interface.
Our in vitro data demonstrate an allele-specific effect on the sensitivity of the mutant actin filaments to cofilin action with no significant differences in the binding of cofilin to the actin ATP or ADP monomers as a result of the mutations. Another possible explanation for the range of cofilin sensitivities displayed by these mutants is altered affinities of the cofilin for the mutant F-actin. Previous studies showed the severing activity of mammalian cofilin was pH-dependent (20, 57). As a result, it is possible to measure the ability of mammalian cofilin to bind to muscle actin filaments by lowering the pH of the solution to pH 6.5 (20), as this condition eliminates/greatly reduces cofilin filament severing activity. However, we could not use this approach because yeast cofilin severing is pH-independent (45). As a result, we have no insight into whether these mutations directly alter either the cofilin binding site and/or alter the inherent topology of the monomer-monomer interface which changes the accessibility of cofilin for its F-actin binding site.
The addition of cofilin to G-actin before induction of polymerization provides a more complex picture. Polymerization depends on the rate of nucleation, which is actin monomer-dependent, and the rate of elongation, which is dependent on both on the concentration of monomer and the number of filament ends available for elongation. Substoichiometric cofilin appears to sever filaments regardless of the mutation. As the cofilin increases, the multiple processes with which it is associated become more predominant, leading to a complex situation in which some of the mutant proteins exhibit a net lag and others show no lag at all. The contribution of each of these processes to the overall effect, however, would be difficult if not impossible to assign based on the bulk solution studies we have performed. The initial sharp increase in light scattering followed by the decrease observed with T89I under stoichiometric cofilin levels might reflect an unusually high critical concentration for ADP-actin in addition to increased severing leading to more rapid filament loss as we and others have observed elsewhere (58–60).
Our results from the hybrid filament experiments indicate that the overall net effect of changing the relative amounts of mutant versus WT actin in the system leads to altered biochemical and physical properties of the filament that gives rise to filament cofilin sensitivities proportional to the fraction of mutant present. Our and other laboratories have already demonstrated that mixtures of purified actins yield heterogeneous filaments (61, 62). Taken together, the proportionality of this effect indicates that the presence of small amounts of mutant actin does not exert dominant long range cooperative effects on the WT actin within the filament. If this were the case, a hyperbolic curve might be expected. A similar effect would be anticipated for the β/γ filaments in hair cells as these filaments will have physical and mechanical properties proportional to the level of mutant actin. At levels of mutant T89I in the hair cell, the hybrid filaments will display a heightened sensitivity to cofilin disassembly compared with WT hair cells or cells containing P332A actin. This situation could easily result in a globally less stable cytoskeletal system within the cell because of inappropriate regulation by an array of actin interacting proteins, not necessarily just one. A less stable cytoskeletal hair cell system in conjunction with the relatively low amount of mutant actin may explain the slow and progressive nature of the hearing loss patients experience.
Our in vitro cofilin results do not directly correlate with the in vivo observations. For example, cofilin hypersensitive mutations produce growth defects ranging from mild to severe, whereas a hyperstable mutation yields a severe growth defect. However, there is a possible solution for this paradox. A properly functioning actin cytoskeleton requires a delicate balance between assembly and disassembly of actin filaments, and this balance is maintained by the combinatorial effects of a large array of regulatory proteins inside the cell, only one of which is cofilin. The presence of the mutant actin might adversely affect the stability of the cytoskeleton because of misregulation by this array of actin regulatory proteins. Decreasing the cofilin-dependent filament-destabilizing system by Aip1p elimination might compensate for this mutant-induced destabilization resulting in rescue of cytoskeletal function as seen for the four mutants with a glycerol phenotype.
In summary, this study makes a number of significant points. First, the deafness-causing actin mutations, although they seem to not drastically affect polymerization, can cause a range of allele-specific effects of the ability of actin to be properly regulated by an actin-binding protein. Second, the presence of the mutant actin at levels comparable in mole fraction to what occurs in the hair cell can affect actin filament behavior. Third, the severe growth phenotypes caused by four of the mutations apparently result in increased cytoskeletal instability, which can be compensated for in vivo by suppression of a major actin filament severing system. Fourth, because of the combinatorial regulation of the actin cytoskeleton in vivo, the effects observed with a single purified regulatory protein in vitro may not directly correlate with the overall severity of the defect caused by the mutation in the cell. Thus, cytoskeletal regulatory instability imparted by these mutations could easily be a central factor in the hair cell malfunction leading to deafness. It is clear that a more complete understanding of the nature of this instability will require the examination of the mutation effects on the interaction of the actin with additional actin-binding proteins both singly and in combination, although the results presented here present a valuable first step to the achievement of this goal.
This work was supported, in whole or in part, by National Institutes of Health Grant DC008803 (from the NIDCD; to P. A. R.) and National Research Service Award Predoctoral Fellowship F31DC008913 (from the NIDCD; to K. E. B.).
- WT
- wild type.
REFERENCES
- 1.Müller U., Littlewood-Evans A. ( 2001) Trends Cell Biol. 11, 334– 342 [DOI] [PubMed] [Google Scholar]
- 2.Drenckhahn D., Engel K., Höfer D., Merte C., Tilney L., Tilney M. ( 1991) J. Cell Biol. 112, 641– 651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tilney L. G., Egelman E. H., DeRosier D. J., Saunder J. C. ( 1983) J. Cell Biol. 96, 822– 834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tilney L. G., Derosier D. J., Mulroy M. J. ( 1980) J. Cell Biol. 86, 244– 259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fettiplace R., Hackney C. M. ( 2006) Nat. Rev. Neurosci. 7, 19– 29 [DOI] [PubMed] [Google Scholar]
- 6.DeRosier D. J., Tilney L. G. ( 1989) J. Cell Biol. 109, 2853– 2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rubenstein P. A. ( 1990) BioEssays 12, 309– 315 [DOI] [PubMed] [Google Scholar]
- 8.Höfer D., Ness W., Drenckhahn D. ( 1997) J. Cell Sci. 110, 765– 770 [DOI] [PubMed] [Google Scholar]
- 9.Furness D. N., Katori Y., Mahendrasingam S., Hackney C. M. ( 2005) Hear. Res. 207, 22– 34 [DOI] [PubMed] [Google Scholar]
- 10.Rendtorff N. D., Zhu M., Fagerheim T., Antal T. L., Jones M., Teslovich T. M., Gillanders E. M., Barmada M., Teig E., Trent J. M., Friderici K. H., Stephan D. A., Tranebjaerg L. ( 2006) Eur. J. Hum. Genet. 14, 1097– 1105 [DOI] [PubMed] [Google Scholar]
- 11.van Wijk E., Krieger E., Kemperman M. H., De Leenheer E. M., Huygen P. L., Cremers C. W., Cremers F. P., Kremer H. ( 2003) J. Med. Genet. 40, 879– 884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu M., Yang T., Wei S., DeWan A. T., Morell R. J., Elfenbein J. L., Fisher R. A., Leal S. M., Smith R. J., Friderici K. H. ( 2003) Am. J. Hum. Genet. 73, 1082– 1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bryan K. E., Wen K. K., Zhu M., Rendtorff N. D., Feldkamp M., Tranebjaerg L., Friderici K. H., Rubenstein P. A. ( 2006) J. Biol. Chem. 281, 20129– 20139 [DOI] [PubMed] [Google Scholar]
- 14.Moore P. B., Huxley H. E., DeRosier D. J. ( 1970) J. Mol. Biol. 50, 279– 295 [DOI] [PubMed] [Google Scholar]
- 15.Holmes K. C., Popp D., Gebhard W., Kabsch W. ( 1990) Nature 347, 44– 49 [DOI] [PubMed] [Google Scholar]
- 16.Pollard T. D., Mooseker M. S. ( 1981) J. Cell Biol. 88, 654– 659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galkin V. E., Orlova A., Lukoyanova N., Wriggers W., Egelman E. H. ( 2001) J. Cell Biol. 153, 75– 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlier M. F., Ressad F., Pantaloni D. ( 1999) J. Biol. Chem. 274, 33827– 33830 [DOI] [PubMed] [Google Scholar]
- 19.Ressad F., Didry D., Xia G. X., Hong Y., Chua N. H., Pantaloni D., Carlier M. F. ( 1998) J. Biol. Chem. 273, 20894– 20902 [DOI] [PubMed] [Google Scholar]
- 20.Hawkins M., Pope B., Maciver S. K., Weeds A. G. ( 1993) Biochemistry 32, 9985– 9993 [DOI] [PubMed] [Google Scholar]
- 21.Galkin V. E., Orlova A., VanLoock M. S., Shvetsov A., Reisler E., Egelman E. H. ( 2003) J. Cell Biol. 163, 1057– 1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pavlov D., Muhlrad A., Cooper J., Wear M., Reisler E. ( 2007) J. Mol. Biol. 365, 1350– 1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlier M. F., Laurent V., Santolini J., Melki R., Didry D., Xia G. X., Hong Y., Chua N. H., Pantaloni D. ( 1997) J. Cell Biol. 136, 1307– 1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bobkov A. A., Muhlrad A., Shvetsov A., Benchaar S., Scoville D., Almo S. C., Reisler E. ( 2004) J. Mol. Biol. 337, 93– 104 [DOI] [PubMed] [Google Scholar]
- 25.Kudryashov D. S., Galkin V. E., Orlova A., Phan M., Egelman E. H., Reisler E. ( 2006) J. Mol. Biol. 358, 785– 797 [DOI] [PubMed] [Google Scholar]
- 26.Andrianantoandro E., Pollard T. D. ( 2006) Mol. Cell 24, 13– 23 [DOI] [PubMed] [Google Scholar]
- 27.Bamburg J. R., Wiggan O. P. ( 2002) Trends Cell Biol. 12, 598– 605 [DOI] [PubMed] [Google Scholar]
- 28.Chen H., Bernstein B. W., Bamburg J. R. ( 2000) Trends Biochem. Sci. 25, 19– 23 [DOI] [PubMed] [Google Scholar]
- 29.DesMarais V., Ghosh M., Eddy R., Condeelis J. ( 2005) J. Cell Sci. 118, 19– 26 [DOI] [PubMed] [Google Scholar]
- 30.Paavilainen V. O., Bertling E., Falck S., Lappalainen P. ( 2004) Trends Cell Biol. 14, 386– 394 [DOI] [PubMed] [Google Scholar]
- 31.Pollard T. D., Blanchoin L., Mullins R. D. ( 2000) Annu. Rev. Biophys. Biomol. Struct. 29, 545– 576 [DOI] [PubMed] [Google Scholar]
- 32.McGough A., Chiu W. ( 1999) J. Mol. Biol. 291, 513– 519 [DOI] [PubMed] [Google Scholar]
- 33.Blanchoin L., Pollard T. D. ( 1998) J. Biol. Chem. 273, 25106– 25111 [DOI] [PubMed] [Google Scholar]
- 34.Maciver S. K., Weeds A. G. ( 1994) FEBS Lett. 347, 251– 256 [DOI] [PubMed] [Google Scholar]
- 35.Cook R. K., Rubenstein P. A. ( 1992) in Practical Approaches in Cell Biology ( Carraway K., Carraway C. C. eds) pp. 99– 122, IRL Press at Oxford University Press, Oxford [Google Scholar]
- 36.Lappalainen P., Fedorov E. V., Fedorov A. A., Almo S. C., Drubin D. G. ( 1997) EMBO J. 16, 5520– 5530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng L., Kim E., Lee W. L., Miller C. J., Kuang B., Reisler E., Rubenstein P. A. ( 1997) J. Biol. Chem. 272, 16829– 16837 [DOI] [PubMed] [Google Scholar]
- 38.Kim E., Miller C. J., Reisler E. ( 1996) Biochemistry 35, 16566– 16572 [DOI] [PubMed] [Google Scholar]
- 39.Stokasimov E., McKane M., Rubenstein P. A. ( 2008) J. Biol. Chem. 283, 34844– 34854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaiser C., Michaelis S., Mitchell A. ( 1994) Methods in Yeast Genetics, pp. 133– 134, Cold Spring Harbor Laboratory Press, Plainview, New York [Google Scholar]
- 41.McGough A., Pope B., Chiu W., Weeds A. ( 1997) J. Cell Biol. 138, 771– 781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Renoult C., Ternent D., Maciver S. K., Fattoum A., Astier C., Benyamin Y., Roustan C. ( 1999) J. Biol. Chem. 274, 28893– 28899 [DOI] [PubMed] [Google Scholar]
- 43.Wen K. K., Rubenstein P. A. ( 2005) J. Biol. Chem. 280, 24168– 24174 [DOI] [PubMed] [Google Scholar]
- 44.Wen K. K., McKane M., Houtman J. C., Rubenstein P. A. ( 2008) J. Biol. Chem. 283, 9444– 9453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pavlov D., Muhlrad A., Cooper J., Wear M., Reisler E. ( 2006) Cell Motil. Cytoskeleton 63, 533– 542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vartiainen M. K., Mustonen T., Mattila P. K., Ojala P. J., Thesleff I., Partanen J., Lappalainen P. ( 2002) Mol. Biol. Cell 13, 183– 194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moon A. L., Janmey P. A., Louie K. A., Drubin D. G. ( 1993) J. Cell Biol. 120, 421– 435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodal A. A., Tetreault J. W., Lappalainen P., Drubin D. G., Amberg D. C. ( 1999) J. Cell Biol. 145, 1251– 1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okada K., Obinata T., Abe H. ( 1999) J. Cell Sci. 112, 1553– 1565 [DOI] [PubMed] [Google Scholar]
- 50.Iida K., Yahara I. ( 1999) Genes Cells 4, 21– 32 [DOI] [PubMed] [Google Scholar]
- 51.Bryan K. E., Rubenstein P. A. ( 2005) J. Biol. Chem. 280, 1696– 1703 [DOI] [PubMed] [Google Scholar]
- 52.Prochniewicz E., Janson N., Thomas D. D., De la Cruz E. M. ( 2005) J. Mol. Biol. 353, 990– 1000 [DOI] [PubMed] [Google Scholar]
- 53.Adams S. B., Reisler E. ( 1994) Biochemistry 33, 14426– 14433 [DOI] [PubMed] [Google Scholar]
- 54.DalleDonne I., Milzani A., Colombo R. ( 1998) Biochem. Cell Biol. 76, 583– 591 [PubMed] [Google Scholar]
- 55.Strzelecka-Gołaszewska H., Mossakowska M., Woźniak A., Moraczewska J., Nakayama H. ( 1995) Biochem. J. 307, 527– 534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Page R., Lindberg U., Schutt C. E. ( 1998) J. Mol. Biol. 280, 463– 474 [DOI] [PubMed] [Google Scholar]
- 57.Yonezawa N., Nishida E., Sakai H. ( 1985) J. Biol. Chem. 260, 14410– 14412 [PubMed] [Google Scholar]
- 58.McKane M., Wen K. K., Meyer A., Rubenstein P. A. ( 2006) J. Biol. Chem. 281, 29916– 29928 [DOI] [PubMed] [Google Scholar]
- 59.Garner E. C., Campbell C. S., Mullins R. D. ( 2004) Science 306, 1021– 1025 [DOI] [PubMed] [Google Scholar]
- 60.Pollard T. D. ( 1986) J. Cell Biol. 103, 2747– 2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wen K. K., Rubenstein P. A. ( 2003) J. Biol. Chem. 278, 48386– 48394 [DOI] [PubMed] [Google Scholar]
- 62.Costa C. F., Rommelaere H., Waterschoot D., Sethi K. K., Nowak K. J., Laing N. G., Ampe C., Machesky L. M. ( 2004) J. Cell Sci. 117, 3367– 3377 [DOI] [PubMed] [Google Scholar]
- 63.Vorobiev S., Strokopytov B., Drubin D. G., Frieden C., Ono S., Condeelis J., Rubenstein P. A., Almo S. C. ( 2003) Proc. Natl. Acad. Sci. U.S.A. 100, 5760– 5765 [DOI] [PMC free article] [PubMed] [Google Scholar]