

Abstract
Malic enzyme has a dimer of dimers quaternary structure in which the dimer interface associates more tightly than the tetramer interface. In addition, the enzyme has distinct active sites within each subunit. The mitochondrial NAD(P)+-dependent malic enzyme (m-NAD(P)-ME) isoform behaves cooperatively and allosterically and exhibits a quaternary structure in dimer-tetramer equilibrium. The cytosolic NADP+-dependent malic enzyme (c-NADP-ME) isoform is noncooperative and nonallosteric and exists as a stable tetramer. In this study, we analyze the essential factors governing the quaternary structure stability for human c-NADP-ME and m-NAD(P)-ME. Site-directed mutagenesis at the dimer and tetramer interfaces was employed to generate a series of dimers of c-NADP-ME and m-NAD(P)-ME. Size distribution analysis demonstrated that human c-NADP-ME exists mainly as a tetramer, whereas human m-NAD(P)-ME exists as a mixture of dimers and tetramers. Kinetic data indicated that the enzyme activity of c-NADP-ME is not affected by disruption of the interface. There are no significant differences in the kinetic properties between AB and AD dimers, and the dimeric form of c-NADP-ME is as active as tetramers. In contrast, disrupting the interface of m-NAD(P)-ME causes the enzyme to be less active than wild type and to become less cooperative for malate binding; the kcat values of mutants decreased with increasing Kd,24 values, indicating that the dissociation of subunits at the dimer or tetramer interfaces significantly affects the enzyme activity. The above results suggest that the tetramer is required for a fully functional m-NAD(P)-ME. Taken together, the analytical ultracentrifugation data and the kinetic analysis of these interface mutants demonstrate the differential role of tetramer organization for the c-NADP-ME and m-NAD(P)-ME isoforms. The regulatory mechanism of m-NAD(P)-ME is closely related to the tetramer formation of this isoform.
Malic enzymes catalyze a reversible oxidative decarboxylation of l-malate to yield pyruvate and CO2 with reduction of NAD(P)+ to NAD(P)H. This reaction requires a divalent metal ion (Mg2+ or Mn2+) for catalysis (1–3). Malic enzymes are found in a broad spectrum of living organisms that share conserved amino acid sequences and structural topology; such shared characteristics reveal a crucial role for the biological functions of these enzymes (4, 5). In mammals, malic enzymes have been divided into three isoforms according to their cofactor specificity and subcellular localization as follows: cytosolic NADP+-dependent (c-NADP-ME),2 mitochondrial NADP+-dependent (m-NADP-ME), and mitochondrial NAD(P)+-dependent (m-NAD(P)-ME). The m-NAD(P)-ME isoform displays dual cofactor specificity; it can use both NAD+ and NADP+ as the coenzyme, but NAD+ is more favored in a physiological environment (6–8). Dissimilar to the other two isoforms, m-NAD(P)-ME binds malate cooperatively, and it can be allosterically activated by fumarate; the sigmoidal kinetics observed with cooperativity is abolished by fumarate (9–12). Mutagenesis and kinetic studies demonstrated that ATP is an active-site inhibitor, although it also binds to the exo sites in the tetramer interface (13–15). Structural studies also revealed an allosteric binding site for fumarate residing at the dimer interface. Mutation in the binding site significantly affects the activating effects of fumarate (11, 16, 17).
The c-NADP-ME and m-NADP-ME isoforms play an important role in lipogenesis by providing NADPH for the biosynthesis of long-chain fatty acids and steroids. Thus, c-NADP-ME together with acetyl-CoA carboxylase, fatty-acid synthase, and glucose-6-phosphate dehydrogenase are classified as lipogenic enzymes (2, 18–21). The m-NAD(P)-ME isoform has attracted much attention because it is involved in glutaminolysis, which is an energy-producing pathway of tumor cells that utilizes glutamine and glutamate. Thus, m-NAD(P)-ME is considered to be a potential target in cancer therapy (22–27).
Various crystal structures of malic enzymes in complex with substrate, metal ion, coenzyme, regulator, and inhibitor are available in the Protein Data Bank (4, 11, 28–32). The overall tertiary structures of these malic enzymes are similar, but there are still some differences that may be significant for catalysis and regulation of the enzyme. Malic enzyme exists as a dimer of dimers with a stronger association of the dimer interface than that of the tetramer interface (Fig. 1A). The dimer interface is formed by subunits A and B or C and D (Fig. 1B), whereas the tetramer interaction consists of contacts between subunits A and D or B and C (Fig. 1C). A hydrophobic interaction is the major driving force for subunit assembly, but hydrogen bonding and ionic interactions also contribute markedly. The homotetramer of the enzyme is composed of four identical monomers each with its own active site. In the structure of human m-NAD(P)-ME, aside from its well defined active site, there are two regulatory sites on the enzyme (Fig. 1A). One of these sites is located at the dimer interface and is occupied by fumarate (Fig. 1B), whereas the other site, which is referred to as the exo site, is located at the tetramer interface and is occupied by either an NAD or an ATP molecule (Fig. 1A). In the ME family, Ascaris suum and human m-NAD(P)-ME were found to be activated by fumarate (11, 15–17, 31). However, the relationship between enzyme regulation and subunit-subunit interaction is still unclear.
FIGURE 1.

Dimer and tetramer interfaces of human m-NAD(P)-ME. A, dimer of dimers quaternary structure of human m-NAD(P)-ME (Protein Data Bank code 1PJ3). The active site, fumarate site, and exo site in each subunit are indicated. B, dimer interface between A and B subunits of m-NAD(P)-ME. C, tetramer interface between A and D subunits of m-NAD(P)-ME. The amino acid residues at the dimer interface, Gln-51, Glu-90, Asp-139, His-142, and Asp-568 and C terminus in the tetramer interface, are represented by ball-and-stick modeling. The amino acid residues 51 and 90 in human c-NADP-ME are His and Asp, respectively. This figure was generated with PyMOL (DeLano Scientific LLC, San Carlos, CA).
Previous studies have shown that the quaternary structure stability of malic enzyme isoforms is diverse. At neutral pH, pigeon c-NADP-ME exists as a unique tetramer with a sedimentation coefficient of ∼10 S (33–35), whereas human m-NAD(P)-ME exists as a mixture of tetramer and dimer of 9.5 S and 6.5 S, respectively (13, 35). Some mutations at the interface will affect the quaternary structure (34–37). Although the crystal structure of human c-NADP-ME has not been resolved, it is believed that it is similar to pigeon c-NADP-ME.
Here we analyze the essential factors governing quaternary structure stability for human c-NADP-ME and m-NAD(P)-ME. Site-directed mutagenesis at the dimer and tetramer interfaces was used to disrupt the tetramer organization to create a series of c-NADP-ME and m-NAD(P)-ME dimers. Combined with the analytical ultracentrifugation data and kinetic analysis of these interface mutants, we demonstrate the differential role of tetramer organization for the c-NADP-ME and m-NAD(P)-ME isoforms. The regulatory mechanism of m-NAD(P)-ME is highly associated with the tetramer formation of this isoform.
MATERIALS AND METHODS
Expression and Purification of Recombinant Malic Enzymes
The expression and purification protocols for human m-NAD(P)-ME have been described in previous reports (4, 6). Briefly, m-NAD(P)-ME was subcloned into the expression vector (pRH281) and transformed into Escherichia coli BL21 cells for overexpression using the inducible trp promoter system. The enzyme was purified by chromatography through an anionic exchange, DEAE-Sepharose (Amersham Biosciences) column, followed by an ATP-agarose affinity column (Sigma).
c-NADP-ME was subcloned into the pET21b vector, which carries a C-terminal His6 tag sequence. This ampicillin-resistant vector was transformed into the BL21(DE3) strain of E. coli. Expression was controlled by an inducible T7 promoter system. The overexpressed enzyme was purified using nickel-nitrilotriacetic acid-Sepharose (Sigma). The lysate/nickel-nitrilotriacetic acid mixture was washed with buffer (10 mm imidazole, 500 mm sodium chloride, 2 mm β-mercaptoethanol, and 30 mm Tris-HCl, pH 7.4) to remove unwanted proteins. Desired c-NADP-ME was recovered from the resin with elution buffer (250 mm imidazole, 500 mm sodium chloride, 2 mm β-mercaptoethanol, and 30 mm Tris-HCl, pH 7.4).
The purified enzyme was dialyzed and concentrated in 30 mm Tris-HCl, pH 7.4, and 2 mm β-mercaptoethanol using a centrifugal filter device (Amicon Ultra-15, Millipore, Billerica, MA) with a molecular mass cutoff of 30 kDa. Enzyme purity was checked by SDS-PAGE, and the protein concentrations were estimated by the Bradford method (38).
Site-directed Mutagenesis
Site-directed mutagenesis was performed using the QuikChangeTM kit (Stratagene, La Jolla, CA) to construct the mutated human c-NADP-ME and m-NAD(P)-ME plasmids. The purified DNA of human c-NADP-ME and m-NAD(P)-ME was used as a template for PCRs along with specific primers containing the desired codons and high fidelity Pfu DNA polymerase. The primers, including the desired mutation sites, were about 25–45 nucleotides in length, which is considered necessary for binding specifically to template DNA. After 16–18 temperature cycles, the mutated plasmids, including staggered nicks, were complete. The PCR products were subsequently treated with DpnI to digest the wild-type human malic enzyme templates. Finally, the nicked DNA with desired mutations was transformed into the XL-1 E. coli strain, and their DNA sequences were checked by autosequencing.
Enzyme Kinetic Analysis
The malic enzyme reaction was measured by tracing NADH or NADPH production. The reaction mixture contained saturated concentrations of NAD+ or NADP+, l-malate, and MgCl2 in 50 mm Tris-HCl, pH 7.4. The absorbance at 340 nm was continuously monitored at 30 °C with a Lambda 25 UV/Vis spectrophotometer (PerkinElmer Life Sciences). An extinction coefficient of 6.22 mm−1 for NAD(P)H was employed in the calculations. Apparent Michaelis constants of the substrate and coenzymes were determined by varying the concentration of one substrate (or coenzymes) around its Km value and keeping the other components constant under saturating concentrations.
Furthermore, the kcat values of c-NADP-ME and m-NAD(P)- ME were calculated by Equation 1,
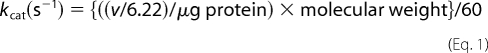 |
where v represents ΔA340/min; 6.22 is the millimolar absorption coefficient of NAD(P)H; 260,000 and 256,000 are the tetramer molecular weights of human c-NADP-ME and m-NAD(P)-ME, respectively; and 60 is the number of seconds in 1 min.
The sigmoidal curves of malate against the initial rate were fitted into the Hill Equation 2. The data were analyzed to calculate the K0.5 value, the substrate concentration at half-maximal velocity, and the Hill coefficient (h), which were utilized to evaluate the degree of cooperativity.
Data were fitted with the Sigma Plot 8.0 program (Jandel, San Rafael, CA).
Quaternary Structure Analysis of Malic Enzyme by Analytical Ultracentrifugation
Sedimentation velocity experiments of c-NADP-ME and m-NAD(P)-ME were carried out using a Beckman Optima XL-A analytical ultracentrifuge. Samples (380 μl) and buffer solutions (400 μl) were loaded into the double sector centerpiece separately and built up in a Beckman An-50 Ti rotor. Experiments were performed at 20 °C and a rotor speed of 42,000 rpm for 3.5–4 h. Protein samples were monitored by UV absorbance at 280 nm in a continuous mode with a time interval of 480 s and a step size of 0.002 cm. Multiple scans of sedimentation velocity data were collected and analyzed using the software SEDFIT 9.4c (39–43). All size distributions were solved with a confidence level of p = 0.95, a best-fitted average anhydrous frictional ratio (f/f0), and a resolution N of 200 sedimentation coefficients between 0.1 and 20.0 S.
To determine precisely the dissociation constants of malic enzyme in monomer-dimer-tetramer equilibrium, sedimentation velocity experiments were performed for three different enzyme concentrations. All sedimentation data were globally fitted to the dimer-tetramer equilibrium model for c-NADP-ME and monomer-dimer-tetramer equilibrium model for m-NAD(P)-ME using the program SEDPHAT to calculate the dissociation constant (Kd) of the enzyme (41). The partial specific volume of the enzyme, solvent density, and viscosity were calculated by the software program SEDNTERP (44).
RESULTS
Kinetic Properties of Human c-NADP-ME WT and Interface Mutant
The kinetic parameters of the human WT and interface mutant c-NADP-ME are shown in Table 1. The Km,NADP and Km,malate values of WT c-NADP-ME are 0.009 and 0.5 mm, respectively. There was no significant difference in Km,NADP and Km,malate among WT and interface mutants, indicating that the active site region is not affected by the mutation at the interface. The kcat value of WT c-NADP-ME was ∼110 s−1. The values for the c-NADP-ME interface mutants were in the range of 95–136 s−1. The similar kcat values for WT and mutant enzymes suggest that the catalytic activity of the enzyme was not changed by disrupting the subunit-subunit interactions.
TABLE 1.
Kinetic parameters of the human WT and interface mutant c-NADP-ME
| c-NADP-ME | Km,NADP | Km,malate | kcat | Quaternary structurea | Kd,24 |
|---|---|---|---|---|---|
| mm | mm | s−1 | μm | ||
| WT | 0.0090 ± 0.0010 | 0.49 ± 0.09 | 110.89 ± 0.9 | T | 0.062 ± 0.001 |
| Dimer interface | |||||
| H51A | 0.0072 ± 0.0013 | 1.10 ± 0.04 | 109.50 ± 1.2 | D/T | 9.95 ± 0.09 |
| D90A | 0.0039 ± 0.0003 | 0.68 ± 0.02 | 94.57 ± 0.5 | D/T | 1.01 ± 0.01 |
| D139A | 0.0048 ± 0.0007 | 0.95 ± 0.03 | 95.53 ± 0.6 | D | 217.8 ± 1.7 |
| H51A/D90A | 0.0037 ± 0.0005 | 0.84 ± 0.01 | 84.83 ± 0.8 | D | 156.3 ± 1.3 |
| H51A/D139A | 0.0040 ± 0.0005 | 1.03 ± 0.04 | 105.29 ± 0.9 | D | 94.2 ± 0.8 |
| Tetramer interface | |||||
| H142A | 0.0040 ± 0.0003 | 0.89 ± 0.04 | 122.72 ± 1.3 | D | 133.1 ± 1.0 |
| D568A | 0.0050 ± 0.0008 | 0.97 ± 0.10 | 136.90 ± 1.4 | D/T | 0.26 ± 0.01 |
| H142A/D568A | 0.0042 ± 0.0005 | 0.92 ± 0.03 | 98.03 ± 1.1 | D | 123.1 ± 1.0 |
| W572A | 0.0050 ± 0.0008 | 0.98 ± 0.04 | 106.04 ± 0.9 | D | 262.4 ± 1.6 |
a The abbreviations used are as follows: T, tetramer; D, dimer; Kd,24, dissociation constant between dimer and tetramer.
Quaternary Structure of Human c-NADP-ME WT and Interface Mutant
The quaternary structures of human c-NADP-ME WT and the interface mutant were examined by analytical ultracentrifugation (Figs. 2 and 3), and the dissociation constants between dimer and tetramer (Kd,24) for the respective enzymes were determined (Table 1). Sedimentation velocity experiments were performed to analyze the differences in the size distribution among WT and mutant enzymes. The WT enzyme primarily exists as a tetramer in solution with the major peak exhibiting a Kd,24 value of 0.062 μm (Fig. 2A). At the dimer interface, Asp-139 of subunit A is hydrogen-bonded with His-51 of subunit B. Asp-90 may be involved in the electrostatic network at the dimer interface. The single mutants, D90A and H51A, existed in a dimer-tetramer equilibrium with Kd,24 values of 1.01 and 9.95 μm, respectively (Fig. 2, B and C). However, the D139A single mutant dissociated more readily according to a Kd,24 value of 217.8 μm (Fig. 2D). The double mutants, H51A/D90A and H51A/D139A, were predominantly dimeric with Kd,24 values of 156.3 and 94.2 μm, respectively (Fig. 2, E and F). These results suggest that the hydrogen bonding formed by Asp-139(A) and His-51(B) and the ion pair network involving Asp-90 at the dimer interface are crucial factors for the stability of the tetrameric enzyme. The dimer, for these dimer interface mutants, should be composed of AD or BC subunit complexes.
FIGURE 2.

Continuous sedimentation coefficient distribution of the WT and dimer interface mutant c-NADP-ME. The enzyme concentrations used in the experiment were 0.1, 0.3, and 0.9 mg/ml in 50 mm Tris-HCl buffer, pH 7.4, at 20 °C. D, dimer; T, tetramer.
FIGURE 3.

Continuous sedimentation coefficient distribution of the WT and tetramer interface mutant c-NADP-ME. The enzyme concentrations were 0.1, 0.3, and 0.9 mg/ml in 50 mm Tris-HCl buffer, pH 7.4, at 20 °C. D, dimer; T, tetramer.
In the tetramer interface, His-142 of subunit A is ion-paired with Asp-568 of subunit D. Trp-572 is in the C-terminal domain involved in hydrophobic interactions with another subunit at the tetramer interface. Mutation of His-142 and Asp-568 to alanine resulted in mixed quaternary structural changes. The D568A enzyme mainly behaved as a tetramer, whereas H142A was primarily in dimeric form (Fig. 3, B and C); their Kd,24 values were 0.26 and 133.1 μm, respectively. The H142A/D568A also behaved as a dimer with a Kd,24 value of 123.1 μm (Fig. 3D), representing the importance of the salt bridge between His-142(A) and Asp-568(D) in preserving the quaternary structure. The C-terminal mutant, W572A, mostly behaved as a dimer with a Kd,24 value of 262.4 μm (Fig. 3E), suggesting that a hydrophobic interaction at the tetramer interface is also essential for the stability of the holoenzyme. The dimer for these tetramer interface mutants should be composed of AB or CD subunit complexes.
Kinetic Properties of Human m-NAD(P)-ME WT and Interface Mutant
Distinct from c-NADP-ME, m-NAD(P)-ME binds l-malate cooperatively; the initial velocities of m-NAD(P)-ME measured in various concentrations of malate exhibited sigmoidal kinetics. Table 2 summarizes the results obtained by fitting these sigmoidal curves to the Hill equation. The half-saturation for l-malate (K0.5,malate) and the degree of cooperativity of malate (h) were estimated. Like c-NADP-ME, the Km,NAD and K0.5,malate values of the m-NAD(P)-ME interface mutant were similar to that of WT, indicating that the active site was not perturbed by the mutations. However, the kcat values of these interface mutants were less than that of WT. The kcat values of WT m-NAD(P)-ME were ∼306 and 239 s−1, with or without fumarate, respectively. The values for the m-NAD(P)-ME interface mutants ranged from 110 to 234 s−1, implying that enzyme activity was altered by disrupting the interactions at the subunit interface.
TABLE 2.
Kinetic parameters of the human WT and interface mutant m-NAD(P)-ME
| m-NAD(P)-MEa | Km,NAD | k0.5,malate | kcat | h | Kd,24b | Kd,12b |
|---|---|---|---|---|---|---|
| mm | mm | s−1 | μm | μm | ||
| WT | ||||||
| − | 0.68 ± 0.16 | 11.81 ± 0.48 | 238.5 ± 2.3 | 2.0 ± 0.15 | 1.38 ± 0.02 | 0.25 ± 0.003 |
| + | 0.32 ± 0.01 | 2.76 ± 0.12 | 306.4 ± 2.5 | 1.1 ± 0.06 | ||
| Dimer interface | ||||||
| Q51A | ||||||
| − | 0.99 ± 0.06 | 16.35 ± 0.46 | 139.1 ± 1.5 | 1.4 ± 0.05 | 3.28 ± 0.04 | 0.36 ± 0.004 |
| + | 0.51 ± 0.03 | 5.97 ± 0.25 | 189.9 ± 1.9 | 1.0 ± 0.04 | ||
| E90A | ||||||
| − | 0.32 ± 0.02 | 2.83 ± 0.17 | 203.0 ± 2.3 | 1.0 ± 0.07 | 1.08 ± 0.01 | 0.22 ± 0.002 |
| + | 0.34 ± 0.04 | 2.95 ± 0.08 | 210.7 ± 2.6 | 1.0 ± 0.03 | ||
| Q51A/E90A | ||||||
| − | 0.45 ± 0.02 | 7.23 ± 0.35 | 140.1 ± 1.5 | 1.1 ± 0.05 | 7.73 ± 0.06 | 0.09 ± 0.001 |
| + | 0.43 ± 0.02 | 6.36 ± 0.30 | 143.3 ± 1.2 | 1.0 ± 0.04 | ||
| D139A | ||||||
| − | 1.06 ± 0.05 | 23.31 ± 1.81 | 114.5 ± 1.0 | 1.1 ± 0.07 | 15.92 ± 0.20 | 1.87 ± 0.024 |
| −+ | 0.76 ± 0.07 | 9.70 ± 1.43 | 115.4 ± 1.6 | 1.0 ± 0.12 | ||
| Tetramer interface | ||||||
| H142A | ||||||
| − | 0.82 ± 0.03 | 18.84 ± 0.76 | 117.9 ± 1.3 | 1.3 ± 0.05 | 8.75 ± 0.10 | 3.63 ± 0.04 |
| + | 0.47 ± 0.02 | 8.06 ± 0.76 | 139.9 ± 1.9 | 1.0 ± 0.08 | ||
| D568A | ||||||
| − | 1.82 ± 0.12 | 28.54 ± 2.13 | 124.0 ± 1.2 | 1.3 ± 0.08 | 12.59 ± 0.16 | 0.57 ± 0.007 |
| −+ | 0.85 ± 0.08 | 13.08 ± 2.39 | 139.6 ± 1.6 | 1.0 ± 0.12 | ||
| H142A/D568A | ||||||
| − | 1.39 ± 0.73 | 29.00 ± 1.72 | 112.8 ± 1.5 | 1.3 ± 0.06 | 11.61 ± 0.18 | 13.06 ± 0.20 |
| + | 0.72 ± 0.05 | 10.55 ± 1.09 | 127.4 ± 2.0 | 1.0 ± 0.06 | ||
| P567A | ||||||
| − | 0.87 ± 0.05 | 21.10 ± 1.03 | 141.1 ± 1.2 | 1.4 ± 0.07 | NDc | |
| + | 0.51 ± 0.02 | 6.80 ± 0.41 | 164.4 ± 1.3 | 1.0 ± 0.05 | ND | |
| Y570A | ||||||
| − | 1.07 ± 0.04 | 21.75 ± 0.51 | 141.9 ± 1.1 | 1.4 ± 0.04 | 12.6 ± 0.10 | 0.89 ± 0.007 |
| + | 0.58 ± 0.03 | 8.33 ± 0.54 | 165.7 ± 1.4 | 1.0 ± 0.06 | ||
| W572A | ||||||
| − | 1.01 ± 0.01 | 20.49 ± 0.84 | 150.4 ± 1.8 | 1.3 ± 0.04 | ND | |
| + | 0.54 ± 0.02 | 6.54 ± 0.31 | 181.9 ± 1.3 | 1.0 ± 0.05 | ND | |
| P573A | ||||||
| − | 1.20 ± 0.10 | 21.08 ± 0.65 | 150.2 ± 1.5 | 1.5 ± 0.06 | ND | |
| + | 0.59 ± 0.03 | 6.97 ± 0.71 | 167.1 ± 1.4 | 1.0 ± 0.09 | ND | |
| P579A | ||||||
| − | 0.79 ± 0.18 | 13.88 ± 0.37 | 234.3 ± 2.1 | 1.7 ± 0.08 | ND | |
| + | 0.39 ± 0.02 | 3.86 ± 0.32 | 284.1 ± 2.5 | 1.0 ± 0.08 | ND | |
| P580A | ||||||
| − | 0.95 ± 0.17 | 13.32 ± 0.46 | 196.5 ± 2.0 | 1.6 ± 0.08 | 2.92 ± 0.03 | 0.85 ± 0.01 |
| + | 0.40 ± 0.02 | 3.18 ± 0.17 | 211.6 ± 2.3 | 1.2 ± 0.07 | ||
| V581A | ||||||
| − | 0.83 ± 0.09 | 13.37 ± 0.60 | 205.8 ± 2.8 | 1.6 ± 0.10 | ND | |
| + | 0.40 ± 0.04 | 2.98 ± 0.14 | 234.4 ± 2.1 | 1.0 ± 0.05 | ND | |
a − indicates no fumarate; + indicates 4 mm fumarate added.
bKd,24 indicates dissociation constant between dimer and tetramer; Kd,12 indicates dissociation constant between monomer and dimer.
c ND indicates not determined.
The h value for WT m-NAD(P)-ME is approximately 2; however, the h value was significantly reduced for the interface mutants. Most mutations at the dimer interface had an h value close to 1, indicating that cooperativity was diminished by interruption of the dimer interface. The mutations at the tetramer interface had h values of 1.3 and 1.7, suggesting that the cooperativity was partially maintained for these mutant enzymes even when the tetramer interface was disrupted. With fumarate, the Km,NAD and K0.5,malate of most of the interface mutant enzymes is reduced; the kcat values of some mutant enzymes can be raised, and the h value can be reduced to 1, indicating that allosteric regulation by fumarate is functioning for some interface mutants. The dimer interface mutants E90A, Q51A/E90A, and D139A are not activated by fumarate, and their h values were near 1, indicating that they have become noncooperative and nonallosteric enzymes.
Size Distribution Analysis of Human m-NAD(P)-ME WT and Interface Mutant
The size distribution of human m-NAD(P)- ME WT and interface mutant is shown in Figs. 4–6. Becausethe WT and interface mutant may exhibit monomer-dimer or dimer-tetramer behavior (Figs. 4–6), the dissociation constants between dimer and tetramer forms (Kd,24) or monomer and dimer forms (Kd,12) were determined (Table 2). The WT enzyme chiefly existed in a dimer-tetramer equilibrium with little monomer present; the Kd,24 and Kd,12 values were 1.38 and 0.25 μm, respectively (Fig. 4A). Similar to c-NADP-ME, the dimer interface of m-NAD(P)-ME positions Asp-139 of subunit A in a hydrogen bond with Gln-51 of subunit B, and Glu-90 may be involved in electrostatic interactions. Mutating these amino acid residues caused the enzyme to be dissociated. The E90A enzyme, similar to WT, also existed in a dimer-tetramer equilibrium with little monomer observable; the Kd,24 and Kd,12 values were 1.08 and 0.22 μm, respectively (Fig. 4C). The D139A single mutant was significantly dissociated into monomers and dimers with Kd,24 and Kd,12 values of 15.9 and 1.87 μm, respectively (Fig. 4D). The Q51A and Q51A/E90A mutants displayed monomer-dimer-tetramer equilibriums (Fig. 4, B and E, respectively) with larger Kd,24 values (3.28 and 7.73 μm, respectively) than that of WT (1.38 μm), indicating that mutation of Gln-51 resulted in greater dissociation of the dimer interface.
FIGURE 4.

Continuous sedimentation coefficient distribution of the WT and dimer interface mutant m-NAD(P)-ME. The enzyme concentrations were 0.1, 0.3, and 0.9 mg/ml in 50 mm Tris-HCl buffer, pH 7.4, at 20 °C. M, monomer; D, dimer; T, tetramer.
FIGURE 5.

Continuous sedimentation coefficient distribution of the WT and tetramer interface mutant m-NAD(P)-ME. The enzyme concentrations were 0.1, 0.3, and 0.9 mg/ml in 50 mm Tris-HCl buffer, pH 7.4, at 20 °C. M, monomer; D, dimer; T, tetramer.
FIGURE 6.

Continuous sedimentation coefficient distribution of the WT and C-terminal mutant m-NAD(P)-ME. The enzyme concentrations for WT, Y570A, and P580A were 0.1, 0.3, and 0.9 mg/ml. The enzyme concentration for the other C-terminal mutants was 0.3 mg/ml. M, monomer; D, dimer; T, tetramer.
At the tetramer interface of m-NAD(P)-ME, the positions of His-142(A) and Asp-568(D) are also conserved and form a salt bridge. Mutation of His-142 and Asp-568 to alanine caused the enzyme to be predominantly dissociated into dimers and monomers with Kd,24 and Kd,12 values of 8.75 and 3.63 μm for H142A and 12.6 and 0.57 μm for D568A, respectively (Fig. 5, B and C). The H142A/D568A double mutant also presented itself as a dimer (Fig. 5D); its Kd,24 and Kd,12 values were 11.6 and 13.1 μm, respectively, again suggesting the significance of the salt bridge between His-142(A) and Asp-568(D) at the tetramer interface.
The hydrophobic amino acid residues in the C-terminal domain are also important for stability of the quaternary structure. The W57A, Y770A, W572A, and P175A mutants were dimers at 0.3 mg/ml protein concentration (Fig. 6, B–E, respectively). For K507A, the Kd,24 and Kd,12 values were 12.6 and 0.89 μm, respectively (Fig. 6C). However, the quaternary structure distribution of P175A, K508A, and V18A (Fig. 6, F–H, respectively) demonstrated a pattern similar to WT. For K508A, the Kd,24 and Kd,12 values were 2.92 and 0.85 μm, respectively (Fig. 6G). These results suggest that the amino acid residues from 579 to 581 are not absolutely required for stability of the tetramer interface as these mutants had kinetic properties similar to those of WT (Table 2).
DISCUSSION
Malic enzyme has a dimer of dimers quaternary structure in which the dimer interface is made up of relatively stronger interactions than the tetramer interface. Furthermore, each subunit of the tetramer has a corresponding active site. The m-NAD(P)-ME isoform exhibits both cooperative and allosteric activity and is in dimer-tetramer flux according to its equilibrium constant (Fig. 4A). On the other hand, c-NADP-ME behaves noncooperatively and nonallosterically and exists as a stable tetramer (Fig. 2A). The correlation between the catalytic efficiency and quaternary structure organization of the enzyme is unclear. This study establishes the functional significance of the dimer and tetramer interface for the enzyme.
Relationship between the Catalytic Efficiency and Quaternary Structure Organization of c-NADP-ME
Our data clearly indicate that the enzyme activity of c-NADP-ME is not affected by the disruption at the interface. There are no significant differences in the kinetic properties between AB and AD dimers. The dimeric enzyme, either AB or AD, is as active as the tetramer, suggesting that the four subunits of the enzyme may not cross-talk with one another, and they may work independently. We also attempted to isolate the monomeric form of the enzyme by mutating several residues at the dimer and tetramer interfaces; however, we have not yet been successful because the monomer aggregates as inclusion bodies. Given these results, we conclude that the functional significance of the quaternary structure organization for c-NADP-ME may be in maintaining the stability of the holoenzyme rather than in communicating among the subunits.
Enzyme Regulation and Subunit-Subunit Interactions of m-NAD(P)-ME
In contrast to c-NADP-ME, disruption of the subunit interfaces of m-NAD(P)-ME causes it to be less active than WT; the kcat values of the mutants decreased with increasing Kd,24 values (Table 2), suggesting that the dissociation of subunits at the dimer or tetramer interface significantly affects the activity of the enzyme. Moreover, these interface mutants become less cooperative for malate binding and less activated by fumarate, suggesting that the tetramer is the fully functional form of the enzyme. There seems to be some differences between AB and AD dimers of this isoform. For some dimer interface mutants, malate cooperativity is nearly abolished, and fumarate activation becomes minimal, suggesting that organization at the dimer interface is critical for proper malate cooperativity and fumarate-induced allosteric regulation. Although fumarate occupies a separate binding site on each subunit, its binding is dependent on the neighboring subunit, which helps to shield the binding site from the solvent and thus stabilize the binding of fumarate at the dimer interface (11). We suggest that the subunit-subunit interactions between A and B (C and D) is important not only for the malate cooperativity but also for the binding of fumarate (supplemental Fig. 1). The AD dimer may exhibit noncooperative and nonallosteric characteristics.
In contrast, for tetramer interface mutants, cooperativity is partially conserved, and fumarate still functions by reducing the Km and increasing the kcat value. Fumarate may still bind to the allosteric site of the tetramer interface mutants to activate the enzyme and reduce the malate cooperativity (Table 2 and supplemental Fig. 2); however, these tetramer interface mutants cannot be reconstituted into tetramers by fumarate (supplemental Fig. 1); thus the enzyme cannot be fully activated by fumarate (Table 2). These results support the hypothesis that tetramer interface organization is associated with malate cooperativity and involved in the fumarate-triggered tetramer reorganization for full activation of the enzyme. The AB dimer, however, may still be a cooperative and allosteric enzyme but less active than the tetramer.
Ionic and Hydrophobic Interactions Stabilize the Tetramer Interface
Size distribution analysis demonstrates that H142A and D568A form dimers in context of c-NADP-ME or m-NAD(P)-ME, suggesting that the ion pair formed by His-142(A) and Asp-568(D) is essential for the stability of the tetramer interface. Hydrophobic interactions in the C-terminal domain are also an important factor for the tetramer interface organization. Mutation of the hydrophobic amino acid residues (Tyr-570, Trp-572, and Pro-573) in the C-terminal domain makes the tetramer interface unstable causing the enzyme to dissociate into dimers.
The stabilities of the tetramer interfaces in c-NADP-ME and m-NAD(P)-ME have unique qualities. The c-NADP-ME isoform exists as a stable tetramer (Fig. 2A), whereas m-NAD(P)-ME exhibits a dimer-tetramer equilibrium (Fig. 4A). The ionic bond formed between His-142(A) and Asp-568(D) and the hydrophobic amino acid residues Tyr-570, Trp-572, and Pro-573 are conserved at the interfaces of both isoforms. Our data suggest that the hydrophobic amino acid residues Pro-579, Pro-580, and Val-581, which lie in the C-terminal tail of m-NAD(P)-ME, are not as important as Tyr-570, Trp-572, and Pro-573 for tetramer interface stability. The C-terminal tail of these two isoforms is not conserved, especially from residues 575 to 584. In this nonconserved region, c-NADP-ME contains additional ionic residues (Glu-575, Lys-578, and Lys-582), where m-NAD(P)-ME contains Ser or Ile. Investigating the role of these ionic residues at the tetramer interface may be helpful in finding additional factors governing the differential stability observed for the tetramer interfaces of these two isoforms.
The D139A mutant is rather unique. Of all the mutants, it produces the highest percentage of monomer relative to other forms. In the structure, Asp-139 participates in the subunit-subunit interactions both at the dimer and tetramer interfaces. Asp-139(A) is hydrogen-bonded with His-51(B) in c-NADP-ME or with Gln-51(B) in m-NAD(P)-ME at the dimer interface. In addition, Asp-139(A) is hydrogen-bonded with Tyr-570(D) and Trp-572(D) at the tetramer interface of c-NADP-ME and m-NAD(P)-ME (supplemental Fig. 3). Thus, the mutant shows the highest percentage of monomer relative to others.
In summary, we demonstrate that the dimeric form of c-NADP-ME has similar enzyme activity to the tetrameric form. The quaternary structure of c-NADP-ME is mainly for protein stability. For m-NAD(P)-ME, dissociation of subunits at the dimer or tetramer interface causes the enzyme to be less active, less cooperative for malate binding, and less responsive to fumarate. Thus the tetramer organization for this isoform is principally for enzyme regulation.
Supplementary Material
This work was supported by National Science Council Grant NSC-96-2311-B-005-005 (to H.-C. H.) and in part by the Ministry of Education, Taiwan, under the Aiming for Top University plan.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3.
- c-NADP-ME
- cytosolic NADP+-dependent malic enzyme
- ME
- malic enzyme
- m-NADP-ME
- mitochondrial NADP+-dependent malic enzyme
- m-NAD(P)-ME
- mitochondrial NAD(P)+-dependent malic enzyme
- WT
- wild type.
REFERENCES
- 1.Hsu R. Y. ( 1982) Mol. Cell. Biochem. 43, 3– 26 [DOI] [PubMed] [Google Scholar]
- 2.Frenkel R. ( 1975) Curr. Top. Cell Regul. 9, 157– 181 [DOI] [PubMed] [Google Scholar]
- 3.Edens W. A., Urbauer J. L., Cleland W. W. ( 1997) Biochemistry 36, 1141– 1147 [DOI] [PubMed] [Google Scholar]
- 4.Xu Y., Bhargava G., Wu H., Loeber G., Tong L. ( 1999) Structure 7, 877– 889 [PubMed] [Google Scholar]
- 5.Chang G. G., Tong L. ( 2003) Biochemistry 42, 12721– 12733 [DOI] [PubMed] [Google Scholar]
- 6.Loeber G., Infante A. A., Maurer-Fogy I., Krystek E., Dworkin M. B. ( 1991) J. Biol. Chem. 266, 3016– 3021 [PubMed] [Google Scholar]
- 7.Mandella R. D., Sauer L. A. ( 1975) J. Biol. Chem. 250, 5877– 5884 [PubMed] [Google Scholar]
- 8.Hsieh J. Y., Liu G. Y., Chang G. G., Hung H. C. ( 2006) J. Biol. Chem. 281, 23237– 23245 [DOI] [PubMed] [Google Scholar]
- 9.Moreadith R. W., Lehninger A. L. ( 1984) J. Biol. Chem. 259, 6222– 6227 [PubMed] [Google Scholar]
- 10.Sauer L. A. ( 1973) Biochem. Biophys. Res. Commun. 50, 524– 531 [DOI] [PubMed] [Google Scholar]
- 11.Yang Z., Lanks C. W., Tong L. ( 2002) Structure 10, 951– 960 [DOI] [PubMed] [Google Scholar]
- 12.Teller J. K., Fahien L. A., Davis J. W. ( 1992) J. Biol. Chem. 267, 10423– 10432 [PubMed] [Google Scholar]
- 13.Hsu W. C., Hung H. C., Tong L., Chang G. G. ( 2004) Biochemistry 43, 7382– 7390 [DOI] [PubMed] [Google Scholar]
- 14.Hung H. C., Chien Y. C., Hsieh J. Y., Chang G. G., Liu G. Y. ( 2005) Biochemistry 44, 12737– 12745 [DOI] [PubMed] [Google Scholar]
- 15.Hsieh J. Y., Liu G. Y., Hung H. C. ( 2008) FEBS J. 275, 5383– 5392 [DOI] [PubMed] [Google Scholar]
- 16.Karsten W. E., Pais J. E., Rao G. S., Harris B. G., Cook P. F. ( 2003) Biochemistry 42, 9712– 9721 [DOI] [PubMed] [Google Scholar]
- 17.Hung H. C., Kuo M. W., Chang G. G., Liu G. Y. ( 2005) Biochem. J. 392, 39– 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loeber G., Dworkin M. B., Infante A., Ahorn H. ( 1994) FEBS Lett. 344, 181– 186 [DOI] [PubMed] [Google Scholar]
- 19.Chang G. G., Wang J. K., Huang T. M., Lee H. J., Chou W. Y., Meng C. L. ( 1991) Eur. J. Biochem. 202, 681– 688 [DOI] [PubMed] [Google Scholar]
- 20.Loeber G., Maurer-Fogy I., Schwendenwein R. ( 1994) Biochem. J. 304, 687– 692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanz N., Díez-Fernández C., Valverde A. M., Lorenzo M., Benito M., Cascales M. ( 1997) Br. J. Cancer 75, 487– 492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fahien L. A., Teller J. K. ( 1992) J. Biol. Chem. 267, 10411– 10422 [PubMed] [Google Scholar]
- 23.Moreadith R. W., Lehninger A. L. ( 1984) J. Biol. Chem. 259, 6215– 6221 [PubMed] [Google Scholar]
- 24.Sauer L. A., Dauchy R. T., Nagel W. O., Morris H. P. ( 1980) J. Biol. Chem. 255, 3844– 3848 [PubMed] [Google Scholar]
- 25.Zołnierowicz S., Swierczyński J., Zelewski L. ( 1988) Biochem. Med. Metab. Biol. 39, 208– 216 [DOI] [PubMed] [Google Scholar]
- 26.Baggetto L. G. ( 1992) Biochimie 74, 959– 974 [DOI] [PubMed] [Google Scholar]
- 27.McKeehan W. L. ( 1982) Cell Biol. Int. Rep. 6, 635– 650 [DOI] [PubMed] [Google Scholar]
- 28.Yang Z., Floyd D. L., Loeber G., Tong L. ( 2000) Nat. Struct. Biol. 7, 251– 257 [DOI] [PubMed] [Google Scholar]
- 29.Yang Z., Zhang H., Hung H. C., Kuo C. C., Tsai L. C., Yuan H. S., Chou W. Y., Chang G. G., Tong L. ( 2002) Protein Sci. 11, 332– 341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coleman D. E., Rao G. S., Goldsmith E. J., Cook P. F., Harris B. G. ( 2002) Biochemistry 41, 6928– 6938 [DOI] [PubMed] [Google Scholar]
- 31.Rao G. S., Coleman D. E., Karsten W. E., Cook P. F., Harris B. G. ( 2003) J. Biol. Chem. 278, 38051– 38058 [DOI] [PubMed] [Google Scholar]
- 32.Tao X., Yang Z., Tong L. ( 2003) Structure 11, 1141– 1150 [DOI] [PubMed] [Google Scholar]
- 33.Chang H. C., Chou W. Y., Chang G. G. ( 2002) J. Biol. Chem. 277, 4663– 4671 [DOI] [PubMed] [Google Scholar]
- 34.Chang H. C., Chang G. G. ( 2003) J. Biol. Chem. 278, 23996– 24002 [DOI] [PubMed] [Google Scholar]
- 35.Chang H. C., Chen L. Y., Lu Y. H., Li M. Y., Chen Y. H., Lin C. H., Chang G. G. ( 2007) Biophys. J. 93, 3977– 3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chou W. Y., Huang S. M., Chang G. G. ( 1997) Protein Eng. 10, 1205– 1211 [DOI] [PubMed] [Google Scholar]
- 37.Chou W. Y., Liu M. Y., Huang S. M., Chang G. G. ( 1996) Biochemistry 35, 9873– 9879 [DOI] [PubMed] [Google Scholar]
- 38.Bradford M. M. ( 1976) Anal. Biochem. 72, 248– 254 [DOI] [PubMed] [Google Scholar]
- 39.Schuck P. ( 2000) Biophys. J. 78, 1606– 1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lebowitz J., Lewis M. S., Schuck P. ( 2002) Protein Sci. 11, 2067– 2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuck P. ( 2003) Anal. Biochem. 320, 104– 124 [DOI] [PubMed] [Google Scholar]
- 42.Schuck P., Perugini M. A., Gonzales N. R., Howlett G. J., Schubert D. ( 2002) Biophys. J. 82, 1096– 1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown P. H., Schuck P. ( 2006) Biophys. J. 90, 4651– 4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laue T. M., Shah B. D., Ridgeway T. M., Pelleter S. L. ( 1992) Analytical Ultracentrifugation in Biochemistry and Polymer Science, The Royal Society of Chemistry, Cambridge, UK [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.