

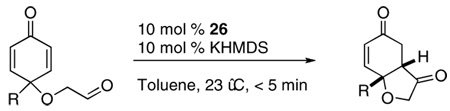
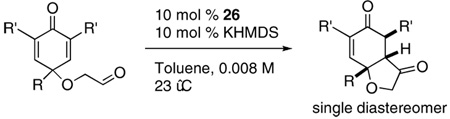
Abstract

A series of cyclohexadienones were synthesized by dearomatization of phenols followed by Dess-Martin oxidation. Asymmetric intramolecular Stetter reactions of these substrates provide hydrobenzofuranones in good to excellent yields and excellent stereoselectivities. Up to three stereocenters as well as quaternary stereocenter are formed from polysubstituted substrates. A scale up experiment demonstrates the utility of this transformation.
Keywords: dearomatization, Stetter, enantioselectivity, diastereoselectivity, triazolium, carbene
Introduction
Introduced by D. Seebach and E. J. Corey, reactivity umpolung1 is a process in which the normal donor and acceptor reactivity of a functional group is inverted to provide unobvious, complementary reactivity in organic synthesis. The Stetter reaction2]is an umpolung process in which an acylanion equivalent, generated from an aldehyde in the presence of a nucleophilic catalyst, is added to a Michael acceptor to form a C-C bond. If the Michael acceptor involves a prochiral alkene, this reaction generates new stereocenters.3 Recently our group developed a family of triazolium catalysts that promote intramolecular Stetter reactions in excellent enantioselectivities and diastereoselectivities.4
As powerful as it may be, any strategy is inherently limited if the requisite substrates are esoteric or difficult to access. In an effort to expand the scope of the asymmetric intramolecular Stetter reaction in order to access more diverse product scaffolds amenable to complex molecule total synthesis, we initiated an effort at using aromatic feedstock starting materials to provide Stetter substrates. Dearomatization of aromatic compounds is a very useful strategy to synthesize alicyclic compounds due to its high economy and simplicity.5 When coupled with a stereoselective process, it has the potential to afford enantioenriched material from commonly available precursors.6 As part of efforts to extend this powerful transformation, we investigated cyclohexadienones, readily available from dearomatization of phenols,7 as substrates for an asymmetric desymmetrizing Stetter reaction (Scheme 1). In a preliminary communication, we reported the asymmetric intramolecular Stetter reaction of phenol derived cyclohexadienones with chiral triazolium salt-based catalysts.8 This reaction allows for a rapid entry to hydrobenzofurans, which are core skeletons found in several natural products.9 Herein we report our full investigation of this transformation, including the optimization of reaction conditions, expansion of the substrate scope and scale up of the reaction.
Scheme 1.

Intramolecular Stetter reaction of phenol derived cyclohexadienones
Results and Discussion
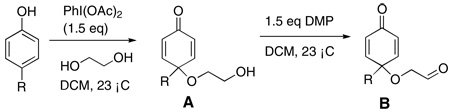
Our initial investigations were focused on substrate 1, which was synthesized in two steps from cresol. (Table 1, entry 1). Firstly, oxidation of cresol using iodobenzenediacetate in the presence of excess ethylene glycol as a trapping agent produces cyclohexadienone 1 in 49% yield. A large excess (30 eq) of glycol is required to suppress formation of undesired byproducts in this reaction, which is rarely a problem given its cost and water solubility, the latter greatly facilitating workup. We have spent some effort at using lower equivalents of alcohol with some success but the above protocol is the most general. Alcohol thus generated is then converted to aldehyde 1 using Dess-Martin reagent (DMP) in 87% yield. Using this general procedure, a series of cyclohexadienones may be synthesized successfully (Table 1).
Table 1.
Syntheses of monosubstituted substrates
 | |||
|---|---|---|---|
| Entry | Phenol | B | Yield (%) A/B |
| 1 | 1 R = Me | 49/87 | |
| 2 | 2 R = Et | 42/78 | |
| 3 | 3 R = iPr | 38/92 | |
| 4 | 4 R = tBu | 31/78 | |
| 5 | 5 R = Ph | 56/64 | |
| 6 | 6 R = 4-BrC6H4 | 26/75 | |
| 7 | 7 R = CH2OAc | 17/52 | |
| 8 | 8 R = CH2CH2OMe | 39/59 | |
| 9 | 9 R = CH2CH2CO2Me | 20/47 | |
| 10 | 10 R = CH2CH2NHBoc | 22/26 | |
| 11 | 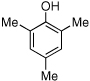 |
11 | 93/56 |
| 12 | 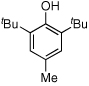 |
12 | 25/54 |
| 13 | 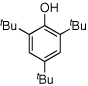 |
13 | 31/89 |
| 14 | 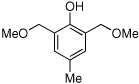 |
14 | 70/74 |
| 15 | 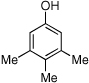 |
15 | 56/62 |
| 16 | 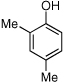 |
16 | 93/56 |
| 17 | 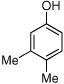 |
17 | 25/54 |
| 18 |  |
18 | 70/74 |
| 19 | 19 | 31/89 | |
As can be seen from Table 1, this approach is remarkably tolerant of arene substitution. Although yields in the arene oxidation vary, most reactions were conducted only once, and are thus unoptimized. The Dess-Martin oxidation was found to be the most appropriate for the second step, with fresh batches of reagent providing highest and most reproducible yields.
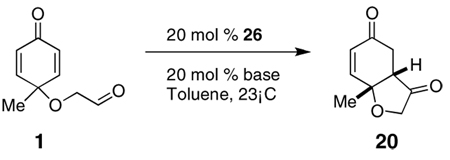
Subjection of 1 to the standard Stetter reaction conditions using previously reported triazolium salts4 as catalyst precursors (Scheme 3) provides the desired benzohydrofuranone 20 in good yields. We found the best catalyst to be 26, an aminoindanol-derived electron-rich triazolium salt, providing the product in 90% yield and 88% ee in 5 minutes. The diastereoselectivity of this transformation is excellent (>95:5 by 1H NMR and GC), favoring formation of the cis-fused hydrobenzofuranone. Surprisingly, electron-tuning of the arene ring was required for optimal selectivities as the sterically identical phenyl and pentafluorophenyl catalysts provide lower selectivities. Although at this time we cannot account for these differences, the tunability of these azolium salts is a hallmark of their design.10
 |
(1) |
A screen of different bases revealed that KHMDS is the best base for this reaction (Table 2). KOt-Bu is equally effective at inducing asymmetry but requires a longer reaction time. Amine bases also require longer reactions times, consistent with their reduced basicity thereby leading to lower concentrations of active carbene catalyst. Somewhat surprising is the reduced selectivity evident when amine bases are used, a situation not generally encountered in other Stetter reactions developed in our laboratory.
Table 2.
Screen of bases
 | ||||
|---|---|---|---|---|
| Entry[a] | Base | Reaction time | Yield (%) | ee (%)[b] |
| 1 | KHMDS | 5 min | 90 | 88 |
| 2 | KOt-Bu | 30 min | 83 | 88 |
| 3 | KH | 16 h | 76 | 76 |
| 4 | iPr2NEt | 21 h | 62 | 70 |
| 5 | Et3N | 32 h | 68 | 66 |
[1] = 0.04 M.
Determined by GC using a chiral stationary phase.
When screening a variety of solvents (Table 3), we observed a large effect on both yield and ee. For example, the reaction results in 16% yield and 67% ee after 3 days when dichloromethane is used as solvent. Overall, toluene was found to be the best solvent for this reaction.
Table 3.
Screen of solvents
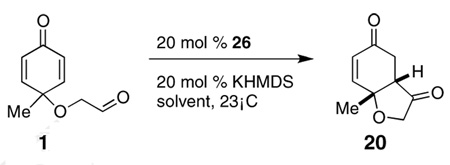
By far the most surprising aspect of this study was the effect of alcoholic solvents on the reaction, which invariably afford the opposite enantiomer using the same series of catalyst (Table 3). There is a clear effect of alcohol size on selectivity with isopropanol providing the highest ee’s. Trifluoroethanol shuts down the reaction, presumably because of its increased acidity.
The profound difference between polar aprotic solvents such as DMF and the alcoholic solvents cannot be accounted for by polarity alone. In order to study the effect of the alcohol further, a mixture of toluene and isopropanol were used as solvent for the reaction. A gradual inversion in selectivity occurs as the volume fraction of isopropanol increases to a plateau of ~60% isopropanol in toluene. We suggest that these effects are most consistent with the involvement of the alcohol in the transition state likely via hydrogen bonding to either the nucleophilic enol or the carbonyl acceptor or both. This hydrogen bonding thus changes the chiral environment, ultimately affecting the stereochemical outcome of the reaction.
A stereochemical rationale to account for the turnover in selectivity is provided in Figure 2. The absolute stereochemistry of 20 is consistent with model C wherein minimization of charge separation is emphasized. We suggest that extensive hydrogen bonding in isopropanol destabilizes this transition state relative to D since the incipient enolate and alkoxide are each hydrogen bonded to solvent. Solvation and electrostatic effects are likely also playing a role.
Figure 2.

Proposed model for turnover in selectivity in i-PrOH
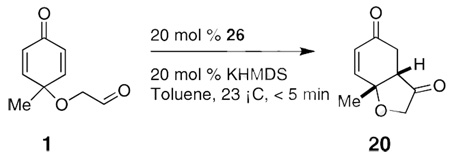
Having evaluated the effect of each component on the reaction, we were faced with a reaction that provided product in 88% ee. A preliminary screen of different substrates (not shown) revealed that the enantioselectivities were invariably <88%. We were particularly intrigued with the effect of alcoholic solvents on enantioselectivity and speculated whether hydrogen bond donors present as intermediates in the reaction could be interfering. In an effort to minimize the contribution of bimolecular events to the stereoselectivity of the process, we evaluated the impact of reaction concentration. When the concentration of 2 decreased from 0.12 to 0.013 M, the ee increased from 79 to 90%. Selectivities were further improved by a serendipitous discovery that the use of an argon purge through the reaction leads to improved ee’s, entries 5 and 7 in Table 4. We suggest that these effects are a consequence of the availability of hydrogen bond donors under conditions involving higher concentrations or adventitious oxygen-derived byproducts, and these lead to competitive transition states similar to D in Figure 2 above.
Table 4.
Effect of concentration
 | |||
|---|---|---|---|
| Entry | Concentration 1 (M) | Yield (%) | ee (%)[a] |
| 1 | 0.12 | 85 | 79 |
| 2 | 0.04 | 90 | 88 |
| 3 | 0.013 | 90 | 90 |
| 4 | 0.008 | 90 | 90 |
| 5[b] | 0.008 | 90 | 93 |
| 6 | 0.005 | 86 | 90 |
| 7[b] | 0.005 | 91 | 96 |
See Table 2.
Ar purge.
With the optimized reaction conditions in hand, we screened a series of mono-substituted dienone substrates. As the steric size of R group is increased from methyl to tert-butyl, the enantioselectivity of the transformation remained largely invariant (Table 5, entries 1–4). Aryl groups result in slightly lower selectivities (Table 5, entries 5–6). Single crystal analysis of 31 (Table 5, entry 6) revealed the absolute configuration of this product, while the rest were assigned by analogy. More functionalized substitutions are also tolerated in the reaction, although with lower ee’s (Table 5, entries 7–10). In light of the detrimental effect of groups capable of hydrogen bonding, it is tempting to invoke an intramolecular hydrogen bond that alters selectivities in the case of substrates 7–10. However, this scenario clearly cannot account for depressed selectivities observed with aryl substituents in substrates 5 and 6. Furthermore, it is difficult to envision an intramolecular hydrogen bonding affecting the transition state when cyclization occurs trans to the R group at the 4 position of the dienone. We suggest, therefore, that this effect may best be rationalized as due to electronics. Every group in substrates 5–10 is sigma withdrawing, and this may have a subtle effect in altering the diastereomeric transition states. Of note is that all the substrates provide products in excellent diastereoselectivities (>95:5 by 1H NMR and GC).
Table 5.
Reaction of monosubstituted substrate
 | |||||
|---|---|---|---|---|---|
| Entry[a] | R | Substrate | Product | Yield (%) | ee (%)[b] |
| 1 | Me | 1 | 20 | 90 | 92 |
| 2 | Et | 2 | 27 | 86 | 94 |
| 3 | iPr | 3 | 28 | 87 | 94 |
| 4 | tBu | 4 | 29 | 86 | 94 |
| 5 | Ph | 5 | 30 | 87 | 88 |
| 6 | 4-BrC6H4 | 6 | 31 | 78 | 85 |
| 7 | CH2OAc | 7 | 32 | 86 | 83 |
| 8 | CH2CH2OMe | 8 | 33 | 86 | 82 |
| 9 | CH2CH2CO2Me | 9 | 34 | 94 | 87 |
| 10 | CH2CH2NHBoc | 10 | 35 | 28 | 64 |
Argon purge, [substrate] = 0.008 M.
Determined by GC or HPLC using a chiral stationary phase.
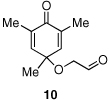
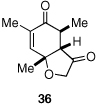
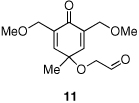
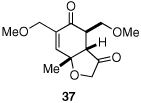
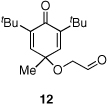
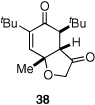
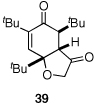
Given our previous success in cyclizing onto trisubstituted Michael acceptors,4d we were intrigued to attempt cyclizations onto dienones derived from trisubstituted phenol starting materials. A number of 2,4,6-trisubsituted phenols are readily available. When the derived dienones are subjected to the optimized reaction conditions, hydrobenzofuranones with three contiguous stereocenters are formed in good yield and excellent selectivities (Table 6). No elimination of methoxy group is observed under the basic reaction conditions (Table 6, entry 2). The tolerance of this reaction to steric bulk is particularly noteworthy. Dienone 13, derived from the ubiquitous and inexpensive antioxidant BHT, provides product 38 in good yield as largely a single enantiomer and diastereomer (Table 6, entry 3) possessing a neopentyl stereocenter. Substrate 14, derived from 2,4,6-tri-tert-butyl phenol, provides product 39 in excellent ee, having three contiguous stereocenters, two of them being neopentyl. Significantly, for each reaction in Table 6, a single diastereomer is observed (>95:5 by 1H NMR, GC and HPLC). Attempted epimerization experiments on products 36 and 38 (Et3N in PhMe at 110 °C for 24 h) failed to provide noticeable evidence of epimeric products. Preliminary semiempirical calculations suggest the major diastereomers are more thermodynamically stable products.11 The relative configuration of the product was also confirmed by nOe experiments (see supporting information).
Table 6.
Reaction of polysubstituted substrates
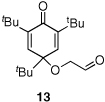
Analogously, in order to test whether this chemistry is able to generate quaternary stereocenters,4c,f cyclohexadienone 15 was synthesized from commercially available 3,4,5-trimethylphenol. Cyclization of 15 provides the desired product 40 with a quaternary stereocenter adjacent to a tertiary ether in good yield and excellent stereoselectivity [Eq. (2)].
 |
(2) |
The above examples all involve the synthesis of hydrobenzofuranones, largely due to the accessibility of the substrate by the above route.12 In order to test whether the oxygen tether was required, we synthesized substrate 41 starting from from 4-(3-hydroxypropyl)phenol. However when 41 was subjected to the optimized conditions, the only isolated product was 6-hydroxy-1-indanone [Eq. (3a)]. We suggest this product is derived from the elimination of the expected product under the reaction conditions. Fortunately, we found that the elimination may be avoided if the reaction of 41 is conducted using the preformed free carbene,[4d] providing the desired carbocycle 42 in 60% yield and 90% ee [Eq. (3b)].
 |
(3a) |
 |
(3b) |
The use of several commercially available disubstituted phenols as starting materials in this chemistry would lead to chiral racemic dienones. Previous work in our laboratory has revealed that kinetic resolutions are not practical using this approach; rather, the catalysts overrides preexisting stereocenters unless they are alpha to the aldehyde.4e However, we were intrigued by the opportunity to investigate the relative propensity of our catalysts to induce cyclization onto disubstituted versus trisubstituted Michael acceptors in an intramolecular competition. As such, chiral substrates 16–19 were assembled and subjected to optimized reaction conditions using catalyst 26 along with an achiral azolium salt. Substrates 16, 17 and 18 provided the products favoring cyclization onto the less-substituted olefin, consistent with expectations. The former provides 43 in 12:1 selectivity over 44 when using an achiral azolium salt. This selectivity is greatly degraded when the chiral catalyst is used, as it exerts its influence to provide only 2:1 selectivity, with the minor adduct formed in high ee, [Eq (8)]. Substrates 17 and 18 provide products 45 and 47 respectively in nearly exclusive selectivity regardless of catalyst, [Eqs (9) and (10)]. However, when we subject substrate 19 to both catalysts, we observe both constitutional isomers formed in identical amounts, eq 11, illustrating no selectivity between cyclization onto a di- versus trisubtituted Michael acceptor. When this reaction is conducted using catalyst 26, both isomers are formed in high ee’s, as the chirality of the catalyst exerts its control.
 |
(4) |
 |
(5) |
 |
(6) |
 |
(7) |
The different behavior of substrates 17 and 18 versus 19 is not easy to rationalize. We speculated that the dichotomy lay in product stability. Semiempirical calculations were carried out and the results are consistent with our suggestion (Scheme 4). Products 45 and 47 are correspondingly more thermodynamically stable than 46 and 48 by ~15 kJ/mol. In contrast, 50 is actually very slightly more stable than 49 (but only ~1.5 kJ/mol), implying the fused five-membered-ring has a profound effect on the outcome of the reaction. We suggest this is due to destabilization of 49 having the fused cyclopentane considerably twisted in the angular tricycle, a situation that is alleviated in 50. We further suggest that this difference in ground state energy is partially reflected in the transition states leading to each of these constitutional isomers, thereby negating the typical selectivities we observe between di- and trisubstituted Michael acceptors. This situation also allows the chiral catalyst to exert control over the newly formed bond providing for parallel kinetic resolution.
Finally, in order to test whether this chemistry could be used on scale, we conducted an experiment using one gram of 11 as the starting material. Although the catalyst loading was reduced to 3 mol % and the concentration (0.1M) is much higher than small scale experiments (0.008M), the reaction proceeds efficiently, providing ent-36 in 82% yield and 96% ee [Eq. (8)].
 |
(8) |
In conclusion, a series of cyclohexadienones were synthesized in a rapid and efficient manner. The asymmetric intramolecular Stetter reactions of these substrates affords hydrobenzofuranones in good yields and excellent selectivities. Up to three contiguous stereocenters as well as quaternary stereocenters can be formed using this transformation. A successful scale-up experiment demonstrates the utility of this methodology.
Experimental Section
General procedure for synthesis of the substrates
A flame-dried 100 ml round bottom flask was charged with cresol (1.08 g, 10 mmol); the flask was purged under vacuum for 5 mins and then refilled with argon and 2 ml CH2Cl2. Ethylene glycol (16.7 ml, 300 mmol) and then PhI(OAc)2 (4.83 g, 15 mmol, dissolved in 40 ml CH2Cl2) was added dropwise over 2 hours. The solution was then allowed to stir at ambient temperature for further 30 mins. The solution was concentrated in vacuo and the residue was subjected to column chromatography (EtOAc : Hexane = 1:1) to provide dienone alcohol (823 mg, 49 %) as an orange oil.
In a flame-dried 50 ml round bottom flask, this alcohol (556 mg, 3.86 mmol) was dissolved in 36 ml CH2Cl2, Dess Martin periodinane (1.80 g, 4.25 mmol) was added to the solution directly and the solution was then allowed to stir at ambient temperature for 1 hour. The solution was filtered through Celite 545 and then concentrated in vacuo and the residue was subjected to column chromatography (EtOAc : Hexane = 1:3) to provide 1 (556 mg, 87 %) as yellow oil.
General procedure for the synthesis of Hydrobenzofuranones
A flame-dried 25 ml round bottom flask was charged with triazolium salt 26 (4.9 mg, 0.012 mmol). The flask was purged under vacuum for 5 mins and then refilled with argon and 12 ml toluene. Argon was bubbled through the solution for 5 mins, and then KHMDS (0.024 ml, 0.012 mmol) was added and the solution was allowed to stir at ambient temperature for 15 minutes. The substrate (around 20 mg, 0.12 mmol) was dissolved in 3 ml toluene and then was added via syringe and the reaction was allowed to stir at ambient temperature. After the reaction was complete (checked by TLC), usually in 5 mins, the reaction mixture was directly purified by flash column chromatography.
Supplementary Material
This information is available free of charge via the Internet at http://pubs.acs.org.
Figure 1.

Effect of isopropanol as solvent
Scheme 2.

Calculation of relative energies
Acknowledgements
We gratefully acknowledge the National Institute of General Medical Sciences (GM72586) and Johnson and Johnson (Focused Funding) for partial support of this research. T.R. thanks Merck Research Laboratories, Eli Lilly, Amgen, and Boehringer Ingelheim for unrestricted support, and the Monfort Family Foundation for a Monfort Professorship. T.R. is a fellow of the Alfred P. Sloan Foundation.
References
- 1.a) Seebach D, Corey EJ. J. Org. Chem. 1975;40:231–237. [Google Scholar]; b) Seebach D. Angew. Chem. Int. Edit. Engl. 1979;18:239. [Google Scholar]
- 2.a) Stetter H. Angew. Chem. Int. Edit. Engl. 1976;15:639. [Google Scholar]; b) Stetter H, Kuhlmann H. Org. React. 1991;40:407. [Google Scholar]
- 3.a) Enders D, Breuer K, Runsink J, Teles JH. Helv. Chim. Acta. 1996;79:1899. [Google Scholar]; b) Enders D, Balensiefer T. Acc. Chem. Res. 2004;37:534. doi: 10.1021/ar030050j. [DOI] [PubMed] [Google Scholar]; c) Christmann M. Angew. Chem. Int. Edit. 2005;44:2632. doi: 10.1002/anie.200500761. [DOI] [PubMed] [Google Scholar]; d) Matsumoto Y, Tomioka K. Tetrahedron Lett. 2006;47:5843. [Google Scholar]
- 4.a) Kerr MS, Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2002;124:10298. doi: 10.1021/ja027411v. [DOI] [PubMed] [Google Scholar]; b) Kerr MS, Rovis T. Synlett. 2003:1934. [Google Scholar]; c) Kerr MS, Rovis T. J. Am. Chem. Soc. 2004;126:8876. doi: 10.1021/ja047644h. [DOI] [PubMed] [Google Scholar]; d) Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2005;127:6284. doi: 10.1021/ja0425132. [DOI] [PubMed] [Google Scholar]; e) Reynolds NT, Rovis T. Tetrahedron. 2005;61:6368. [Google Scholar]; f) Moore JL, Kerr MS, Rovis T. Tetrahedron. 2006;62:11477. [Google Scholar]
- 5.a) Woodward RB. J. Am. Chem. Soc. 1940;62:1208. [Google Scholar]; b) Waring AJ. Advances in Alicyclic Chemistry. New York: Academic Press; 1966. [Google Scholar]; c) Corey EJ, Girotra NN, Mathew CT. J. Am. Chem. Soc. 1969;91:1557. [Google Scholar]; d) Wiesner K, Tsai TYR, Nambiar KP. Can. J. Chem. 1978;56:1451. [Google Scholar]; e) Semmelhack MF, Harrison JJ, Thebtaranonth Y. J. Org. Chem. 1979;44:3275. [Google Scholar]; f) Corey EJ, Dittami JP. J. Am. Chem. Soc. 1985;107:256. [Google Scholar]; g) Mander LN. Synlett. 1991:134. [Google Scholar]; h) Kopach ME, Harman WD. J. Am. Chem. Soc. 1994;116:6581. [Google Scholar]; i) Maruoka K, Ito M, Yamamoto H. J. Am. Chem. Soc. 1995;117:9091. [Google Scholar]; j) Villar M, Kolly-Kovac T, Equey O, Renaud P. Chem.-Eur. J. 2003;9:1566. doi: 10.1002/chem.200390180. [DOI] [PubMed] [Google Scholar]; h) Clive DLJ, Fletcher SP, Liu DZ. J. Org. Chem. 2004;69:3282. doi: 10.1021/jo030364k. [DOI] [PubMed] [Google Scholar]; i) Canesi S, Bouchu D, Ciufolini MA. Org. Lett. 2005;7:175. doi: 10.1021/ol048094v. [DOI] [PubMed] [Google Scholar]; j) Jones SB, He LW, Castle SL. Org. Lett. 2006;8:3757. doi: 10.1021/ol0613564. [DOI] [PubMed] [Google Scholar]; k) Marsini MA, Gowin KM, Pettus TRR. Org. Lett. 2006;8:3481. doi: 10.1021/o10610993. [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Monje P, Grana P, Paleo MR, Sardina FJ. Org. Lett. 2006;8:951. doi: 10.1021/ol053130r. [DOI] [PubMed] [Google Scholar]
- 6.a) Pearson AJ, Zhu PY, Youngs WJ, Bradshaw JD, McConville DB. J. Am. Chem. Soc. 1993;115:10376. [Google Scholar]; b) Pearson AJ, Milletti MC, Zhu PY. Chem. Commun. 1995:853. [Google Scholar]; c) Schultz AG. Chem. Commun. 1999:1263. [Google Scholar]; d) Imbos R, Minnaard AJ, Feringa BL. J. Am. Chem. Soc. 2002;124:184. doi: 10.1021/ja017200a. [DOI] [PubMed] [Google Scholar]; e) Hayashi Y, Gotoh H, Tamura T, Yamaguchi H, Masui R, Shoji M. J. Am. Chem. Soc. 2005;127:16028. doi: 10.1021/ja055740s. [DOI] [PubMed] [Google Scholar]; f) Studer A, Schleth F. Synlett. 2005:3033. [Google Scholar]; g) Hoarau C, Pettus TRR. Org. Lett. 2006;8:2843. doi: 10.1021/ol061000s. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Zhu JL, Porco JA. Org. Lett. 2006;8:5169. doi: 10.1021/ol062233m. [DOI] [PubMed] [Google Scholar]
- 7.a) Pelter A, Elgendy SMA. J. Chem. Soc. Perkin Trans. 1. 1993:1891. [Google Scholar]; b) Moriarty RM, Prakash O. Org. React. 2001;57:327. [Google Scholar]; c) Tran-Huu-Dau ME, Wartchow R, Winterfeldt E, Wong YS. Chem.-Eur. J. 2001;7:2349. doi: 10.1002/1521-3765(20010601)7:11<2349::aid-chem23490>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]; d) Canesi S, Bouchu D, Ciufolini MA. Org. Lett. 2005;7:175. doi: 10.1021/ol048094v. [DOI] [PubMed] [Google Scholar]
- 8.Liu Q, Rovis T. J. Am. Chem. Soc. 2006;128:2552. doi: 10.1021/ja058337u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Burke SD, Cobb JE, Takeuchi K. J. Org. Chem. 1985;50:3420. [Google Scholar]; b) Chumoyer MY, Danishefsky SJ, Schulte GK. J. Am. Chem. Soc. 1994;116:11213. [Google Scholar]; c) Guth H. Helv. Chim. Acta. 1996;79:1559. [Google Scholar]; d) Yao SL, Johannsen M, Hazell RG, Jorgensen KA. J. Org. Chem. 1998;63:118. doi: 10.1021/jo971528y. [DOI] [PubMed] [Google Scholar]; e) Jonasson C, Ronn M, Backvall JE. J. Org. Chem. 2000;65:2122. doi: 10.1021/jo991787i. [DOI] [PubMed] [Google Scholar]; f) Germain J, Deslongchamps P. J. Org. Chem. 2002;67:5269. doi: 10.1021/jo025873l. [DOI] [PubMed] [Google Scholar]; g) Takao K, Tsujita T, Hara M, Tadano K. J. Org. Chem. 2002;67:6690. doi: 10.1021/jo0203140. [DOI] [PubMed] [Google Scholar]; h) Taber DF, Neubert TD, Rheingold AL. J. Am. Chem. Soc. 2002;124:12416. doi: 10.1021/ja027882h. [DOI] [PubMed] [Google Scholar]; i) Booker-Milburn KI, Hirst P, Charmant JPH, Taylor LHJ. Angew. Chem. Int. Edit. 2003;42:1642. doi: 10.1002/anie.200250507. [DOI] [PubMed] [Google Scholar]; j) Clive DLK, Fletcher SP. Chem. Commun. 2003:2464. doi: 10.1039/b307937f. [DOI] [PubMed] [Google Scholar]; k) Yamashita M, Ohta N, Shimizu T, Matsumoto K, Matsuura Y, Kawasaki I, Tanaka T, Maezaki N, Ohta S. J. Am. Chem. Soc. 2003;68:1216. doi: 10.1021/jo020619e. [DOI] [PubMed] [Google Scholar]
- 10.Kerr MS, Read de Alaniz J, Rovis T. J. Org. Chem. 2005;70:5725. doi: 10.1021/jo050645n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For example, 36 is ~9.9 kJ/mol more stable than the diasteomer having the methyl group on the endo face of the bicycle while 38 is ~36 kJ/mol more stable than the diastereomer having the bulky tert-butyl in that position.
- 12.The use of propylene glycol in the starting material synthesis affords homologous substrates. These substrates undergo efficient Stetter reaction using achiral catalyst, but are much more prone to base-induced Michael cyclization in the presence of our suite of chiral catalysts. Efforts to remedy this situation are currently underway.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This information is available free of charge via the Internet at http://pubs.acs.org.