

Abstract
The development of a new method for the synthesis of 1,1-dioxido-1,2-benzisothiazoline-3-acetic acid by a domino process is reported whereby a classical Heck reaction is coupled to an intramolecular aza-Michael reaction. Ultimately, this method has been expanded to a one-pot, sequential three-component protocol to generate diverse benzofused γ-sultams from a range of commercially available α-bromobenzenesulfonyl chlorides, amines and Michael acceptors.
Keywords: Domino reactions, Sultams, Heck reaction, Aza-Michael reaction, Multicomponent reactions
Introduction
The development of facile methods to heterocyclic scaffolds is critical to the drug-discovery process.[1] Cascade or domino reactions are highly efficient pathways that allow for the synthesis of complex molecules from simple substrates and encompass a variety of transformations such as Mannich and Robinson annulation reactions to name a few.[2] Ultimately, domino strategies require compatibility of reagents with all products, along with efficient individual steps to afford an overall high yielding process. Several approaches have emerged that couple transition-metal-catalyzed reactions and classical protocols to achieve domino processes in a single one-pot operation.[3] The Heck reaction is well suited in this regard revealing the original functionality of its olefinic partner for additional transformations. In particular, acrylates are ideal partners, because they can undergo facile Heck coupling, and still retain Michael accepting capabilities.[4] Despite successes in this area, the existence of a domino process coupling Heck and aza-Michael methodologies as a means to generate new heterocycles is restricted to two examples in the literature, both limited by low yields and/or long reaction times.[5] To the best of our knowledge, no examples of a domino Heck–aza-Michael approach towards the synthesis of sultams exist in the literature. Herein, we report the synthesis of 1,1-dioxido-1,2-benzisothiazoline-3-acetic acid by a domino process whereby a classical Heck reaction is coupled to an intramolecular aza-Michael reaction (HaM), ultimately resulting in a one-pot, sequential three-component protocol from commercially available starting materials (Scheme 1).[6]
Scheme 1.

Domino Heck–aza-Michael (HaM).
Compounds containing the sulfonamide moiety have gained wide popularity due to their extensive chemical and biological profiles.[7] Sultams (cyclic sulfonamide analogs) have emerged as privileged structures in drug discovery due to their wide biological profile. A number of benzofused sultams have recently surfaced that display potent activity including anti-inflammatory,[8] anti-malarial,[9] anti-HIV[10] and lipoxygenase inhibition (Figure 1),[11] to name a few. Traditionally, syntheses of sultams have been accomplished by classical cyclization protocols such as Friedel–Craft,[12] dianion,[13] [3+2] cycloadditions,[14] and Diels–Alder reactions.[15] Recently, however, a number of transition-metal-catalyzed approaches to sultams have emerged, including use of ring-closing metathesis (RCM),[16] Heck,[17] as well as Au-,[18] Cu-,[19] and Rh-catalyzed[20] cyclizations. Interest in the development of new routes to structurally diverse heterocycles has led to the titled domino process, which is now reported.[21]
Figure 1.

Representative biologically active sultams.
Results and Discussion
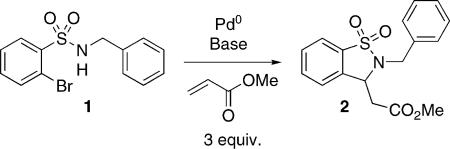
Initial reaction screening in this study was conducted on preformed sulfonamide 1 where a variety of bases and Pd0-sources were studied for the synthesis of sultam 2 (Table 1). Sulfonamide 1 (0.1 m in DMF) was heated at 110 °C in the presence of methyl acrylate, Pd0-catalysts and base for 14 h. Under these conditions, sulfonamide 1 underwent facile Heck coupling, followed by intramolecular aza-Michael cyclization,[22] to afford 2 in an overall domino process. These preliminary studies showed that the combination of Et3N with either Pd(OAc)2/PPh3 or Pd2(dba)3·CHCl3 gave the best results, with the presence of additive essential for high yields (Table 1, Entry 1 vs. Entries 5/6). Moreover, it was found that use of Pd2(dba)3·CHCl3 complex was preferred over the use of Pd(OAc)2/PPh3 which produced reaction mixtures containing Pd-mirror. Comparable results were achieved using microwave irradiation, however, a higher catalyst load (10−20 mol-%) was required (Table 1, Entries 7/8).
Table 1.
Domino Heck–aza-Michael.[a]
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Additive | % Conv.[b] |
| 1 | Pd(OAc)2/PPh3 | Et3N | – | 34 |
| 2 | Pd(OAc)2/PPh3 | Cs2CO3 | – | 15 |
| 3 | PdCl2(PPh)3 | Cs2CO3 | – | 13 |
| 4 | Pd2(dba)3·CHCl3 | Et3N | – | 35 |
| 5 | Pd(OAc)2/PPh3 | Et3N | Bu4NCl | 91 |
| 6 | Pd2(dba)3·CHCl3 | Et3N | Bu4NCl | 95 |
| 7 | Pd(OAc)2/PPh3 | Et3N | – | 94[c] |
| 8 | Pd(OAc)2/PPh3 | Et3N | Bu4NCl | 95[d] |
Reaction conditions: 1 (0.12 mmol), Pd (2 mol-%), methyl acrylate (0.36 mmol), base (0.36 mmol) in DMF at 110 °C for 14 h.
Conversion by G.C.
Pd(OAc)2 (20 mol-%), PPh3 (40 mol-%) in microwave 110 °C for 2 h.
Pd(OAc)2 (10 mol-%), PPh3 (20 mol-%) in microwave 110 °C for 2 h.
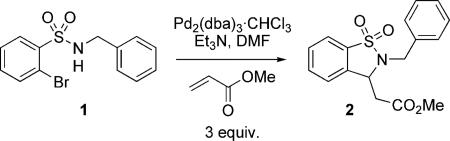
In the initial optimization phase, wet Bu4NCl was utilized to promote the domino process. Further investigation into this effect revealed that anhydrous Bu4NCl promoted the reaction, however with diminished efficiency (Table 2). On further study of this empirical result, we found that the addition of 10 mol-% of H2O to the reaction enhanced the yield when using either anhydrous Bu4NCl or Bu4NI. Though a true understanding of how the addition of water enhances yield is not entirely reached, it has been documented that higher yields can be achieved for Pd-catalyzed reactions that use water as a co-solvent.[23]
Table 2.
Domino Heck–aza-Michael.[a]
 | |||
|---|---|---|---|
| Entry | Additive | Water [mol-%] | % Conv.[b] |
| 1 | – | – | 35 |
| 2 | Bu4NCl | – | 72 |
| 3 | Bu4NCl | 10 mol-% | 95 |
| 4 | Bu4NI | – | 68 |
| 5 | Bu4NI | 10 mol-% | 94 |
Reaction conditions: 1 (0.12 mmol), Pd (2 mol-%), methyl acrylate (0.36 mmol), Et3N (0.36 mmol) and Bu4NCl (0.12 mmol) in DMF at 110 °C for 14 h.
Conversion by G.C.
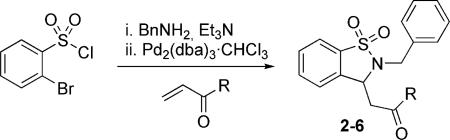
The scope of this chemistry was next examined with an array of commercially available Michael acceptors, yielding products bearing a variety of functional handles (Table 3). In the presence of Pd2(dba)3·CHCl3, several Michael acceptors were subjected to the reaction conditions, affording the desired compounds in good to high yield (Table 3, Entries 1−4). Attempts utilizing methyl methacrylate were unsuccessful, while the use of acrylonitrile and dimethyl vinyl phosphonate gave low yields. Of notable importance is the successful use of both acrylic acid and acrylamide to yield the corresponding sultam bearing carboxylic or amide handles, which are primed for further diversification.
Table 3.
Scope of possible Michael acceptors.[a]
 | |||
|---|---|---|---|
| Entry | R | Product | % Yield[b] |
| 1 | OMe | 2 | 95 |
| 2 | OEt | 3 | 92 |
| 3 | OH | 4 | 94 |
| 4 | Me | 5 | 79 |
| 5 | NH2 | 6 | 81 |
Reaction conditions: 1 (0.12 mmol), Pd2(dba)3·CHCl3 (2mol-%), methyl acrylate (0.36 mmol), Bu4NCl (0.12 mmol) in DMF at 110 °C for 14 h.
Isolated yields.
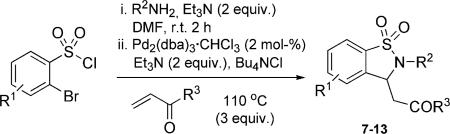
The use of the Heck–aza-Michael reaction in a one-pot, sequential three-component process was next demonstrated. In this protocol, a variety of α-bromobenzenesulfonyl chlorides were coupled with a range of aromatic, cyclic and alkylamines to yield intermediate sulfonamides. After two hours, Et3N, Bu4NCl, Pd2(dba)3·CHCl3 and the corresponding Michael acceptor were added to the reaction mixture, which was subsequently heated to 110 °C and subjected to workup after 14 h.[24] A small subset of 1,1-dioxido-1,2-benzisothiazoline-3-acetic acid was made using this one-pot protocol (Table 4). Reactions were typically carried out on the 0.27 mmol scale, affording products in good isolated yields (79−90%).[25]
Table 4.
Demonstration of a one-pot domino protocol.[a]
 | |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Product | % Yield[b] |
| 1 | H | c-C5H9 | Me | 7 | 74 |
| 2 | H | c-C6H11 | OH | 8 | 86 |
| 3 | 4-F | Bn | OEt | 9 | 84 |
| 4 | 4-F | C8H17 | OH | 10 | 90 |
| 5 | H | 4-ClPh | OEt | 11 | 81 |
| 6 | 4-F | c-C6H11 | OH | 12 | 84 |
| 7 | H | 4-MeOPh | OMe | 13 | 84 |
Reaction conditions: 1 (0.12 mmol), Pd2(dba)3·CHCl3 (2mol-%), methyl acrylate (0.36 mmol), Bu4NCl (0.12 mmol) in DMF at 110 °C for 14 h.
Isolated yields.
Further application of this multicomponent protocol was demonstrated utilizing the commercially available 2,5-dibromosulfonyl chloride 15 (Table 5). In this method, 1,1-dioxido-1,2-benzisothiazoline-3-acetic acid 16−19 possessing both saturated and unsaturated side chains were generated in good to excellent yields (85−93%). In all cases the intramolecular aza-Michael reaction proceeded with high yield forming the desired γ-sultam.
Table 5.
Application of domino HAM protocol with 15.[a]
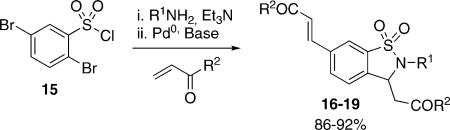 | ||||
|---|---|---|---|---|
| Entry | R1 | R2 | Product | % Yield[b] |
| 1 | 4-MeOPh | Me | 16 | 88 |
| 2 | C8H17 | OMe | 17 | 84 |
| 3 | 4-MeOPh | OMe | 18 | 93 |
| 4 | 2-MeOBn | OEt | 19 | 86 |
Reaction conditions: 1 (0.12 mmol), Pd2(dba)3·CHCl3 (2mol-%), methyl acrylate (0.36 mmol), Bu4NCl (0.12 mmol) in DMF at 110 °C for 14 h.
Isolated yields. Compounds purified by mass-directed fractionation.
The versatility of this protocol was next examined with a variety of other attractive substrates (20−22, Scheme 2). When submitted to the domino Heck–aza-Michael procedure, amide 20 remained unreacted, which further substantiates the aforementioned amide studies by Dyker and coworkers.[5b,26] In addition to 20, sulfonamide derivatives 21 and 22 were subjected to this protocol and, to our surprise, yielded only unreacted starting material. A plausible explanation for the unreactive cases involving sulfonamides 21 and 22 is the possibility of the free NH coordinating to palladium after the oxidative addition step and deactivating it for further chemistry.
Scheme 2.

In order to further probe the results shown in Scheme 2, N-(o-bromobenzyl)-2-bromobenzenesulfonamide (23), possessing both electron-poor and electron-rich bromoaryl moieties, was assembled and subjected to the domino Heck–aza-Michael conditions. In theory, this reaction can produce two possible products, 24 and 25, depending on which Heck–aza-Michael pathway is operative. According to the data presented in Table 4, we would expect an initial facile oxidative addition of Pd0 at the more electron-deficient aryl bromide. In the case of 23, this would be the bromide position ortho to the sulfonamide. Following the initial Heck addition, a facile aza-Michael reaction proceeds forming sultam 24. A second Heck reaction then proceeds on the remaining aryl bromide since no free NH is present to deactivate the Pd0 (as in the case of 21 and 22), resulting in the formation of only 24 (Scheme 3). When 23 was submitted to the standard HaM reaction conditions, one sole product was isolated as predicted along with starting material. Despite inconclusive NMR spectroscopic data due to the similar characteristics of 24 and 25, results discussed have allowed for tentative assignment of sultam 24 as the isolated product.
Scheme 3.

In addition to the aforementioned biologically active sultams outlined in Figure 1,[8-11] a number of related 1-oxoisoindol-3-acetamide derivatives have been reported to possess potent sedative-hypnotic activity, and are NHE1 inhibitors as well as allosteric modulators of glucokinase (Figure 2).[27]
Figure 2.

Biologically active 1-oxoisoindol-3-acetamides.
This promising bioactivity prompted use of the titled protocol to generate the analogous sultam derivatives in a one-pot procedure. The corresponding sulfonamide 26 and acrylamide 27 were thus generated in situ, followed by combination and submission to the domino HaM reaction conditions (Scheme 4). In the case of 28, none of the desired product was isolated from the reaction, only the product resulting from simple Heck reaction was observed in the crude mixture. Despite the utilization of a variety of both 1° and 2° amines, none of the desired sultam products were isolated. A plausible explanation is that the corresponding acrylamides are poorer Michael acceptors in comparison to their acrylate counterparts, further demonstrating how subtle electronic effects are operative in these systems.
Scheme 4.

To circumvent this problem, the sultam product derived from use of acrylic acid was coupled with the corresponding amine utilizing a high-load, soluble polymeric EDC variant OACC, derived from ring-opening metathesis polymerization (ROMP) (Scheme 5).[28] To this effect, some 1,1-dioxido-1,2-benzisothiazoline-3-acetamide derivatives 29−31 bearing both saturated and aromatic side chains were synthesized. This was accomplished by a one-pot protocol whereby the corresponding sultam acid was generated, followed by OACC coupling with the corresponding amine.
Scheme 5.

As previously discussed, the formation of the sultam carboxylic acids requires the use of excess acrylic acid. This excess reagent itself can undergo coupling with the free amine to form the corresponding amide. To circumvent this problem, the crude mixture was concentrated in situ to remove the excess acrylic acid. This is followed by resolvation and formation of the corresponding amide by coupling utilizing 2GOACC50.[29] Attempts to develop a “true” one-pot process starting from the corresponding sulfonamide were investigated. However, it was discovered that lowering the equivalents of acrylic acid from 3 to 1−1.5 significantly reduced the amount of cyclic acid formed and hence the amount of corresponding amide isolated.
Conclusions
In conclusion, a new method has been developed for the rapid synthesis of 1,1-dioxido-1,2-benzisothiazoline-3-acetic acid via a domino Heck–aza-Michael reaction. Expansion of the method to a one-pot, sequential three-component protocol to generate diverse benzofused sultams from commercially available α-bromobenzenesulfonyl chlorides, amines and Michael acceptors has been accomplished. This method has been utilized to synthesize a small demonstration library of 1,1-dioxido-1,2-benzisothiazoline-3-acetic acids bearing both saturated and unsaturated side chains. With this method in hand, a number of sultam derivatives of bioactive related isoindol-1-one amides were synthesized in a one-pot manner utilizing a ROMP-derived coupling reagent OACC. All of the compounds reported are currently undergoing broad biological screening through the NIH Molecular Library Screening Network (NIH-MLSCN) and the results will be reported in due course.
Experimental Section
General Procedures
All air- and moisture-sensitive reactions were carried out in flame- or oven-dried glassware under argon using standard gastight syringes, cannulas, and septa. Stirring was achieved with oven-dried, magnetic stir bars. CH3CN was purified by passage through the Solv-Tek purification system employing activated Al2O3.[30] Et3N was purified by passage over basic alumina and stored over KOH. Flash column chromatography was performed with SiO2 from Sorbent Technology (30930M-25, Silica Gel 60A, 40−63 um). Thin-layer chromatography was performed on silica gel 60F254 plates (EM-5717, Merck). Deuterated solvents were purchased from Cambridge Isotope laboratories. 1H and 13C NMR spectra were recorded with a Bruker DRX-400 NMR spectrometer operating at 400 MHz, 100 MHz, or 162 MHz, respectively; or a Bruker Avance operating at 500 MHz or 125 MHz, respectively. High-resolution mass spectrometry (HRMS) and FAB spectra were obtained in one of two manners: (i) on a VG Instrument ZAB double-focusing mass spectrometer and (ii) on a LCT Premier Spectrometer (Micromass UK Limited) operating on ESI (MeOH).
N-Benzyl-2-bromobenzenesulfonamide (1)
Into a flame-dried flask under argon was added benzylamine (1.87 mL, 17.2 mmol) and dry CH2Cl2 (36 mL). Et3N (4.35 mL, 31.2 mmol) was added and the reaction flask stirred at r.t for 15 min at which time N-benzyl-2-bromobenzene (4.0 g, 15.6 mmol) was added and the reaction mixture stirred for 2 h. The crude reaction mixture was concentrated and the resulting slurry was suspended in EtOAc (20 mL). The organic salts were removed by filtration and the solvent removed under reduced pressure. Flash chromatography (EtOAc/hexane, 1:1) provided 5.02 g (98% yield) of the title compound as a white solid, m.p. 93−96 °C. FTIR (neat): . 1H NMR (500 MHz, CDCl3: δ = 8.12 (dd, J = 7.8, 1.8 Hz, 1 H), 7.69 (dd, J = 7.8, 1.2 Hz, 1 H), 7.44 (dt, J = 7.6, 1.26 Hz, 1 H), 7.39 (dt, J = 5.6, 1.8 Hz, 1 H), 7.27−7.25 (m, 3 H), 7.18−7.23 (m, 2 H), 5.41−5.43 (m, 1 H), 4.10 (d, J = 6.2 Hz, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 138.7, 135.6, 135.0, 133.7, 131.4, 128.7, 128.2, 128.0, 127.6, 119.7, 47.5 ppm. HRMS calculated for C13H12BrNO2SNa [M + Na] 347.9670; found 347.9700 (TOF MS ES+).
Methyl (2-Benzyl-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-3-yl)-acetate (2)
Into a microwave vial was added 1 (39.1 mg, 0.12 mmol), Et3N (50 μL, 0.36 mmol), Pd(OAc)2 (20 mol-%, 5.4 mg, 0.024 mmol), PPh3 (40 mol-%, 12.6 mg, 0.048 mmol) and CH3CN (0.6 mL). After stirring for 5 min, methyl acrylate (32 μL, 0.36 mmol) was added and the reaction placed in the microwave oven at 120 °C for 2 h. Then the reaction was concentrated under reduced pressure and purified using flash chromatography (hexane/EtOAc, 6:4) to provide 37.7 mg (95%) of the title compound as a yellow oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.85 (d, J = 7.2 Hz, 1 H), 7.52−7.63 (m, 2 H), 7.42 (d, J = 7.2 Hz, 1 H), 7.28−7.36 (m, 5 H), 4.83 (t, J = 6.0 Hz, 1 H), 4.61 (d, J = 15.6 Hz, 1 H), 4.49 (d, J = 15.6 Hz, 1 H), 3.59 (s, 3 H), 2.86 (dd, J = 16.3, 5.4 Hz, 1 H), 2.70 (dd, J = 16.3, 6.7 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 170.5, 137.3, 135.3, 132.9, 131.0, 130.3, 129.5, 128.8, 128.4, 128.0, 124.1, 121.4, 119.5, 57.0, 52.0, 46.6, 38.9 ppm. HRMS calculated for C17H18NO4S [M + H]+ 332.0956; found 332.0958 (TOF MS EI+).
General Procedure (A) for the Synthesis of 1,1-Dioxido-1,2-benzisothiazoline-3-acetic Acid Derivatives Using a Range of Michael Acceptors from 1
Into a 1-dram vial was added 1 (40 mg, 0.12 mmol), Et3N (51 μL, 0.36 mmol), Bu4NCl (30 mg, 0.12 mmol), Pd2(dba)3·CHCl3 (2 mol-%, 2.5 mg, 0.0024 mmol) and dry DMF (1.22 mL). After stirring for 5 min at room temp., Michael acceptor (0.36 mmol) was added and the reaction vial was placed immediately into a preheated reaction block. The reaction mixture was stirred at 110 °C for 14 h after which time the reaction was cooled and concentrated under reduced pressure. The crude product was purified using flash chromatography (hexane/EtOAc, 6:4).
Ethyl (2-Benzyl-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-3-yl)acetate (3)
Prepared using general method (A) with ethyl acrylate (39.9 μL, 0.366 mmol) to yield 38 mg of 3 (92%) as a yellow oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.84 (dd, J = 6.9, 1.2 Hz, 1 H), 7.53−7.60 (m, 2 H), 7.43 (d, J = 7.2 Hz, 2 H), 7.30−7.38 (m, 4 H), 4.82 5(t, J = 6.1 Hz, 1 H), 4.62 (d, J = 15.6 Hz, 1 H), 4.52 (d, J = 15.6 Hz, 1 H), 4.03−4.06 (m, 2 H), 2.86 (dd, J = 16.3, 5.2 Hz, 1 H), 2.70 (dd, J = 16.3, 3.6 Hz 1 H), 1.14 (t, J = 5.6 Hz 3 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 170.0, 137.3, 135.3, 134.6, 132.8, 129.5, 128.7, 128.4, 128.0, 124.18, 121.40, 61.1, 57.0, 46.4, 39.0, 13.9 ppm. HRMS calculated for C18H19NO4SNa [M + Na]+ 368.0932; found 368.0930 (TOF MS EI+).
(2-Benzyl-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-3-yl)acetic Acid (4)
Prepared using general method (A) with acrylic acid (25.0 μL, 0.366 mmol) to give 36 mg of 4 (94%) as a white solid, m.p. 120−122 °C. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.84 (d, J = 7.2 Hz, 1 H), 7.55−7.62 (m, 2 H), 7.38−7.42 (m, 3 H), 7.27−7.34 (m, 3 H), 4.82 (t, J = 5.4 Hz, 1 H), 4.62 (d, J = 15.6 Hz, 1 H), 4.51 (d, J = 15.6 Hz, 1 H), 2.91 (dd, J = 16.6, 5.0 Hz, 1 H), 2.70 (dd, J = 16.6, 6.9 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 174.8, 137.0, 135.0, 134.6, 133.0, 129.6, 128.8, 128.5, 128.1, 124.16, 121.49, 56.6, 46.7, 38.6 ppm. HRMS calculated for C16H16NO4S [M + H]+ 318.0800; found 318.0817 (TOF MS EI+).
1-(2-Benzyl-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-3-yl)propane-2-one (5)
Prepared using general method (A) with methyl vinyl ketone (25.0 μL, 0.366 mmol) to give 30 mg of 5 (79%) as a clear oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.83 (m, 1 H), 7.51−7.58 (m, 2 H), 7.40 (d, J = 7.2 Hz, 2 H), 7.25−7.36 (m, 4 H), 4.98 (t, J = 5.9 Hz, 1 H), 4.56 (d, J = 14.7 Hz, 1 H), 4.42 (d, J = 15.6 Hz, 1 H), 2.97 (dd, J = 18.0, 5.6 Hz, 1 H), 2.77 (dd, J = 18.0, 6.3 Hz, 1 H), 1.90 (s, 3 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 205.4, 138.1, 135.8, 134.6, 132.95, 129.3, 128.7, 128.5, 127.9, 124.2, 121.3, 56.3, 48.2, 30.27 ppm. HRMS calculated for C17H17NO3SNa [M + Na]+ 338.0827 found 338.0800 (TOF MS EI+).
(2-Benzyl-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-3-yl)acetamide (6)
Prepared using general method (A) with acrylamide (26 mg, 0.366 mmol) to yield 31 mg of 6 (81%) as a white solid. FTIR (neat): . 1H NMR (500 MHz, [D4]MeOD): δ = 7.81 (d, J = 7.7 Hz, 1 H), 7.65 (t, J = 7.5 Hz, 1 H), 7.59 (t, J = 7.2 Hz, 1 H), 7.51 (d, J = 7.7 Hz, 1 H), 7.47 (d, J = 7.3 Hz, 2 H), 7.28−7.38 (m, 3 H), 4.92 (t, J = 6.6 Hz, 1 H), 4.87 (br. s, 2 H), 4.64 (d, J = 15.6 Hz, 1 H), 4.50 (d, J = 15.6 Hz, 1 H), 2.84 (dd, J = 15.1, 6.1 Hz, 1 H), 2.61 (dd, J = 15.1, 7.0 Hz, 1 H) ppm. 13C NMR (125 MHz, [D4]MeOD): δ = 174.8, 139.7, 137.5, 136.3, 134.6, 131.2, 130.3, 130.2, 130.1, 129.1, 59.5, 48.1, 41.6 ppm. HRMS calculated for C16H17N2O3S [M + H]+ 317.0960 found 317.0932 (TOF MS EI+).
General Procedure (B) for the Synthesis of 1,1-Dioxido-1,2-benzisothiazoline-3-acetic Acid Derivatives
Into a 1-dram vial was added amine (0.273 mmol), Et3N (0.547 mmol) and dry DMF (0.59 mL) and reaction mixture was stirred at r.t. for 15 min. Then, α-bromobenzenesulfonyl chlorides (0.273 mmol) were added and the reaction mixture was stirred for 2 h. To the reaction vial was added Et3N (76 μL, 0.548 mmol), Bu4N (74.5 mg, 0.273 mmol), Pd2(dba)3·CHCl3 (2 mol-%, 25.6 mg, 0.0056 mmol) and dry DMF (1.4 mL). After stirring for 5 min at room temp., Michael acceptor (0.819 mmol) was added and the reaction vial was placed immediately into a preheated reaction block. The reaction mixture was stirred at 110 °C for 14 h after which time the reaction was cooled and concentrated under reduced pressure. The crude was purified using flash chromatography (hexane/EtOAc, 6:4).
1-(2-Cyclopentyl-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-3-yl)propane-2-one (7)
Prepared using general method (B) using benzenesulfonyl chloride, cyclopentylamine and methyl vinyl ketone to afford 52 mg of 7 (76%) as a yellow oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.67 (d, J = 7.1 Hz, 1 H), 7.40−7.51 (m, 2 H), 7.32 (d, J = 7.6 Hz, 1 H), 5.02 (dd, J = 8.7, 3.7 Hz, 1 H), 3.15 (dd, J = 17.9, 5.7 Hz, 1 H), 2.86 (dd, J = 17.9, 7.6 Hz, 1 H), 2.11 (s, 3 H), 1.92−1.98 (m, 2 H), 1.64−1.75 (m, 4 H), 1.51−1.54 (m, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 206.3, 139.3, 135.9, 132.8, 130.2, 129.7, 124.5, 121.1, 57.6, 55.3, 48.7, 31.0, 30.85, 28.9, 24.1, 22.7 ppm. HRMS calculated for C15H23N2O3S [M + NH4]+ 311.1429; found 311.1446 (TOF MS EI+).
(2-Cyclohexyl-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-3-yl)acetic Acid (8)
Prepared using general method (B) using α-bromobenzenesulfonyl chloride, cyclohexylamine and acrylic acid to yield 76 mg of 8 (86%) as a white solid, m.p. 121−124 °C. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 9.08−9.48 (br. s, 1 H), 7.74 (d, J = 7.6 Hz, 1 H), 7.57 (td, J = 7.5, 1.0 Hz, 1 H), 7.51 (t, J = 7.5 Hz, 1 H), 7.46 (d, J = 7.6 Hz, 1 H), 5.06 (dd, J = 8.1, 4.2 Hz, 1 H), 3.58−3.64 (m, 1 H), 3.08 (dd, J = 16.5, 4.2 Hz, 1 H), 2.84 (dd, J = 16.5, 8.1 Hz, 1 H), 1.94−2.04 (m, 4 H), 1.79−1.85 (m, 2 H), 1.59−1.68 (m, 2 H), 1.28−1.36 (m, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 173.4, 136.4, 130.9, 127.5, 122.2, 119.0, 54.6, 52.8, 38.8, 30.3, 28.2, 24.1, 23.9, 23.4 ppm. HRMS calculated for C15H23N2O4S [M + NH4]+ 327.1379; found 327.1399 (TOF MS EI+).
Ethyl (2-Benzyl-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-3-yl)acetate (9)
Prepared using general method (B) using, 4-fluoro-2-bromobenzenesulfonyl chloride, benzylamine and ethyl acrylate to afford 92 mg of 9 (84%) as a yellow oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.82 (dd, J = 8.6, 4.8 Hz, 1 H), 7.40−7.42 (m, 2 H), 7.30−7.36 (m, 3 H), 7.22−7.26 (m, 1 H), 7.09 (dd, J = 8.4, 2.1 Hz, 1 H), 4.78 (dd, J = 6.8, 6.7 Hz, 1 H), 4.60 (d, J = 15.6 Hz, 1 H), 4.48 (d, J = 15.6 Hz, 1 H), 4.02−4.10 (m, 2 H), 2.86 (dd, J = 16.4, 4.9 Hz, 1 H), 2.68 (dd, J = 16.4, 7.1 Hz, 1 H), 1.15 (t, J = 7.2 Hz, 3 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 167.9, 164.5 (d, JC-F = 250.5 Hz), 138.6, 133.1, 127.2, 126.7, 126.4, 126.1, 121.7, 115.6, 109.9, 59.3, 54.7, 44.7, 36.7, 12.0 ppm. HRMS calculated for C18H22FN2O4S [M + NH4]+ 381.1284; found 381.1295 (TOF MS EI+).
(2-Octyl-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-3-yl)acetic Acid (10)
Prepared using general method (B) using 4-fluoro-2-bromobenzenesulfonyl chloride, octylamine and acrylic acid to yield 87 mg of 10 (90%) as a pale yellow solid, m.p. 123−127 °C. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 9.02−9.16 (br. s, 1 H), 7.79 (dd, J = 8.6, 4.7 Hz, 1 H), 7.24−7.26 (m, 1 H), 7.16 (dd, J = 8.3, 2.0 Hz, 1 H), 4.86 (m, 1 H), 3.30 (t, J = 15.3 Hz, 2 H), 3.06 (dd, J = 21.6, 4.7 Hz, 1 H), 2.83 (dd, J = 16.9, 7.4 Hz, 1 H), 1.66−1.75 (m, 2 H), 1.23−1.39 (m, 10 H), 0.85 (t, J = 6.8 Hz, 2 H). 13C NMR (125 MHz, CDCl3)δ = 173.5, 163.3 (d, JC-F = 254.9 Hz), 138.4, 129.1, 121.8, 115.7, 109.7, 54.8, 41.8, 36.7, 29.8, 27.2, 26.1, 25.0, 20.7, 12.16 ppm. HRMS calculated for C17H28FN2O4S (M + NH4)+ 375.1754; found 375.1769 (TOF MS EI+).
Ethyl (4-Chlorophenyl-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-3-yl)acetate (11)
Prepared using general method (B) using 2-bromobenzenesulfonyl chloride, 4-chlorobenzylamine and ethyl acrylate to afford 73 mg of 11 (81%) as a yellow oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.77 (d, J = 7.1 Hz, 1 H), 7.47−7.56 (m, 2 H), 7.30−7.34 (m, 3 H), 7.25 (d, J = 8.5 Hz, 2 H), 4.77 (t, J = 5.9 Hz, 1 H), 4.48 (q, J = 15.9 Hz, 2 H), 3.98−4.06 (m, 2 H), 2.78 (dd, J = 16.4, 5.8 Hz, 1 H), 2.66 (dd, J = 16.4, 6.2 Hz, 1 H), 1.09 (t, J = 7.2 Hz, 3 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 170.0, 137.1, 134.6, 134.1, 133.1, 133.0, 129.8, 129.2, 128.7, 124.1, 121.4, 61.2, 57.3, 46.2, 39.4, 14.2 ppm. HRMS calculated for C18H19ClNO4S [M + H]+ 380.0723; found 380.0739 (TOF MS EI+).
(2-Cyclohexyl-6-fluoro-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-3-yl)acetic Acid (12)
Prepared using general method (B) using 4-fluoro-2-bromobenzenesulfonyl chloride, cyclohexylamine and acrylic acid to yield 65 mg of 12 (84%) as a yellow oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.73−7.76 (m, 1 H), 7.17−7.30 (m, 3 H), 5.01−5.08 (m, 1 H), 3.62 (t, J = 11.9 Hz, 1 H), 3.10 (d, J = 16.8 Hz, 1 H), 2.86 (dd, J = 16.8, 8.5 Hz, 2 H), 2.04 (d, J = 11.9 Hz, 1 H), 1.95 (d, J = 11.6 Hz, 1 H), 1.84 (t, J = 15.0 Hz, 1 H), 1.58−1.68 (m, 3 H), 1.30−1.38 (dd, J = 12.8, 13.1 Hz, 2 H), 1.08−1.17 (m, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 174.7, 165.1 (d, JC-F = 254.6 Hz), 141.4, 132.4, 123.3, 117.4, 111.6, 56.7, 54.0, 40.6, 32.1, 26.0, 25.8, 25.3 ppm. HRMS calculated for C15H18FNO4S [M + H]+ 328.1019; found 328.1036 (TOF MS EI+).
Methyl [2-(4-Methoxyphenyl)-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-3-yl]acetate (13)
Prepared using general method (B) using benzenesulfonyl chloride, p-methoxybenzylamine and methyl acrylate to produce 71 mg of 13 (84%) as a yellow oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.85 (d, J = 7.1 Hz, 1 H), 7.51−7.59 (m, 2 H), 7.42 (d, J = 8.5 Hz, 3 H), 6.86 (d, J = 8.6 Hz, 2 H), 4.80 (t, J = 6.4 Hz, 1 H), 4.57 (d, J = 12.4 Hz, 1 H), 4.41 (d, J = 15.4 Hz, 1 H), 3.80 (s, 3 H), 3.63 (s, 3 H), 2.87 (dd, J = 16.3, 5.3 Hz, 1 H), 2.69 (dd, J = 16.3, 6.9 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 170.6, 159.4, 137.3, 134.8, 132.0, 130.2, 129.3, 127.0, 124.0, 121.4, 114.0, 56.6, 55.3, 46.0 ppm. HRMS calculated for C18H23N2O5S [M + NH4]+ 379.1328; found 379.1337 (TOF MS EI+).
Ethyl 3-[2-(N-tert-Butylsulfamoyl)-5-fluorophenyl]propanoate (14)
Prepared using general method (A) using 4-fluoro-2-bromobenzenesulfonyl chloride, tert-butylamine and ethyl acrylate to afford 41 mg of 14 (48%) as a clear oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 8.38 (dd, J = 15.9, 0.65 Hz, 1 H), 8.15 (dd, J = 11.2, 5.6 Hz, 1 H), 7.30 (dd, J = 9.2, 2.6 Hz, 1 H), 7.14 (ddd, J = 10.3, 7.7, 2.6 Hz, 1 H), 6.35 (d, J = 21.5 Hz, 1 H), 4.68 (s, 1 H), 4.28 (q, J = 7.1 Hz, 2 H), 1.33 (t, J = 7.1 Hz, 3 H), 1.16 (s, 9 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 165.8, 163.63, 140.1, 137.5, 136.4, 132.6, 123.7, 116.4, 114.9, 61.3, 55.1, 30.1, 14.1 ppm. HRMS calculated for C15H21FNO4S [MH]+ 330.1175; found 330.1193 (TOF MS EI+).
Compound 16
Prepared using general method (B) using 2,5-dibromobenzenesulfonyl chloride, p-methoxybenzylamine and methyl vinyl ketone to yield 86 mg of 16 (88%) as a yellow oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.95 (s, 1 H), 7.67 (dd, J = 8.1, 1.4 Hz, 1 H), 7.51 (d, J = 16.2 Hz, 1 H), 7.34 (d, J = 8.1 Hz, 1 H), 7.30 (d, J = 6.7 Hz, 2 H), 6.62 (d, J = 11.3 Hz, 2 H), 6.77 (d, J = 10.9 Hz, 1 H), 4.94 (m, 1 H), 4.43 (d, J = 3.84 Hz, 2 H), 3.78 (s, 3 H), 2.98 (dd, J = 18.1, 5.3 Hz, 1 H), 2.78 (dd, J = 18.1, 6.7 Hz, 1 H), 2.39 (s, 3 H), 1.98 (s, 3 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 205.5, 197.8, 159.4, 140.9, 139.8, 136.1, 136.0, 132.44, 129.9, 129.0, 126.9, 125.0, 120.4, 114.1, 55.8, 55.2, 47.8, 46.7, 30.3, 28.0 ppm. HRMS calculated for C22H24NO5S [M + H]+ 414.1375; found 414.1364 (TOF MS EI+).
Methyl (2E)-3-[2-Octyl-3-(2-methoxy-2-oxoethyl)-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-6-yl]acrylate (17)
Prepared using general method (B) using 2,5-dibromobenzenesulfonyl chloride, p-methoxybenzylamine and methyl acrylate to produce 86 mg of 17 (84%) as a yellow oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.91 (s, 1 H), 7.67−7.74 (m, 2 H), 7.46 (d, J = 8.1 Hz, 1 H), 6.51 (d, J = 16.0 Hz, 2 H), 4.92 (t, J = 5.5 Hz, 1 H), 3.81 (s, 3 H), 3.73 (s, 3 H), 3.30 (t, J = 7.5 Hz, 2 H), 2.99 (dd, J = 16.4, 5.3 Hz, 1 H), 2.77 (dd, J = 16.4, 7.1 Hz, 1 H) 1.66−1.74 (m, 2 H), 1.25−1.37 (s, 12 H), 0.86 (t, J = 5.5 Hz, 3 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 170.7, 166.6, 142.0, 138.9, 136.2, 136.0, 132.4, 124.8, 120.7, 120.4, 119.9, 57.4, 53.7, 51.5, 43.7, 38.8, 31.7, 29.5, 28.9, 26.4, 26.6, 14.1 ppm. HRMS calculated for C22H32NO6S [M + H]+ 438.1950; found 438.1964 (TOF MS EI+).
Methyl (2E)-3-[2-(4-Methoxyphenyl)-3-(2-methoxy-2-oxoethyl)-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-6-yl]acrylate (18)
Prepared using general method (B) using 2,5-Dibromobenzenesulfonyl chloride, p-methoxybenzylamine and methyl acrylate to yield 98 mg of 18 (93%) as a yellow oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.95 (s, 1 H), 7.65−7.69 (m, 2 H), 7.38 (d, J = 8.1 Hz, 1 H), 7.33 (d, J = 11.5 Hz, 2 H), 6.86 (d, J = 9.6 Hz, 2 H), 6.52 (d, J = 16.0 Hz, 1 H), 4.79 (dd, J = 6.9, 5.1 Hz, 1 H), 4.59 (d, J = 15.3 Hz, 1 H), 4.40 (d, J = 15.3 Hz, 1 H), 3.82 (s, 3 H), 3.79 (s, 3 H), 3.63 (s, 3 H), 2.88 (dd, J = 16.3, 5.0 Hz, 1 H), 2.70 (dd, J = 16.3, 7.0 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 170.3, 166.6, 159.4, 141.1, 138.7, 136.2, 135.9, 132.3, 129.9, 126.7, 124.8, 120.7, 120.2, 114.1, 56.4, 55.2, 52.1, 46.0, 38.5 ppm. HRMS calculated for C22H24NO7S [M + H]+ 446.1273; found 446.1269 (TOF MS EI+).
Ethyl (2E)-3-[3-(2-Ethoxy-2-oxoethyl)-2-(2-methoxybenzyl)-1,1-dioxido-2,3-dihydro-1,2-benzisothiazol-6-yl]acrylate (19)
Prepared using general method (B) using 2,5-dibromobenzenesulfonyl chloride, 2-methoxyphenylamine and ethyl acrylate to produce 107 mg of 19 (86%) as a yellow oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.99 (s, 1 H), 7.76 (dd, J = 8.1, 1.5 Hz, 1 H), 7.71 (d, J = 16.0 Hz, 1 H), 7.56 (d, J = 8.1 Hz, 1 H), 7.35 (d, J = 8.1 Hz, 1 H), 7.05−7.08 (m, 1 H), 7.36 (t, J = 2.2 Hz, 1 H), 6.88 (dd, J = 8.4, 2.5 Hz, 1 H), 6.53 (d, J = 16.0 Hz, 1 H), 5.56 (dd, J = 7.9, 4.1 Hz, 1 H), 4.28 (q, J = 7.9 Hz, 1 H), 4.08 (q, J = 11.4 Hz, 1 H), 3.83 (s, 3 H), 2.97 (dd, J = 16.3, 4.2 Hz, 1 H), 2.76 (dd, J = 16.4, 8.0 Hz, 1 H), 1.34 (t, J = 9.8 Hz, 1 H), 1.16 (t, J = 11.4 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 170.0, 169.18, 160.6, 141.6, 137.5, 136.6, 135.5, 135.0, 134.8, 132.6, 130.8, 124.9, 121.4, 120.4, 117.4, 113.2, 111.0, 61.3, 60.9, 58.1, 55.2, 38.5, 14.2, 13.8 ppm. HRMS calculated for C23H26NO7S [M + H]+ 460.1430; found 460.1442 (TOF MS EI+).
N-Benzyl-2-bromobenzamide (20)
To a round-bottomed flask under Ar was added benzylamine (0.51 cm3, 4.66 mmol), dry DCM (0.46 m, 10 cm3) and Et3N (1.29 cm3, 9.33 mmol). The reaction was cooled to 0 °C and after stirring for 5 min, 2-bromobenzyl bromide (0.547 cm3, 4.19 mmol) was added. The reaction was warmed to r.t and stirred for 2 h. Then the crude reaction was concentrated in situ and suspended in EtOAc (10 cm3). The resulting suspension was filtered through a silica plug and the solvent removed to yield 1.1 g of 20 (95%) as a white solid. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.58 (dd, J = 8.0, 1.0 Hz, 1 H), 7.56 (dd, J = 7.6, 1.7 Hz, 1 H), 7.33−7.40 (m, 4 H), 7.31 (dt, J = 7.0, 1.5 Hz, 1 H), 7.27 (td, J = 8.1, 1.8 Hz, 2 H), 6.24 (br. s, 1 H), 4.66 (d, J = 5.7 Hz, 2 H) ppm. 13C NMR (125 MHz, CDCl3)δ = 167.5, 137.6, 137.5, 133.4, 131.7, 129.6, 128.8, 128.0, 127.7, 127.6, 119.3, 44.2 ppm. HRMS calculated for C14H13BrNO [M + H]+ 290.0181; found 290.0179 (TOF MS EI+).1H and 13C NMR spectroscopic data corresponded to that reported by Catellani and coworkers.[31]
N-(2-Bromobenzyl)-4-methylbenzenesulfonamide (21)
To a round-bottomed flask under Ar was added 2-bromobenzylamine (0.5 g, 2.68 mmol), dry DCM (0.46 m, 5.8 cm3) and Et3N (0.75 cm3, 5.36 mmol). The reaction was cooled to 0 °C and after stirring for 5 min, p-toluenesulfonyl chloride (0.51 g, 2.68 mmol) was added. The reaction was warmed to r.t and stirred for 2 h. Then the crude reaction was concentrated in situ and suspended in EtOAc (10 cm3). The resulting suspension was filtered through a silica plug and the solvent removed to yield 0.87 g of 21 (96%) as a white solid. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.60 (d, J = 8.2 Hz, 2 H), 7.45 (d, J = 7.9 Hz, 1 H), 7.19−7.30 (m, 4 H), 7.10 (dt, J = 9.3, 1.5 Hz, 1 H), 4.92 (m, 1 H), 4.22 (d, J = 6.5 Hz, 2 H), 2.40 (s, 3 H) ppm. 13C NMR (125 MHz, CDCl3)δ = 162.1, 143.5, 136.8, 135.4, 132.8, 130.6, 129.7, 129.6, 127.7, 127.6, 127.1, 123.5, 47.5, 21.5 ppm. 1H and 13C NMR spectroscopic data corresponded to that reported by Malacria and coworkers.[32]
N-(2-Bromobenzyl)methanesulfonamide (22)
To a round-bottomed flask under Ar was added 2-bromobenzylamine (0.5 g, 2.68 mmol), dry DCM (0.46 m, 5.8 cm3) and Et3N (0.75 cm3, 5.36 mmol). The reaction was cooled to 0 °C and after stirring for 5 min, methanesulfonyl chloride (0.22 cm3, 2.95 mmol) was added. The reaction was warmed to r.t and stirred for 2 h. Then the crude reaction was concentrated in situ and suspended in EtOAc (10 cm3). The resulting suspension was filtered through a silica plug and the solvent removed to yield 0.65 g of 22 (92%) as a white solid. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.59 (d, J = 7.9 Hz, 1 H), 7.45 (d, J = 7.6 Hz, 1 H), 7.33 (t, J = 7.5 Hz, 1 H), 7.21 (dt, J = 7.5, 1.5 Hz, 1 H), 4.90 (m, 1 H), 4.41 (d, J = 6.4 Hz, 2 H), 2.81 (s, 3 H). 13C NMR (125 MHz, CDCl3)δ = 162.1, 135.8, 133.2, 130.7, 130.0, 128.0, 123.7, 47.63, 41.3; 1H and 13C NMR spectroscopic data corresponded to that reported by Malacria and coworkers.[32]
2-Bromo-N-(2-bromobenzyl)benzenesulfonamide (23)
To a round-bottomed flask under Ar was added 2-bromobenzylamine (0.5 g, 2.68 mmol), dry DCM (0.46 m, 5.8 cm3) and Et3N (0.75 cm3, 5.36 mmol). The reaction was cooled to 0 °C and after stirring for 5 min, 2-bromobenzenesulfonyl chloride (0.61 g, 2.41 mmol) was added. The reaction was warmed to r.t and stirred for 2 h. Then the crude reaction was concentrated in situ and suspended in EtOAc (10 cm3). The resulting suspension was filtered through a silica plug and the solvent removed to yield 0.90 g of 23 (93%) as a white solid. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 8.80 (dd, J = 7.8, 1.7 Hz, 1 H), 7.58 (dd, J = 7.8, 1.1 Hz, 1 H), 7.45 (dd, J = 6.9, 2.2 Hz, 1 H), 7.40 (dt, J = 7.7, 1.1 Hz, 1 H), 7.33 (dt, J = 7.6, 1.7 Hz, 1 H), 7.12 (dd, J = 7.7, 1.5 Hz, 1 H), 7.05−7.08 (m, 2 H), 5.79 (m, 1 H), 4.26 (d, J = 6.6 Hz, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.1, 139.1, 134.9, 133.5, 132.7, 131.4, 130.6, 129.8, 129.6, 127.6, 127.4, 123.8, 119.9, 47.7 ppm. HRMS calculated for C13H11Br2NO2SNa [M + Na]+ 425.8775; found 425.8781 (TOF MS EI+).
Methyl (E)-3-(2-{[3-(2-methoxy-2-oxoethyl)-1,1-dioxido-1,2-benzisothiazol-2(3H)-yl]methyl}phenyl)acrylate (24)
To a 1-dram vial was added 23 (76 mg, 0.19 mmol), Et3N (79 μL, 0.56 mmol), Bu4NCl (70 mg, 0.19 mmol), Pd2(dba)3·CHCl3 (2 mol-%, 3.9 mg, 0.0038 mmol) and dry DMF (1.89 mL). After stirring for 5 min at room temp., methyl acrylate (51 μL, 0.56 mmol) was added and the reaction vial was placed immediately into a preheated reaction block. The reaction mixture was stirred at 110 °C for 14 h after which time the reaction was cooled and concentrated under reduced pressure. The crude was purified by column chromatography (1:1 hexane/EtOAc) to yield 59 mg of 24 (76%) as a yellow oil. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 8.10 (d, J = 15.7 Hz, 1 H), 7.83 (d, J = 7.3 Hz, 1 H), 7.61 (ddd, J = 7.5, 1.2 Hz, 2 H), 7.55 (ddd, J = 5.3, 0.7 Hz, 2 H), 7.32−7.40 (m, 3 H), 6.35 (d, J = 15.7 Hz, 1 H), 4.87 (t, J = 6.3 Hz, 1 H), 4.76 (d, J = 15.9 Hz, 1 H), 4.60 (d, J = 15.9 Hz, 1 H), 3.80 (s, 3 H), 3.54 (s, 3 H), 2.87 (dd, J = 16.6, 5.5 Hz, 1 H), 2.70 (dd, J = 16.6, 6.9 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3)δ = 170.5, 166.9, 140.8, 137.2, 134.5, 134.3, 133.7, 133.3, 130.3, 129.6, 128.9, 127.1, 126.2, 124.2, 121.5, 120.9, 58.3, 52.0, 51.8, 45.0, 39.1 ppm. HRMS calculated for C21H21NO6SNa [M + Na]+ 438.0987; found 438.0981 (TOF MS EI+).
General Procedure (C) for the Synthesis of 1,1-Dioxido-1,2-benzisothiazol-3-acetamide Derivatives 29−31
Into a 1-dram vial was added amine (2.73 × 10−5 mol), Et3N (5.47 × 10−5 mol) and dry DMF (0.1 mL) were the reaction mixture was stirred at room temp. for 15 min. Then, α-bromobenzenesulfonyl chlorides (2.73 × 10−5 mol) were added and the reaction mixture was stirred for 2 h. Then, Et3N (5.482.73 × 10−5 mol), Bu4N (7.45 mg, 2.73 × 10−5 mol), Pd2(dba)3·CHCl3 (2 mol-%, 2.56 mg, 0.056 × 10−5 mol) and dry DMF (0.14 mL) were added to the reaction vial. After stirring for 5 min at room temp., acrylic acid (8.19 × 10−5 mol mmol) was added and the reaction vial was placed immediately into a preheated reaction block. The reaction mixture was stirred at 110 °C for 14 h after which time the reaction was cooled and concentrated. The crude was then solvated in dry DCM (0.1 mL), to which N-methylmorpholine (35 μL, 4.09 × 10−5 mol) was added. After stirring for 5 min a solution of OACC50 (17 mg, 5.46 × 10−5 mol) in dry DCM (0.1 mL) was added and the reaction mixture stirred for 10 h at room temp. Then, the crude was absorbed onto silica and purified by flash chromatography (hexane/EtOAc).
Compound 29
Prepared using general method (C) using benzenesulfonyl chloride, cyclopentylamine, acrylic acid and morpholine. The crude material was purified by flash chromatography (hexane/EtOAc, 1:1) to yield 5.92 mg of 30 (56%) as a white solid. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.75 (d, J = 8.1 Hz, 1 H), 7.54−7.57 (m, 1 H), 7.49−7.54 (m, 2 H), 5.13−5.17 (m, 1 H), 3.96 (h, J = 7.6 Hz, 1 H), 3.71−3.74 (m, 1 H), 3.67 (t, J = 4.7 Hz, 2 H), 3.54−3.58 (m, 2 H), 3.40−3.56 (m, 2 H), 3.29−3.33 (m, 1 H), 3.04 (dd, J = 15.3, 5.4, Hz, 1 H), 2.71 (dd, J = 15.3, 7.9, Hz, 1 H), 2.02−2.08 (m, 1 H), 1.91−1.96 (m, 1 H), 1.71−1.78 (m, 2 H), 1.56−1.68 (m, 4 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 168.4, 139.6, 136.3, 132.8, 129.3, 124.8, 121.0, 66.7, 58.4, 57.1, 46.2, 42.1, 38.8, 30.8, 28.3, 23.4, 22.8 ppm. HRMS calculated for C18H24N2O4SNa [M + Na]+ 387.1354; found 387.1354 (TOF MS EI+).
Compound 30
Prepared using general method (C) using benzenesulfonyl chloride, cyclohexylamine, acrylic acid and pyridin-3-amine. The crude material was purified by flash chromatography (EtOAc) to yield 4.3 mg of 31 (43%) as a off white solid. FTIR (neat): . 1H NMR (500 MHz, MeOD): δ = 8.74 (s, 1 H), 8.28 (d, J = 3.2 Hz, 1 H), 8.12 (dt, J = 8.4, 1.5 Hz, 1 H), 7.73 (d, J = 7.7 Hz, 1 H), 7.64 (dt, J = 7.6, 1.0 Hz, 1 H), 7.57 (t, J = 7.5 Hz, 1 H), 7.54 (t, J = 7.5 Hz, 1 H), 7.41 (dd, J = 8.3, 4.8 Hz, 1 H), 5.26 (t, J = 6.6 Hz, 1 H), 3.57 (tt, J = 11.8, 3.6 Hz, 1 H), 3.07 (dd, J = 14.9, 6.1 Hz, 1 H), 3.07 (dd, J = 14.9, 6.1 Hz, 1 H), 2.85 (dd, J = 14.9, 7.1 Hz, 1 H), 1.97 (d, J = 12.6 Hz, 1 H), 1.90 (d, J = 12.3 Hz, 1 H), 1.69−1.83 (m, 4 H), 1.63 (d, J = 12.7 Hz, 1 H), 1.23−1.37 (m, 2 H), 1.18 (tt, J = 12.7, 2.9 Hz, 1 H) ppm. 13C NMR (125 MHz, MeOD): δ = 171.0, 165.2 (d, JC-F = 254.1 Hz), 145.6, 142.0, 140.4, 137.8, 134.3, 130.8, 129.2, 125.8, 121.7,58.7, 57.4, 44.9, 32.8, 31.4, 27.3, 27.2, 26.6 ppm. HRMS calculated for C20H24N3O3S [M + H]+ 386.1538; found 386.1547 (TOF MS EI+).
Compound 31
Prepared using general method (C) using 4-fluorobenzenesulfonyl chloride, 4-methoxybenzylamine, acrylic acid and piperidine. The crude material was purified by flash chromatography (hexane/EtOAc, 1:1) to yield 6.46 mg of 32 (52%) as a white solid. FTIR (neat): . 1H NMR (500 MHz, CDCl3): δ = 7.79 (dd, J = 8.4, 4.7 Hz, 1 H), 7.33 (d, J = 8.6 Hz, 2 H), 7.21 (d, J = 8.6 Hz, 2 H), 6.85 (d, J = 8.7 Hz, 2 H), 5.06 (dd, J = 7.8, 5.0 Hz, 1 H), 4.47 (d, J = 3.7 Hz, 2 H), 3.65 (s, 3 H), 3.47−3.58 (m, 2 H), 3.11−3.21 (m, 2 H), 2.81 (dd, J = 16.2, 5.0 Hz, 1 H), 2.57 (dd, J = 16.2, 7.9 Hz, 1 H), 1.57−1.62 (m, 2 H), 1.48−1.54 (m, 2 H), 1.34−1.42 (m, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 167.1, 165.2 (d, JC,F = 2.1 Hz), 159.39, 131.0, 130.0, 127.3, 123.4, 123.3, 117.2, 117.0, 114.2, 112.7, 112.5, 55.3, 47.0, 46.5, 42.9, 38.2, 26.2, 25.5, 24.3 ppm. HRMS calculated for C22H25FN2O4SNa [M + Na]+ 455.1417; found 455.1422 (TOF MS EI+).
Supplementary Material
Acknowledgments
This work was generously supported by funds provided by the National Institutes of Health (NIH), Center for Chemical Methodologies and Library Development at the University of Kansas (KU-CMLD) (P50 GM069663), Pilot Scale Libraries Program (P41 GM076302) and NIH COBRE award (P20 RR015563). Undergraduate funding was provided via an NIH K-INBRE award (P20 R016475) (to K. Y.) and the KU Center of Research (to K. Y.).
Footnotes
Supporting information for this article is available on the WWW under http://www.eurjoc.org or from the author.
Supporting Information (see also the footnote on the first page of this article): Copies of NMR spectra.
References
- 1.For reviews on domino processes, see: Padwa A, Bur SK. Tetrahedron. 2007;63:5341–5378. doi: 10.1016/j.tet.2007.03.158.; Enders D, Grondal C, Hüttl MRM. Angew. Chem. Int. Ed. 2007;46:1570–1581. doi: 10.1002/anie.200603129.;Tietze LF. Chem. Rev. 1996;96:115–136. doi: 10.1021/cr950027e.;Tietze LF, Beifuss U. Angew. Chem. Int. Ed. Engl. 1993;32:131–163.
- 2.A domino reaction is defined by Tietze as a “transformation of two or more bond-forming reactions under identical reaction conditions, in which the latter transformations take place at the functionalities obtained in the former bond forming reactions.” Tietze LF, Brasche G, Gericke KM, editors. Domino Reactions in Organic Synthesis. Wiley-VCH; Weinheim, Germany: 2006.
- 3.a Fustero S, Jiménez D, Sánchez-Roselló M, del Pozo C. J. Am. Chem. Soc. 2007;129:6700–6701. doi: 10.1021/ja0709829. [DOI] [PubMed] [Google Scholar]; b Bi H-P, Liu X-Y, Gou F-R, Guo L-N, Duan X-H, Shu X-Z, Liang Y-M. Angew. Chem. Int. Ed. 2007;46:7068–7071. doi: 10.1002/anie.200702238. [DOI] [PubMed] [Google Scholar]; c Gai X, Grigg R, Khamnaen T, Rajviroongit S, Sridharan V, Zhang L, Collard S, Keep A. Tetrahedron Lett. 2003;44:7441–7443. [Google Scholar]; d Zeng Y, Reddy DS, Hirt E, Aubé J. Org. Lett. 2004;6:4993–4995. doi: 10.1021/ol047809r. [DOI] [PubMed] [Google Scholar]; e Kirschbaum S, Waldmann H. Tetrahedron Lett. 1997;38:2829–2832. [Google Scholar]
- 4.a Bandini M, Eichholzer A, Monari M, Piccinelli F, Umani-Romcho A. Eur. J. Org. Chem. 2007;18:2917–2920. [Google Scholar]; b Perdicchia D, Jørgensen KA. J. Org. Chem. 2007;72:3565–3568. doi: 10.1021/jo0626717. [DOI] [PubMed] [Google Scholar]; c Balme G, Bouyssi N. Metal-catalyzed cascade reactions. In: Müller TJJ, editor. Topics in Organometallic Chemistry. Vol. 19. Springer Verlag; Berlin: 2006. pp. 115–149. [Google Scholar]
- 5.a Barr N, Bartley JP, Clark PW, Dyke P, Dunstan SF. J. Organomet. Chem. 1986;302:117. [Google Scholar]; b Dyker G, Grundt P. Tetrahedron Lett. 1996;37:619–622. [Google Scholar]; c Khan Md. W., Reza AFGM. Tetrahedron. 2005;61:11204–11210. [Google Scholar]
- 6.Rao BK, Hamor GH. Pharm. Sci. 1969:628–630. doi: 10.1002/jps.2600580528. [DOI] [PubMed] [Google Scholar]
- 7.a Drews J. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]; b Scozzafava A, Owa T, Mastrolorenzo A, Supuran CT. Curr. Med. Chem. 2003;10:925–953. doi: 10.2174/0929867033457647. [DOI] [PubMed] [Google Scholar]
- 8.Levy L. Drugs Future. 1992;17:451–454. [Google Scholar]
- 9.Valente C, Guedes RC, Moreira R, Iley J, Gut J, Rosenthal PJ. Bioorg. Med. Chem. Lett. 2006;16:4115–4119. doi: 10.1016/j.bmcl.2006.04.079. [DOI] [PubMed] [Google Scholar]
- 10.Zhuang L, Wai JS, Embrey MW, Fisher TE, Egbertson MS, Payne LS, Guare JP, Jr., Vacca JP, Hazuda DJ, Felock PJ, Wolfe AL, Stillmock KA, Witmer MV, Moyer G, Schleif WA, Gabryelski LJ, Leonard YM, Lynch JJ, Jr., Michelson SR, Young SD. J. Med. Chem. 2003;46:453–456. doi: 10.1021/jm025553u. [DOI] [PubMed] [Google Scholar]
- 11.Misu Y, Togo H. Org. Biomol. Chem. 2003;1:1342–1346. doi: 10.1039/b301330h. [DOI] [PubMed] [Google Scholar]
- 12.a Bravo RD, Canepa AA. Synth. Commun. 2002;32:3675–3680. [Google Scholar]; b Orazi OO, Corral RA, Bravo R. Heteroat. Chem. 1986;23:1701–1708. [Google Scholar]; c Katritzky AR, Wu J, Rachwal S, Rachwal B, Macomber DW, Smith TP. Org. Prep. Proced. Int. 1992;24:463–467. [Google Scholar]
- 13.Lee J, Zhong Y-L, Reamer RA, Askin D. Org. Lett. 2003;5:4175–4177. doi: 10.1021/ol0356183. [DOI] [PubMed] [Google Scholar]
- 14.Chiacchio U, Corsaro A, Rescifina A, Bkaithan M, Grassi G, Piperno A, Privitera T, Romeo TG. Tetrahedron. 2001;57:3425–3433. [Google Scholar]
- 15.a Metz P, Seng D, Fröhlich R. Synlett. 1996:741–742. [Google Scholar]; b Plietker B, Seng D, Fröhlich R, Metz P. Tetrahedron. 2000;56:873–879. [Google Scholar]; c Rogatchov VO, Bernsmann H, Schwab P, Fröhlich R, Wibbeling B, Metz P. Tetrahedron Lett. 2002;43:4753–4756. [Google Scholar]; d Greig IR, Tozer MJ, Wright PT. Org. Lett. 2001;3:369–371. doi: 10.1021/ol006863e. [DOI] [PubMed] [Google Scholar]
- 16.a McReynolds MD, Dougherty JM, Hanson PR. Chem. Rev. 2004;104:2239–2258. doi: 10.1021/cr020109k. [DOI] [PubMed] [Google Scholar]; b Moriggi J-M, Brown LJ, Castro JL, Brown RCD. Org. Biomol. Chem. 2004;2:835–844. doi: 10.1039/b313686h. [DOI] [PubMed] [Google Scholar]; c Freitag D, Schwab P, Metz P. Tetrahedron Lett. 2004;45:3589–3592. [Google Scholar]; d Karsch S, Freitag D, Schwab P, Metz P. Synthesis. 2004;10:1696–1712. [Google Scholar]; e Jiménez-Hopkins M, Hanson PR. Org. Lett. 2008;10:2223–2226. doi: 10.1021/ol800649n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a Merten S, Fröhlich R, Kataeva O, Metz P. Adv. Synth. Catal. 2005;347:754–758. [Google Scholar]; b Vasudevan A, Tseng PS, Djuric SW. Tetrahedron Lett. 2006;47:8591–8593. [Google Scholar]; c Paquette LA, Dura RD, Fosnaugh N, Marshall S. J. Org. Chem. 2006;71:8438–8445. doi: 10.1021/jo061404y. [DOI] [PubMed] [Google Scholar]; d Paquette LA, Barton WRS, Gallucci JC. Org. Lett. 2004;6:1313–1315. doi: 10.1021/ol049679s. [DOI] [PubMed] [Google Scholar]
- 18.Liu X-Y, Li C-H, Che C-M. Org. Lett. 2006;8:2707–2710. doi: 10.1021/ol060719x. [DOI] [PubMed] [Google Scholar]
- 19.a Dauban P, Dodd RH. Org. Lett. 2000;2:2327–2329. doi: 10.1021/ol000130c. [DOI] [PubMed] [Google Scholar]; b Dauban P, Sanière L, Tarrade A, Dodd RH. J. Am. Chem. Soc. 2001;123:7707–7708. doi: 10.1021/ja010968a. [DOI] [PubMed] [Google Scholar]; c Sherman ES, Chemler SR, Tan TB, Gerlits O. Org. Lett. 2004;6:1573–1575. doi: 10.1021/ol049702+. [DOI] [PubMed] [Google Scholar]; d Zeng W, Chemler SR. J. Am. Chem. Soc. 2007;129:12948–12949. doi: 10.1021/ja0762240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a Liang J-L, Yuan S-X, Chan PWH, Che C-M. Org. Lett. 2002;4:4507–4510. doi: 10.1021/ol0270475. [DOI] [PubMed] [Google Scholar]; b Padwa A, Flick AC, Leverett CA, Stengel T. J. Org. Chem. 2004;69:6377–6386. doi: 10.1021/jo048990k. [DOI] [PubMed] [Google Scholar]
- 21.Zhou A, Hanson PR. Org. Lett. 2008;10:2551–2554. [Google Scholar]
- 22.Sulfonamide 1 was submitted to the one pot conditions at room temp. without Pd2(dba)3·CHCl3, giving the aza-Michael adduct after 2 hours.
- 23.a Artok L, Bulut H. Tetrahedron Lett. 2004;45:3881–3884. [Google Scholar]; b Botella L, Najera C. Tetrahedron Lett. 2004;45:1833–1836. [Google Scholar]; c Gittins ID, Caruso F. Angew. Chem. Int. Ed. 2001;40:3001–3004. doi: 10.1002/1521-3773(20010817)40:16<3001::AID-ANIE3001>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 24.Workup consisted of removal of DMF, suspension of the crude reaction mixture in EtOAc and filtration through a SiO2 SPE to remove Pd and organic salts.
- 25.It is worth noting that in a limited number of cases the acyclic intermediate resulting from the Heck reaction was observed in the crude mixture in small quantities (14: R1 = H, R2 = tBu, R3 = OMe); experimental data for 14 is provided.
- 26.As previously noted in the introductory paragraph, it has been reported that this reaction does proceed but requires days and ultimately gave low yield unless the iodobenzamides were utilized instead of the aryl bromides.
- 27.a Kanamitsu N, Osaki T, Itsuji Y, Yoshimura M, Tsujimoto H, Soga M. Chem. Pharm. Bull. 2007;55:1682–1688. doi: 10.1248/cpb.55.1682. [DOI] [PubMed] [Google Scholar]; b Dudash J, Rybczynski P, Urbanski M, Xiang A, Zeck R, Zhang X, Zhang Y. U. S. Pat. Appl. Publ. 2007:65. [Google Scholar]; c Schubert G, Rieke-Keil J, Zapp J, Kleemann H-W, Hanna R, Huang B-G, Wu X-D, Gourand Y. U. S. Pat. Appl. Publ. 2005:24. [Google Scholar]
- 28.Zhang M, Vedantham P, Flynn DL, Hanson PR. J. Org. Chem. 2004;69:8340–8344. doi: 10.1021/jo048870c. [DOI] [PubMed] [Google Scholar]
- 29.Attempts to develop a “true” one-pot process starting from the corresponding sulfonamide were investigated. However it was discovered that lowering the equivalents of acrylic acid from 3 to 1−1.5 significantly reduced the amount of cyclic acid formed and hence the amount of corresponding amide isolated.
- 30.Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 31.Ferraccioli R, Carenzi D, Rombola O, Catellani M. Org. Lett. 2004;6:4759–4762. doi: 10.1021/ol0479949. [DOI] [PubMed] [Google Scholar]
- 32.Marion F, Coulomb J, Servais A, Courillon C, Fensterbank L, Malacria M. Tetrahedron. 2006;62:3856–3871. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.