

Abstract
The recombinant, catalytically active light chain of botulinum toxin type A was evaluated as a potential vaccine candidate. Previous studies have shown that the light chain can elicit protective immunity in vivo. [5], but the underlying basis for this observation was not determined. In the present study, antibodies directed against the light chain were shown to act at three different sites in the body to produce neutralization. Firstly, these antibodies acted to block toxin absorption into the body. This was demonstrated in vitro, in studies on binding and transport of toxin across epithelial monolayers, and in vivo, in studies on inhalation poisoning. Secondly, anti-light chain antibodies acted to promote clearance of toxin from the general circulation. This was demonstrated in vivo in studies on toxin levels in blood and in parallel studies on toxin accumulation in liver and spleen. Finally, anti-light chain antibodies acted to protect cholinergic nerves from botulinum toxin action. This was demonstrated in two types of in vitro assays: rate of paralysis of murine phrenic nerve-hemidiaphragm preparations and extent of binding to Neuro-2a cells. When taken together, these data show that anti-light chain antibodies can evoke three layers of protection against botulinum toxin.
Keywords: Botulinum toxin, Botulism vaccine, Neutralization
1. Introduction
Botulinum toxin is believed to pose a threat as a bioweapon that can be used against civilian (bioterrorism) and military (biological warfare) populations [1]. Understandably, this has led to efforts to develop a vaccine that will have clinical utility. Most of this effort has focused on the use of recombinant polypeptides that: a.) are devoid of the neurotoxicity that is characteristic of the holotoxin, but b.) are immunogenic and can evoke neutralizing antibodies that inactivate the holotoxin [2].
Botulinum toxin is a protein that has a molecular mass of approximately 150,000 Da. The protein is synthesized as a single chain molecule of negligible toxicity. During post-translational processing the toxin undergoes proteolysis (“nicking”) to yield a heavy chain polypeptide (mw ca. 100,000 Da) and a light chain polypeptide (mw ca. 50,000 Da) linked by a disulfide bond. It is the dichain molecule that is extraordinarily potent and that causes blockade of cholinergic transmission [3,4].
In the recent past, it has been shown that the botulinum holotoxin can be expressed as a mutant that does not poison cholinergic nerve endings, but does evoke an immune response [5]. Thus, an inactive variant of the holotoxin was shown to possess the properties of a vaccine when administered by either the mucosal (oral) or parenteral (subcutaneous) routes. Both routes of administration evoked a circulating titer of antibodies adequate to neutralize large challenge doses of native toxin [5].
One potential drawback to the use of a recombinant holotoxin vaccine is the size of the expression product. It is a common observation that large proteins are expressed at yields lower than those of smaller proteins. Hence, it is logical to determine whether a recombinant polypeptide that represents only a portion of the holotoxin could serve as an efficient vaccine.
Several laboratories have now generated a substantial number of synthetic and recombinant polypeptides that have been tested as vaccines [2], and one of these appears to have emerged as a favored candidate. The carboxyterminal half of the heavy chain (variously referred to as HC, HC50 and C-fragment) has been shown to be an efficacious vaccine when administered by the parenteral [6,7] or mucosal [8] routes. This polypeptide can evoke protection against challenge doses of toxin as high as 1 × 105 mouse LD50.
At the time that this polypeptide was first introduced as a potential vaccine, there were several reasons that appeared to justify the choice. One of the more important of these relates to structure-function relationships. The carboxyterminal half of the heavy chain possesses the structural determinants responsible for binding to cholinergic nerve endings [3]. This suggests that antiserum directed against this polypeptide should possess one or more clonal populations of antibodies that would associate with and occlude the receptor binding domain. Occlusion of this domain would be a plausible mechanism by which antibodies could abolish the cholinergic blocking properties of the holotoxin.
There is ample evidence to demonstrate that antibodies directed against the carboxyterminal half of the heavy chain can block the neuroparalytic actions of botulinum toxin [2]. However, this important observation must be seen in the context that occlusion of the receptor binding domain is not the only site at which antibodies can act. It is now known that mucosal antibodies can block toxin absorption into the body [8,9], and systemic antibodies can enhance toxin clearance from the general circulation [10]. These two mechanisms prevent toxin from reaching peripheral nerve endings, and hence they diminish the need for occlusion of the neuronal receptor binding domain.
Given the multiple sites and mechanisms of action of neutralizing antibodies, one can reasonably ask whether the carboxyterminal half of the heavy chain is a uniquely efficacious vaccine, or alternatively, whether it is merely one of several potentially efficacious vaccines. To address this question, the authors evaluated the opposite end of the toxin molecule, the recombinant light chain, as a putative vaccine candidate. This decision was based on two previous observations. Firstly, the light chain component of the toxin has already been shown to evoke neutralizing antibodies, although the underlying mechanisms were not determined [5]. Secondly, the light chain has recently been shown to evoke antibodies that possess the counterintuitive property of blocking toxin action at nerve endings [11]. These combined findings form a rational basis for seeking to determine where and how anti-light chain antibodies can neutralize botulinum toxin. This in turn could provide essential information for deciding whether the light chain or any related polypeptide should be viewed as an authentic vaccine candidate.
2. Materials and methods
2.1. Botulinum toxin
Botulinum toxin types A, B and E (homogeneous neurotoxins) were purchased from Metabiologics (Madison, WI). Specific toxicity of the material was typically in the range of 2.2 to 2.4 × 108 mouse intraperintoneal LD50/mg.
An aliquot of the pure toxin was iodinated with Bolton-Hunter reagent, as previously reported [9,10], and retention of toxicity was confirmed, also as previously described. The concentration of the labeled toxin was determined spectrophotometrically at 278 nm using the following relationship: 1.63 A278 = 1 mg/ml [12].
2.2. Plasmid construction and bacterial expression of recombinant polypeptides
Gene segments encoding the carboxyterminal half of the heavy chain (HC50 domain) of botulinum toxin type A (strain 62A, accession no. M30196, amino acids 865 – 1296) were cloned into 6xHis vector pQE30 (Qiagen; Valencia, CA) yielding the expression plasmid pQE-HC50/A. Gene segments encoding the light chain fragment (LC) of the toxin (strain 62A, amino acids 01 – 437) were cloned into 6xHis vector pET30a+ (Novagen; Gibstown, NJ) yielding the expression plasmid pET-LC/A.
Escherichia coli [BL21-codon plus (DE3)-RIL (Stratagene; La Jolla, CA] was used as host strain for expression of recombinant polypeptides. Cells which were transformed with plasmid pQE-HC50/A or pET-LC/A were grown in Terrific broth (1.2% peptone, 2.4% yeast extract, 0.94% K2HPO4 and 0.22% KH2PO4) (Difco; Sparks, MD) at 37° C, with shaking to an A600 of 0.6 to 0.8. Isopropyl ß-D-thiogalactopyranoside at a final concentration of 0.5 mM was added and incubation was continued for ca. 12 hr at 20° C. Bacteria from 1 liter of induced culture were harvested by centrifugation (6,000 × g, 15 min) at 4° C.
2.3. Purification of protein
Bacterial cells were suspended in 200 ml of bacterial protein extract reagent, B-PER (Pierce; Rockford, IL) at 4° C. Lysozyme (Sigma; St. Louis, MO) at a final concentration of 0.1 mg/ml, DNAse (Sigma) at a final concentration of 0.01 mg/ml, and protease inhibitor cocktail tablet (Roche; Manheim, Germany) were added to the cell suspension and incubated on a rotating shaker for 2 hr. Four hundred ml of 50 mM sodium phosphate containing 300 mM NaCl, pH 8.0, was added to the lysed cell suspension and allowed to stand for 30 min. The suspension was centrifuged at 27,000 × g for 40 min to remove precipitate.
The clear supernatant was loaded onto a 5 ml column of Ni-NTA superflow (Qiagen) which was equilibrated with 50 mM sodium phosphate containing 300 mM NaCl, pH 8.0. The column was washed with 50 volumes of washing buffer (50 mM sodium phosphate containing 300 mM NaCl, and 20 mM imidazole, pH 8.0). Bound protein was eluted from the column with a gradient of increasing imidazole (100 ml of 50 mM sodium phosphate containing 300 mM NaCl and 20 mM imidazole, and 100 ml of 50 mM sodium phosphate containing 300 mM NaCl and 250 mM imidazole, pH 8.0). The active fractions (at ~80 mM imidazole) of HC50/A or LC/A were pooled.
HC50/A was further purified by cation exchange column chromatography, CM Sepharose fast flow (Amersham Bioscience; Piscataway, NJ). Active fractions from Ni-NTA chromatography were dialyzed against 50 mM sodium phosphate, pH 6.8. The dialysate was centrifuged at 27,000 × g for 30 min to remove precipitate. The clear supernatant was loaded onto a 4 ml CM Sepharose column equilibrated with 50 mM sodium phosphate, pH 6.8, then washed with 50 volumes of the same buffer. Bound protein was eluted from the column with a gradient of increasing NaCl (50 ml of 50 mM sodium phosphate and 50 ml of 50 mM sodium phosphate with 500 mM NaCl, pH 6.8). The active fractions (at ~200 mM NaCl) were pooled and dialyzed against 50 mM sodium phosphate, pH 7.4.
LC/A was purified further by anion exchange column chromatography, Q Sepharose fast flow (Amersham Bioscience; Piscataway, NJ). Active fractions from Ni-NTA chromatography were dialyzed against buffer A (50 mM Tris/HCl, 100 mM NaCl and 5 mM ß-mercaptoethanol; pH 9.0). The dialysate was centrifuged at 27,000 × g for 30 min to remove precipitate. The clear supernatant was loaded onto a 4 ml Q Sepharose column equilibrated and washed with buffer A. Bound protein was eluted from the column using higher concentrations of NaCl in buffer A. The active fractions were pooled and dialyzed against 25 mM HEPES with 5 mM DTT, pH 8.5.
The purity of HC50/A and LC/A were confirmed on 10% SDS polyacrylamide gel electrophoresis and found to be more than 98% homogeneous. Approximately 15-20 mg of pure HC50/A or LC/A protein was obtained from each 1 L of bacterial culture.
2.4. SDS-Page and immunoblot analysis
SDS-PAGE gel electrophoresis was performed according to the methods described by Laemmli [13]. The holotoxin or recombinant polypeptides derived from the toxin were run on 4 to 15% separating gels, then visualized by staining with Commassie Blue.
For Western blotting, separated proteins were transferred to electroblot nitrocellulose membranes that had been treated with 5% skim milk in phosphate buffered saline containing 0.05% Tween-20 to block nonspecific binding sites. Membranes were probed with rabbit primary antibody against either heavy chain or light chain (dilutions 1: 5,000). After repeated washing, the membranes were counter-probed with rabbit IgG-HRP conjugated antibodies (dilutions 1: 10,000) and washed to remove unbound secondary antibodies. The proteins were subsequently visualized by developing membranes with Pierce SuperSignal ECL detection system. Films were scanned and edited using Adobe Photoshop.
2.5. Animals
New Zealand white rabbits (female; 2-3 kg; antibody experiments) were purchased from Covance (Denver, PA). Swiss Webster mice (female; 20-25 g; pharmacokinetic experiments) were purchased from Ace Animals (Boyertown, PA). Rabbits and mice were housed separately in the animal facility at Thomas Jefferson University, and procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee.
2.6. Parenteral vaccination with light chain
Antiserum was obtained by immunizing rabbits. An initial injection of Alum-adsorbed antigen was given subcutaneously to rabbits (50 μg); three subsequent injections of adsorbed material (50 μg each) were given at 2, 4 and 8 weeks. Fourteen days after the final booster, aliquots of blood were drawn and the circulating titer of IgG was determined by enzyme-linked immunosorbent assay (ELISA). Briefly, flat-bottom, 96-well Corning plates were coated with the light chain of botulinum toxin type A (300 ng/well) and incubated at 4°C overnight, followed by washing with phosphate-buffered saline containing 0.1% Tween-20 (pH 7.4). The plates were blocked with 1% bovine serum albumin. Two-fold serial dilutions of serum samples (40 μl) were added to the plates and incubated at 37°C for 60 min.
IgG titers were determined using peroxidase-conjugated goat anti-rabbit IgG. Secondary antibodies were diluted 1:1,000 in phosphate-buffered saline. The primary and secondary antibodies were incubated for 30 min at 37°C, after which 2, 2-azinobis (3-ethylbenthiazoline-6-sulfonic acid) was added as a substrate, and the plates were incubated for an additional 30 min at 37°C. The end point titers were the reciprocals of the last dilutions yielding an absorbance at 405 nm that was greater than the control value (pre-immune serum).
2.7. Blood and organ levels of toxin
125I-Botulinum toxin was administered intravenously to mice via the tail vein, and at various times thereafter specimens were obtained by retro-orbital bleeding. Experiments were done in control animals (toxin only) and in experimental animals (toxin preincubated with antiserum from vaccinated animals). The goal of the experiments was to determine the effect of antibodies on the rate of toxin clearance from the general circulation. The amount of antibody that was used was adequate to completely neutralize the administered dose of toxin (see Results).
A related set of experiments was performed to determine the extent of toxin accumulation in tissues. As mentioned above, experiments were done in control animals (minus antiserum) and in experimental animals (plus antiserum). Sixty minutes after toxin administration, mice were anesthetized by administration of Nembutal® sodium (50 mg/kg) and the thorax was opened to expose the heart. A butterfly needle (23G) was inserted into the left ventricle, an incision was made in the right ventricle, and the body was perfused with a heparinized solution of phosphate buffered saline (50-60 ml). Livers and spleens were removed from the body, and the accumulation of iodinated toxin in each organ was determined.
2.8. Immunoprecipitation and autoradiography
To ensure homogeneity of the injected material, iodinated toxin was subjected to gel chromatography (G-25) prior to administration, thus eliminating free iodine or small degradation products. Biological samples obtained from injected animals were also subjected to gel chromatography. In addition, a combination of studies that entailed immunoprecipitation, enzymatic assays and monitoring of neuromuscular transmission were done to ensure the structural and functional integrity of the toxin.
2.9. In vitro assays
Two in vitro assays were used as models to study the ability of antibodies to block toxin binding to cell membranes. Immortalized human gut epithelial cells (T-84) were used as models for binding, transcytosis and release of toxin that is associated with the absorption process. Murine phrenic nerve-hemidiaphragms were used as models for binding, internalization and catalytic activity that is associated with toxin-induced neuromuscular blockade.
The techniques for measuring [8,10,14] and visualizing [15] the movement of botulinum toxin across epithelial monolayers have been described in detail. In the present study, monolayers were grown until transepithelial electrical resistance was at or near an asymptote (ca. 12 to 15 days). Botulinum toxin type A (1 × 10-8 M) in the continuous absence or presence of antibody was added to the solution bathing the apical surfaces of cells for a period of 18 hr. The solution bathing the basal surface was collected, concentrated, and then run in Western gels to detect toxin that had penetrated epithelial monolayers.
The techniques for quantifying onset of toxin-induced neuromuscular blockade have also been described in detail [8,9,16]. For this study, murine phrenic nerve-hemidiaphragm preparations were excised and suspended in tissue baths containing physiological medium. Phrenic nerves were stimulated, and hemidiaphragm twitch responses were monitored. In a typical experiment, neurogenic responses were evoked for ca. 30 min to achieve a stable magnitude of twitch. Toxin (1 × 10-11M) in the absence or presence of antibody was added to tissues, and the time necessary for 90% reduction in twitch responses was monitored.
2.10. Visualization of cell binding
The binding of botulinum toxin in the absence or presence of antibodies was studied on Neuro-2a cells. Neural cells were plated on poly-D-lysine glass coverslips in the medium recommended by the supplier (ATCC, Manasses, VA) at densities of 1 - 2 × 104 cells/cm2. On day 3, cells were washed with phosphate-buffered saline, then resuspended in the same medium. For binding assays, culture wells were placed on an ice bath for 30 min. Toxin alone or toxin that had been preincubated with antiserum was added to cells, and incubation on ice was continued for an additional 60 min. After exposure to toxin or toxin plus antibody, cells were washed 3x with phosphate-buffered saline and fixed with 4% paraformaldehyde.
Two paradigms were used to determine the effects of antibodies. In the first, toxin was preincubated with antibodies directed against the carboxyterminal half of the heavy chain (HC50 domain; 60 min; room temperature). This mixture was then incubated with Neuro-2a cells as just described. The extent of binding was determined by probing cells with anti-light chain antibodies. In the second paradigm the order of antibody use was reversed. Thus, toxin was preincubated with anti-light chain antibodies, and the extent of subsequent binding was probed with anti-HC50 antibodies.
Immunochemical staining was essentially as described by Joshi [17]. In brief, fixed cells on coverslips were blocked with 5% bovine serum albumin in phosphate buffered saline for 30 min at room temperature, then washed 3x in the same medium. Cells were incubated with primary antibody for 45 min, followed by 3 washes with phosphate buffered saline. Cells were probed with secondary antibody conjugated with FITC. After additional washing, cells were mounted with Fluoromount and examined under confocal microscopy with a Zeiss LSM-510 META system. The final images were edited in Photoshop CS.
2.11. Enzyme assay
The endoprotease activity of the holotoxin was measured using a fluorescent resonance energy transfer (FRET) assay that employed a synthetic substrate that has been previously described [60-SELDDRADALQAGASQFETSAAKLKRKYWWKNLK-95; cleavage site, Q76-F77; 18]. Botulinum toxin (10 μl) or recombinant light chain (10 μl) was added to reaction buffer (final volume, 100 μl; 20 mM Hepes, pH 7.5; 0.25 mM ZnCl2, 1.25 mM DTT, 0.05% Tween-20) that contained the fluorescent substrate (10 μM; gift from Dr. James Schmidt, USARIID, Fort Detrick, MD). The mixture was incubated at 37°C for 2 hr, after which the reaction was terminated by addition of trifluoracetic acid to a final concentration of 0.5%. The resulting fluorescence was measured at room temperature in a Luminescence Spectrometer LS 50B (Perkin Elmer, Norwalk, CT) using FL Winlab software. Measurements were made at Ex390 and Em480, with an integration time of 3 sec. The results were expressed as arbitrary fluorescence units.
3. RESULTS
3.1. Parenteral vaccination
The light chain component of botulinum toxin type A was administered to rabbits, as described under Methods. At the end of the immunization protocol, an aliquot of serum was used for ELISA assays. The results indicated that the 50% dilution titer for the antiserum was ca. 3 × 104.
3.2. Specificity of the antiserum
Western blots were generated using botulinum serotypes A, B and E as antigens (Figure 1A). Antiserum against light chain recognized intact serotype A, but not serotypes B or E. Westerns were also run with recombinant polypeptides representing the HC50 domain and the light chain of serotype A. As expected, antiserum recognized only the latter (Figure 1B).
Figure 1.
Anti-light chain serum was examined for its ability to interact with pure holotoxins under non-reducing conditions (Part A). The antiserum recognized serotype A (lane a), but not serotypes B (lane b) or E (lane c). The antiserum was also examined for its ability to interact with recombinant polypeptides (Part B). The antiserum did not recognize the HC50 polypeptide (lane a) but it did react with the light chain (lane b).
3.3. Anti-light chain antibodies neutralize the toxicity of botulinum toxin type A
Various amounts of antiserum (0.1 to 10.0 μl) were administered i.v. to mice (group n=5 per dose). After 60 min, animals were challenged i.v. with 5 ng (ca 1 × 103 LD50) of pure toxin. As shown in Figure 2, anti-light chain serum produced dose-dependent loss of toxicity. At doses of 0.1 μl and 0.3 μl there were no survivors. At doses of 3.0 μl and 10.0 μl all of the animals survived. The apparent EC50 for antibody-mediated loss of toxicity was ca 2.0 μl.
Figure 2.
Mice (n=5 per data point) were injected i.v. with various amounts of anti-light chain antiserum (0.1 to 10 microliters). After an interval of 60 min., to allow for distribution of antibodies throughout the circulation, mice were injected i.v. with a multilethal dose of pure botulinum toxin type A (ca 1×103 LD50). The antiserum produced dose-dependent loss of toxicity, with an apparent EC50 of 2 microliters.
3.4. Anti-light chain antibodies antagonize transcytosis across epithelial cells
Botulinum toxin has been shown to bind and penetrate human gut and airway epithelial barriers. Most of the work on epithelial transcytosis has been done on T-84 cells, so these cells were selected for the present study.
Monolayers of immortalized human epithelial cells were grown in a Transwell apparatus, and transepithelial electrical resistance was measured to ensure patency of the monolayers. When resistance approached asymptote (350-400 Ohm; ca. 13 days), transcytosis experiments were done. Toxin (1 × 10-8 M) was added to the apical surface of cells, and accumulation of toxin on the basal side of cells was monitored. The experiments were done in the absence or presence of anti-light chain antiserum (40 μl). Antiserum directed against the HC50 domain of botulinum toxin type A was used as a positive control.
Structure-function relationship studies have shown that the HC50 domain possesses the minimum essential determinants for toxin binding and transcytosis across epithelial cells [14]. As shown in Figure 3, this polypeptide was able to cross epithelial monolayers in the absence, but not in the presence, of anti-HC50 antibodies. Obviously the holotoxin also possesses the determinants for penetration of epithelial monolayers. As shown in Figure 3, the holotoxin was able to bind and cross epithelial barriers in the absence, but not in the presence, of anti-light chain antibodies. Thus, antibodies directed against either end of the type A toxin molecule prevented migration from the apical to the basal surface of epithelial cells.
Figure 3.
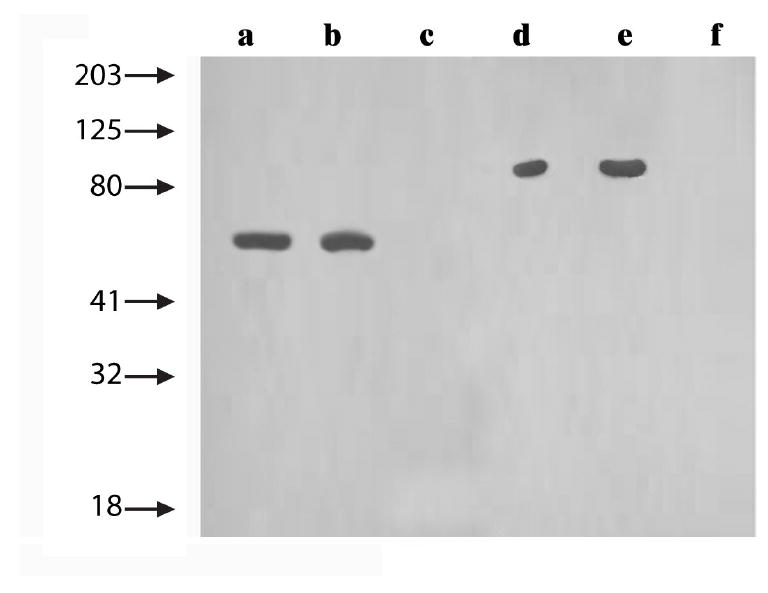
The ability of antiserum to block binding and transcytosis of pure botulinum toxin was studied in T-84 human epithelial monolayers. The Figure illustrates two experiments, one of which involved a positive control (anti-HC50 serum; lanes a to c), and one of which involved the antiserum of interest (anti-light chain serum; lanes d to f). In the first experiment, an aliquot of HC50 (1 × 10-8 M for all conditions) had been applied to the apical surface of cells (lane a), an aliquot of HC50 that had undergone binding and transcytosis to the basal side of cells (lane b), and an aliquot of HC50 that had been exposed to anti-HC50 antiserum (40 microliters) before binding, transcytosis and collection on the basal side of cells (lane c), were examined in Western blots. In the second experiment, an aliquot of type A toxin (1 × 10-8 M for all conditions) that had been applied to the apical surface of cells (lane d), an aliquot of toxin that had undergone binding and transcytosis to the basal side of cells (lane e), and an aliquot of toxin that had been exposed to anti-light chain serum (40 microliters) before binding, transcytosis and collection on the basal side of cells (lane f), were examined in Western blots. As expected, antiserum directed against the binding domain of the toxin (HC50) blocked binding and transcytosis of the polypeptide (lane c). Interestingly, antiserum directed against the light chain of the toxin blocked binding and transcytosis of the holotoxin (lane f).
3.5. Anti-light chain antibodies antagonize mucosal challenge with toxin
In an initial series of experiments, various amounts of anti-light chain antiserum were tested in an effort to identify a dose that would produce complete neutralization of an i.n. challenge by toxin of 50 ng. A dose of 18 μl produced the desired outcome. Thus, control animals that received only the i.n. challenge with toxin died in ca. 480 min, whereas experimental animals that received i.n. challenge with toxin that had been preincubated with antiserum (60 min; room temperature) survived (Figure 4).
Figure 4.
Experiments were done to assess the ability of antibodies given by the i.n. route to neutralize toxin challenge. In the first set of experiments, botulinum toxin type A (50 ng) was administered by the i.n. route in the absence or presence of anti-light chain antibodies (18 μl), also given by the i.n. route. Animals (n = 5 per group) that received toxin alone died in ca. 8 hours; animals that received toxin plus anti-light chain antiserum survived. In the second set of experiments, an equiactive amount of botulinum toxin type A was administered by the i.v. route in the absence or presence of anti-light chain antibodies (18 μl) given by the i.n. route. In this case, none of the animals was protected against challenge.
In a related set of experiments, the amount of antiserum that had produced neutralization of an i.n. challenge was administered by itself by the i.n. route. After a delay of 60 min, mice were challenged with toxin by the i.v. route, using a dose that was equiactive with that used above in the i.n. challenge experiments. All of the control animals that received only the i.v. challenge with toxin died. All of the experimental animals that had received antiserum by the i.n. route and then were challenged by the i.v. route also died. These results make it unlikely that protection observed in the i.n. challenge experiment (i.e., Figure 4) was due to absorption of antibodies that acted in the general circulation to promote clearance or at the neuromuscular junction to block toxin binding (see below, and see reference 19 for experiments in which both toxin and antibodies were administered by the i.v. route). More realistically, neutralization observed in the i.n. challenge experiment was due to a local action of antibodies at the surface of epithelial cells to block absorption.
3.6. Anti-light chain antibodies promote clearance of toxin from the general circulation and accumulation of toxin in liver and spleen
For clearance experiments, 125I-botulinum toxin type A (5 ng per animal) was administered i.v. to mice (n=6 per group), and at various times thereafter (10 min to 100 min) animals were sacrificed by exsanguination. Aliquots of serum were used to quantify the circulating levels of toxin. As shown in Figure 5, the retention of toxin in blood was relatively stable over the timecourse of the experiment, which is in keeping with the reported half-life of the toxin in the general circulation of rodents [10,19].
Figure 5.
125I-Botulinum toxin (5 ng) was administered to mice (n=6 per data point), and at various times thereafter animals were sacrificed and blood was collected. The levels of toxin in serum were determined for control mice (■) and for experimental mice that received a neutralizing dose of anti-light chain antibodies (10 μl; ◆). As the Figure illustrates, there was an initial and dramatic reduction in the circulating levels of toxin in mice that received antiserum. This clearance-driven phenomenon occurred within less than 10 min.
In a variation on this experiment, anti-light chain antiserum (10 μl) was administered i.v. 60 min prior to toxin. Once again, animals were sacrificed at various times after i.v. injection of toxin, and the retention of toxin in blood was measured. A comparison of toxin levels in control mice and in experimental (antiserum) mice showed that levels in the latter were much lower. These data indicated a clearance-driven mechanism with a very rapid onset (< 10 min).
Clearance phenomena are typically associated with enhanced accumulation of antibody~antigen complexes in liver and spleen. As a test for this outcome, groups of control mice and experimental mice were sacrificed 20 minutes after administration of toxin. The fractional accumulation of toxin in the livers and spleens of antibody-pretreated mice were found to be much higher than those in control mice (Figure 6). The magnitude of the enhanced accumulation in liver was particularly striking.
Figure 6.
125I-Botulinum toxin (5 ng) was administered to control mice (hatched bars) or to experimental mice that had been pretreated (60 min) with anti-light chain antiserum (10 μl; dark bars). Twenty minutes after toxin challenge (n=5 animals per group), mice were sacrificed and livers and spleens were excised. The results indicated that anti-light chain antiserum promoted accumulation of toxin in both organs.
3.7. Anti-light chain antibodies antagonize the neuromuscular blocking properties of botulinum toxin
Dose-response experiments were conducted in which various amounts of anti-light chain antiserum were incubated with toxin for 60 min at room temperature. These mixtures were then added to murine phrenic nerve-hemidiaphragm preparations to produce a final toxin concentration of 3 × 10-12 M. The amount of time necessary for onset of neuromuscular blockade was monitored.
As shown in Figure 7, botulinum toxin alone paralyzed transmission (90% reduction in twitch amplitude) in 100 to 120 min. By contrast, anti-light chain antiserum produced complete protection against botulinum toxin. At a dose of 10 μl there was no onset of paralysis over a period of 300 min, which is more than twice the amount of time needed for control preparations to paralyze. Equivalent amounts of pre-immune serum provided no protection against toxin-induced paralysis.
Figure 7.
The murine phrenic nerve-hemidiaphragm was used to assay the ability of anti-light chain antibodies to neutralize the actions of botulinum toxin type A (final concentration, 3 × 10-12 M). When botulinum toxin alone was added to control tissues (○;n=6), evoked responses decayed and a 90% reduction in twitch amplitude occurred within ca. 120 min. Essentially the same outcome was obtained when toxin was pre-incubated with pre-immune serum before being added to tissue baths (▲;n=3). When toxin was pre-incubated with anti-light chain antiserum (10 μl; ■;n=3), the actions of the toxin were blocked during the entire observation period. The standard error of the mean for each data point in the figure was equal to or less than 4% of the mean for the data point
3.8. Anti-light chain antibodies block toxin binding to the surface of neuronal cells
Immunohistochemical studies were done to visualize toxin association with neuronal cells (Neuro-2a). Botulinum toxin (1 × 10-8M) was incubated alone or in the presence of anti-light chain antibodies (20 μl; 60 min; room temperature). The mixtures were added to neuronal cells for 60 min at 4°C. Low temperature was used to retard active processes such as endocytosis, and thus maintain bound toxin at the cell surface. Tissues were then washed and processed for immunostaining as described under Methods. An identical set of experiments was done using anti-HC50 antibodies as a positive control.
As expected, the images (Figure 8) confirmed that the toxin could bind to the surface of neuronal cells. When toxin was pre-incubated with antiserum directed against the HC50 domain, virtually all binding was eliminated. This outcome is in keeping with the fact that the HC50 domain possesses the region known to bind to nerve endings. Interestingly, when toxin was preincubated with antiserum directed against light chain, this too virtually eliminated binding. In this case blockade of binding could not have been due to antibody-mediated occlusion of the toxin receptor-binding domain, although it could be due to steric hindrance (see Discussion).
Figure 8.
Immunohistochemical experiments were done to determine the effects of antibodies on botulinum toxin type A binding to neuronal cells (Neuro-2a). In one paradigm (Part A), toxin (1 × 10-8 M) was preincubated with antiserum directed against the binding domain of the toxin (HC50). This mixture was then added to cells for 60 min at 4° C. After incubation, cells were washed and then probed with antibodies directed against the light chain. In the obverse paradigm, toxin (1 × 10-8 M) was preincubated with antiserum directed against the light chain (Part B). This mixture was applied to cells for 60 min at 4° C. After incubation, cells were washed and then probed with antibodies directed against the HC50 domain. In both paradigms, images were obtained with differential interface contrast microscopy (DIC) to establish orientation, with fluorescence microscopy (FITC) to detect toxin binding to cells, and with an overlay of the two images. As shown in Part A, untreated toxin bound to the surface of neuronal cells, whereas toxin that had been treated with anti-HC50 serum displayed almost no binding activity. Part B illustrates a similar outcome. Thus, untreated toxin was associated with the surface of neuronal cells, whereas toxin that had been treated with anti-light chain serum was essentially devoid of binding activity. These results demonstrate that antibodies directed against either the binding domain of the toxin or the catalytic domain of the toxin can block toxin association with neuronal cells.
The antagonistic effect on toxin binding was not observed when antibodies were incubated with cells, followed by washing and addition of free toxin. Thus, the reduction in binding illustrated in Figure 8 was due to a specific interaction between antibodies and antigen, and not due to a non-specific interaction between antibodies and the cell surface.
3.9. Anti-light chain antibodies block the metalloendoprotease activity of the light chain
The catalytic activity of the recombinant light chain was measured using a FRET assay. For comparison, the catalytic activity of the progenitor toxin complex and the pure neurotoxin were also assayed. The results, which are shown in Figure 9, demonstrate that all three preparations containing the light chain possessed catalytic activity. In addition, the activity of the recombinant light chain was in the same range as that of the wildtype proteins.
Figure 9.
The ability of anti-light chain antibodies to block the endoprotease activity of the toxin was determined using a FRET assay. The extent of catalytic activity was quantified (arbitrary fluorescene units; AFU) using substrate solution alone (SSA), or substrate solution in the presence of progenitor toxin complex, pure neurotoxin, or recombinant light chain (LCA). As shown in the Figure, all three protein preparations possessed catalytic activity. In addition, anti-light chain antibodies (10, 20 or 30 microliters) produced dose-dependent reductions in catalytic activity of all three preparations.
The ability of the three light chain preparations to express enzymatic activity was re-examined after incubation with various amounts of anti-light chain antiserum (60 min; room temperature). In all three cases, the tested doses of antiserum produced dose-dependent reductions in light chain endoprotease activity.
4. Discussion
Efforts to develop a human vaccine against botulinum toxin have been underway since the early part of the last century. During this lengthy period of time, many laboratories have been involved and a wide variety of techniques have been tested. However, there are two approaches to vaccine development that have overshadowed all others [2]. During the early decades of research, almost all effort was focused on a chemical toxoid. This product was generated by: a.) growing toxigenic organisms until there was an appreciable titer of toxin in the culture, b.) removing a portion of the bacterial debris, which produced some enrichment of toxin, and c.) treating the enriched material with formalin to produce total loss of toxicity but only partial loss of immunogenicity. This approach was used to generate a vaccine that is currently distributed for occupational safety use by the Centers for Disease Control and Prevention under Investigational New Drug status.
The second approach evolved more recently, and it is based on the techniques of molecular biology. According to one variation, a recombinant holotoxin can be expressed that possesses point mutations that render the molecule non-toxic, yet fully immunogenic [5]. According to another variation, recombinant polypeptides can be expressed that represent an incomplete toxin [2,19,20]. In the absence of one or more functional domains, these recombinant polypeptides have no ability to produce the neuroparalysis that is characteristic of the holotoxin. However, these polypeptides contain a critical mass of epitopes that can evoke neutralizing antibodies. In an interesting and unanticipated way, these two methods to generate recombinant vaccines have given rise to the present report.
The first study in which a recombinant holotoxin was expressed and demonstrated to be an efficacious vaccine was that of Kiyatkin et al [5]. The work involved the linking of two constructs: a mutated version of the toxin light chain in which the histidine motif (which is the zinc binding region) was altered, and a wildtype version of the heavy chain. Mutation in the histidine motif was selected because: a.) this motif is essential for light chain endoprotease activity, which accounts for the toxicity of botulinum toxin, and b.) all serotypes/subtypes of the toxin possess this motif, so this approach can be applied to all serotype/subtype holotoxin vaccines.
When tested as a vaccine, the mutated holotoxin was shown to evoke both anti-light chain and anti-heavy chain antibodies, and it produced a high level of resistance to the holotoxin. An additional finding was that a recombinant version of the light chain, in the absence of any portion of the heavy chain, evoked production of only anti-light chain antibodies, and this too led to resistance to the holotoxin [5].
Simultaneously with this work, other investigators were examining the potential value of recombinant polypeptides representing portions of the heavy chain, and particularly the carboxyterminal half of the heavy chain [7,20]. This polypeptide was selected because it is structurally analogous to a polypeptide domain within tetanus toxin that had previously been shown to be an effective vaccine, and because it is an epitope-rich region of the BoNT molecule. A final reason for its selection is especially important. This region of the toxin molecule is known to bind to receptors on vulnerable nerve cells. This led to the reasonable assumption that antibodies directed against it could neutralize toxin action by blocking toxin binding to nerve membranes.
The concept that administration of the binding domain of the toxin can evoke production of neutralizing antibodies is absolutely correct. However, the concept that blockade of toxin binding to nerve membranes is the only mechanism – or even the most important mechanism – for antibody-mediated resistance may not be correct. Recent work that seeks to mate the pharmacokinetics of the toxin molecule with the sites and mechanisms of action of medical countermeasures such as vaccines indicates that there are three principal ways in which neutralizing antibodies can act [8, 10,18]. First, mucosal IgA can act in the lumen of the gut or airway to block toxin absorption into the body. It is intuitively clear that this is the most desirable way to block poisoning. Secondly, circulating IgG can decorate the toxin and mark it for enhanced clearance from the circulation, due to enhanced accumulation in liver and spleen. This will prevent any toxin that is absorbed from ever reaching nerve endings. Finally, circulating IgG can associate with the toxin in a way that blocks binding and/or internalization by nerve endings. This in turn will prevent the toxin from reaching the cytosol and expressing its endoprotease activity.
When past work to characterize anti-heavy chain antibodies is placed beside more current work showing that other portions of the holotoxin can serve as vaccines, this raises an obvious question. How do antibodies directed against other portions of the holotoxin, such as the light chain, produce their neutralizing effects?
4.1 Mechanism of action of anti-light chain antibodies
Neither the original studies demonstrating that light chains can evoke resistance to holotoxin [5,21] nor any subsequent study has explained the underlying mechanisms for resistance. There are reports that anti-light chain antibodies can block the enzymatic activity of the toxin in cell-free systems (as in this study) and in cells with permeablized membranes, but this is not likely to explain antitoxin activity in vivo. Antibodies do not possess efficient mechanisms for gaining access to the cytosol of peripheral neurons, so antibodies would be unlikely to reach light chain that is in the vicinity of its substrate. A more likely scenario is that anti-light chain antibodies exert their effects in one or more extracellular compartments.
It is reasonable to predict that at least one mechanism of extracellular action would be enhanced clearance from the circulation. The ability of antibodies to associate with antigen and mark it for hepatic and splenic uptake is not a function of the structure-activity relationships within the toxin molecule. Therefore, antibody-mediated clearance should be just as applicable to the catalytic domain of the toxin (e.g., aminoterminus of the holotoxin) as it is to the binding domain (e.g., carboxyterminus of the holotoxin). This expectation was supported by the data. Animals that were passively vaccinated with light chain displayed enhanced clearance as well as enhanced hepatic and splenic uptake of toxin. These findings make clear that antibody-driven clearance is at least one mechanism by which anti-light chain antibodies can neutralize botulinum toxin.
The ability of antibodies directed against the carboxyterminus of the heavy chain to block absorption across epithelial cells and to block onset of paralysis at the neuromuscular junction is likely due to blockade of toxin binding at both sites [8]. One can easily envision that antibodies directed against this portion of the toxin can act directly by covering the binding domain or indirectly by causing steric hindrance near the binding domain. This view may appear to suggest that antibodies directed against the light chain, which is on the opposite end of the holotoxin, should have little or no ability to block epithelial or neuronal binding. However, the data reveal an entirely different outcome. Anti-light chain antibodies acted on epithelial cells to block binding and transcytosis and on neuronal cells to block binding and onset of paralysis. This effect was especially clear in immunohistochemistry studies on Neuro-2a cells. In this case, blockade of toxin binding to the neuronal cell surface could be visualized.
It is not possible that anti-light chain antibodies can associate with the toxin at its carboxyterminal binding domain to cause occlusion of binding. Some other mechanism must be involved, with the most obvious being steric hindrance. The size of an antibody molecule is approximately the same as that of a toxin molecule. This means that, depending on the exact sites of light chain epitopes, there could be an antibody~light chain configuration that places some portion of the antibody in a position that can hinder heavy chain binding to an epithelial cell or to a nerve membrane. Alternatively, the binding of anti-light chain antibodies to holotoxin could induce conformational changes that distort the binding domain and thus block binding to membranes.
Regardless of whether one is seeking to clarify the molecular actions of anti-light chain antibodies or anti-heavy chain antibodies, the path to resolution will likely be the same. For both classes of antibodies there is a pressing need to clarify the 3-dimensional structures of antibody~toxin complexes. This is the only way to establish with certainty whether occlusion, steric hindrance, induced conformational changes, or some other type of phenomenon accounts for neutralization of toxin activity.
4.2. Composition of an ideal vaccine
The finding that anti-light chain antibodies possess similar actions to those of anti-heavy chain antibodies raises two questions: 1.) is one polypeptide superior to the other as a vaccine candidate, or 2.) is a chimeric polypeptide that incorporates the two superior to either one individually? As a prelude to answering these questions, one should note that neither of the individual polypetitdes in their wildtype variants is an ideal vaccine. Each could be improved by introducing point mutations that alter neurobiologic activity. In the case of a heavy chain antigen, the preferred candidate would be one that has been modified to abolish its neuronal binding activity. In the case of a light chain antigen, the preferred candidate would be one that has been altered to abolish catalytic activity.
In terms of actually comparing individual and chimeric antigens, one would have to perform a large number of equivalency studies and dose optimization studies. This would require a substantial amount of work, and the end result might not be particularly gratifying. It is entirely plausible that the individual polypeptides and the chimeric polypeptide will each prove so efficacious that any differences between or among them will not attain the level of clinical significance. If, for example, all three can protect against challenge doses in the range of 104 to 105 LD50, this may already exceed any level of protection that would be needed in a clinical setting.
To the extent that these polypeptides differ in their utility, it may not be in the realm of evoked titers of antibody. Instead, it may be in ancillary areas such as ease of expression, ease of formulation, or ease of administration. For example, the heavy chain component of the toxin, including the carboxyterminal half of the heavy chain, retains the ability to bind and penetrate epithelial barriers [14]. If this characteristic is essential to the ability of a mucosally-administered vaccine to evoke all of the known layers of protection, then a vaccine that incorporates the carboxyterminus of the heavy chain, either alone or as part of a chimera, may be preferred.
Acknowledgments
This work was supported in part by NIH grants NO1-AI30028 and GM57345 and by DTRA contract 1-07-0032.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arnon SS, Schechter R, Inglesby TV. Botulinum toxin as a biological weapon: medical and public health management. JAMA. 2001;285(8):1059–70. doi: 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- 2.Simpson LL. Rational development of a vaccine against botulinum toxin. In: Barrett Alan DT, Stanberry Lawrence R., editors. Vaccines for Biodefense and Emerging and Neglected Diseases. Elsevier; San Diego: in press. [Google Scholar]
- 3.Simpson LL. Identification of the major steps in botulinum toxin action. Ann Rev Pharmacol Toxicol. 2004;44:167–93. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 4.Rossetto O, Montecucco C. Presynaptic neurotoxins with enzymatic activities. Handb Exp Pharmacol. 2008;184:129–70. doi: 10.1007/978-3-540-74805-2_6. [DOI] [PubMed] [Google Scholar]
- 5.Kiyatkin N, Maksymowych AB, Simpson LL. Induction of immune response by oral administration of recombinant botulinum toxin. Infect Immun. 1997;65(11):4586–91. doi: 10.1128/iai.65.11.4586-4591.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith LA. Development of recombinant vaccines for botulinum neurotoxin. Toxicon. 1998;36(11):1539–48. doi: 10.1016/s0041-0101(98)00146-9. [DOI] [PubMed] [Google Scholar]
- 7.Byrne MP, Smith LA. Development of vaccines for prevention of botulism. Biochimie. 2000;82:955–66. doi: 10.1016/s0300-9084(00)01173-1. [DOI] [PubMed] [Google Scholar]
- 8.Ravichandran E, Al-Saleem FH, Ancharski DM, Elias M, Singh A, Gong Y, et al. A trivalent vaccine against botulinum toxin (serotypes A,B, and E) that can be administered by the mucosal route. Infect Immun. 2007;75:3043–54. doi: 10.1128/IAI.01893-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maksymowych AB, Simpson LL. Binding and transcytosis of botulinum neurotoxin by polarized human colon carcinoma cells. J Biol Chem. 1998;273(34):21950–7. doi: 10.1074/jbc.273.34.21950. [DOI] [PubMed] [Google Scholar]
- 10.Ravichandran E, Gong Y, Al-Saleem FH, Ancharski DM, Joshi SG, Simpson LL. An initial assessment of the systemic pharmacokinetics of botulinum toxin. J Pharmacol Exp Ther. 2006;318(3):1343–51. doi: 10.1124/jpet.106.104661. [DOI] [PubMed] [Google Scholar]
- 11.Simpson L, Al-Saleem F, Ancharski D, Elias M, Takahaski T, Joshi S, Singh A. Identification of the mechanisms that account for antibody-induced neutralization of botulinum toxin. Interagency Botulism Research Coordinating Committee Meeting; Philadelphia, PA. 2008. Abstract VA-6. [Google Scholar]
- 12.DasGupta BR, Sathyamoorthy V. Purification and amino acid composition of type A botulinum neurotoxin. Toxicon. 1984;22(3):415–24. doi: 10.1016/0041-0101(84)90085-0. [DOI] [PubMed] [Google Scholar]
- 13.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 14.Maksymowych AB, Simpson LL. Structural features of the botulinum neurotoxin molecule that govern binding and transcytosis across polarized human intestinal epithelial cells. J Pharmacol Exp Ther. 2004;310(2):633–41. doi: 10.1124/jpet.104.066845. [DOI] [PubMed] [Google Scholar]
- 15.Ahsan CR, Hajnoczky G, Maksymowych AB, Simpson LL. Visualization of binding and transcytosis of botulinum toxin by human intestinal epithelial cells. J Pharmacol Exp Ther. 2005;315:1–8. doi: 10.1124/jpet.105.092213. [DOI] [PubMed] [Google Scholar]
- 16.Simpson LL, Maksymowych AB, Hao S. The role of zinc binding in the biological activity of botulinum toxin. J Biol Chem. 2001;276(29):27034–41. doi: 10.1074/jbc.M102172200. [DOI] [PubMed] [Google Scholar]
- 17.Joshi SG, Francis CW, Silverman DJ, Sahni SK. Nuclear factor kappa B protects against host cell apoptosis during Rickettsia rickettsii infection by inhibiing activation of apical and effector caspases and maintaining mitochondrial integrity. Infect Immunol. 2003;71:4127–36. doi: 10.1128/IAI.71.7.4127-4136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt JJ, Stafford RG. Fluorigenic substrates for the protease activities of botulinum neurotoxins, serotypes A, B, and F. Appl Environ Microbiol. 2003;69(1):297–303. doi: 10.1128/AEM.69.1.297-303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Saleem FH, Ancharski DM, Ravichandran E, Joshi SG, Singh AK, Gong Y, et al. The role of systemic handling in the pathophysiologic actions of botulinum toxin. J Pharmacol Exp Ther. 2008;326(3):856–863. doi: 10.1124/jpet.108.136242. [DOI] [PubMed] [Google Scholar]
- 20.Smith LA, Rusnak JM. Botulinum neurotoxin vaccines: past, present, and future. Crit Rev Immunol. 2007;27(4):303–18. doi: 10.1615/critrevimmunol.v27.i4.20. [DOI] [PubMed] [Google Scholar]
- 21.Jensen MJ, Smith TJ, Ahmed SA, Smith LA. Expression, purification, and efficacy of the type A botulinum neurotoxin catalytic domain fused to two translocation domain variants. Toxicon. 2003;41:691–701. doi: 10.1016/s0041-0101(03)00042-4. [DOI] [PubMed] [Google Scholar]