

Abstract
The deacetylation of UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine (UDP-3-O-acyl-GlcNAc) by LpxC is the committed reaction of lipid A biosynthesis. CHIR-090, a novel N-aroyl-l-threonine hydroxamic acid, is a potent, slow, tight-binding inhibitor of the LpxC deacetylase from the hyperthermophile Aquifex aeolicus, and it has excellent antibiotic activity against P. aeruginosa and E. coli, as judged by disk diffusion assays. We now report that CHIR-090 is also a two-step slow, tight-binding inhibitor of Escherichia coli LpxC with Ki = 4.0 nM, Ki* = 0.5 nM, k5 = 1.9 min-1 and k6 = 0.18 min-1. CHIR-090 at low nM levels inhibits LpxC orthologues from diverse Gram-negative pathogens, including Pseudomonas aeruginosa, Neisseria meningitidis, and Helicobacter pylori. In contrast, CHIR-090 is a relatively weak competitive and conventional inhibitor (lacking slow, tight-binding kinetics) of LpxC from Rhizobium leguminosarum (Ki = 340 nM), a Gram-negative plant endosymbiont that is resistant to this compound. The KM (4.8 μM) and the kcat (1.7 s-1) of R. leguminosarum LpxC with UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine as the substrate are similar to values reported for E. coli LpxC. R. leguminosarum LpxC therefore provides a useful control for validating LpxC as the primary target of CHIR-090 in vivo. An E. coli construct in which the chromosomal lpxC gene is replaced by R. leguminosarum lpxC is resistant to CHIR-090 up to 100 μg/mL, or 400 times above the minimal inhibitory concentration for wild-type E. coli. Given its relatively broad spectrum and potency against diverse Gram-negative pathogens, CHIR-090 is an excellent lead for the further development of new antibiotics targeting the lipid A pathway.
The emergence of multi-drug resistant bacteria in hospital and community clinics has created an urgent need for new antibiotics (1, 2). About half of the multi-drug resistant bacteria are Gram-negative pathogens (2), including strains of Escherichia coli, Pseudomonas aeruginosa (1), and Acinetobacter baumannii (3). Inhibitors that exploit traditional antibiotic targets, such as peptidoglycan, DNA replication or protein biosynthesis (4), are becoming less effective (2). These obstacles could be overcome by developing inhibitors of novel targets required for bacterial growth (5, 6).
The biosynthesis of the lipid A component of lipopolysaccharide (LPS), a unique, outer-membrane lipid that shields Gram-negative bacteria from environmental stresses (7, 8), is a promising target for new antibiotic development (9-12). The lipid A moiety of LPS is a hexa-acylated disaccharide of glucosamine (7, 8) (Figure 1). Although inhibition of any one of the first six enzymes of lipid A biosynthesis is lethal to E. coli (8), the most promising target identified to date is LpxC (9-12), a unique deacetylase that is selective for UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine (UDP-3-O-acyl-GlcNAc) (13-16) (Figure 1). Although LpxC catalyzes the second reaction of the lipid A pathway, it represents the committed step (Figure 1).
Figure 1. Reaction catalyzed by LpxC and structure of E. coli lipid A.

The biosynthesis of lipid A begins with the 3-O-acylation of UDP-GlcNAc by the cytosolic enzyme LpxA (8). In the first irreversible (committed) reaction of the pathway, the deacetylase LpxC unblocks the nitrogen at the 2 position of the glucosamine ring for subsequent acylation by LpxD (8). Six downstream enzymes produce Kdo2-lipid A (8), the hydrophobic membrane anchor of LPS and a potent toll-like receptor 4 agonist (44).
LpxC is a zinc-dependent amidase with a catalytic mechanism related to that of carboxypeptidase and thermolysin (17-19). Aquifex aeolicus LpxC, the structure of which was recently solved by both NMR spectroscopy (18, 20) and X-ray crystallography (21, 22), displays a novel “β-α-α-β sandwich” fold (Figure 2A). A unique feature of LpxC is a hydrophobic passage, leading away from the active site zinc ion, which accommodates the acyl chain of the substrate-mimetic inhibitor TU-514 (Figure 2A) and is presumed to capture the acyl chain of the substrate (18, 20, 22). The terminal methyl group of the TU-514 acyl chain protrudes through an opening at the surface of the enzyme (18, 20).
Figure 2. Structures of A. aeolicus LpxC and of various LpxC inhibitors.
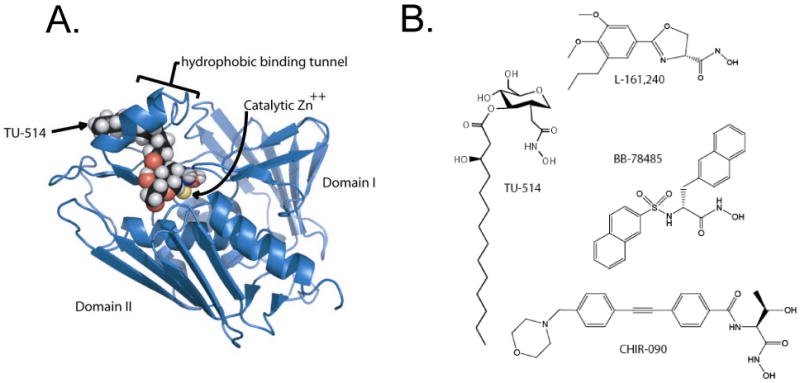
Panel A. The three-dimensional structure of LpxC is characterized by a unique “β-α-α-β sandwich” fold (18, 20-22). The NMR structure of LpxC was solved with one bound molecule of the substrate analog TU-514 (18, 20). LpxC contains an unusual hydrophobic tunnel that encapsulates the acyl chain of TU-514 (18, 20, 22). This figure was generated with PyMol using the protein data base accession code 1XXE. Panel B. All relevant LpxC inhibitors contain a zinc-chelating hydroxamate moiety. Structures of LpxC complexes including other inhibitors are not yet available.
Most of the available LpxC inhibitors that display significant antibiotic activity resemble TU-514 in that they contain a hydroxamic acid moiety to coordinate the catalytic zinc ion (9-12, 23, 24) (Figure 2B). However, many LpxC inhibitors, like L-161,240 and BB-78485 (Figure 2B), which kill E. coli, have little or no effect on P. aeruginosa (10, 12). This anomaly is explained by the fact that P. aeurginosa LpxC is only 55 % identical to E. coli LpxC, and its activity is much less susceptible to inhibition by L-161,240 and BB-78485 (25).
McClerren et al. recently reported CHIR-090 (Figure 2B), a very potent, slow, tight-binding inhibitor of Aquifex aeolicus LpxC (11), the sequence of which is 31 % identical to E. coli LpxC. McClerren et al. also showed that CHIR-090 has remarkable antibiotic activity against E. coli and P. aeruginosa, comparable to ciprofloxacin, as judged by disk diffusion assays (11). However, the inhibition of LpxC from these and other pathogenic bacteria by CHIR-090 was not fully evaluated. It also remains unclear whether or not LpxC is the sole target of CHIR-090 in vivo. We now report that CHIR-090 is a slow, tight-binding inhibitor of E. coli LpxC with Ki = 4.0 nM, Ki* = 0.5 nM, k5 = 1.9 min-1, and k6 = 0.18 min-1. CHIR-090 inhibits LpxC orthologues from several other important Gram-negative pathogens at low nM concentrations. In contrast, however, both growth and LpxC activity of the Gram-negative plant endosymbiont Rhizobium leguminosarum are insensitive to CHIR-090. Replacement of E. coli lpxC by R. leguminosarum lpxC renders E. coli fully resistant at 400 times the minimal inhibitory concentration (MIC) for wild-type cells, demonstrating that LpxC is the primary target for CHIR-090 in vivo.
Experimental Procedures
Materials, Strains and Reagents
PEI-cellulose TLC plates were purchased from EMD Chemicals, Gibbstown, NJ. Bovine serum albumin (BSA), imidazole, HEPES and sodium phosphate were obtained from Sigma-Aldrich, St. Louis MO, and [α-32P]-UTP was purchased from NEN DuPont. Ni-NTA agarose resin and plasmid miniprep kits were purchased from Qiagen, Valencia, CA. Coomassie-Plus protein reagent and GelCode blue staining reagent were purchased from Pierce, Rockford, IL. E. coli BLR(DE3)/pLysS, E. coli XL1-Blue competent cells, and Pfu polymerase were purchased from Stratagene, La Jolla, CA. The vector pET21b was purchased from Novagen (an EMD Chemicals Company). Primers were purchased from MWG Biotech, High Point, NC. Restriction endonucleases NdeI and BamHI were purchased from New England Biolabs, Ipswich, MA. Genomic DNA from Rhizobium leguminosarum strain 3841 was prepared as described previously (26), and DNA from Helicobacter pylori was provided by Dr. M. Stephen Trent, East Tennessee State University, Johnson City, TN. Genomic DNA from Neisseria meningiditis (ATCC 700532D) was purchased from the American Type Culture Collection, Manassas, VA. P. aeruginosa lpxC cloned into pET21b was constructed as previously described (27). The LpxC inhibitor CHIR-090 was prepared at the Duke University Small Molecule Synthesis Facility according to published procedures (28).
Cloning of LpxC Genes from H. pylori, N. meningiditis and R. leguminosarum
The lpxC genes from H. pylori, N. meningitidis, and R. leguminosarum were amplified from genomic DNA by PCR, using the primers listed in Table S1, and cloned into a pET21b vector in-frame with the C-terminal His tag. The presence of the correct insert was confirmed by DNA sequencing.
Overexpression and Purification of H. pylori, N. meningiditis and R. leguminosarum LpxC
The purification and specific activity of each LpxC orthologue is summarized in Table S2. Plasmids containing C-terminal His tagged lpxC genes from H. pylori, N. meningiditis and R. leguminosarum were transformed into E. coli BLR (DE3)/pLysS competent cells for overexpression. Overnight cultures (5 mL) were grown from single colonies at 37 °C in LB broth (29), containing 100 μg/mL ampicillin and 25 μg/mL chloramphenicol, and used to inoculate 50 mL of LB broth, supplemented with the same antibiotics. These cultures were grown to an A600 of 0.5. LpxC expression was induced with 1 mM isopropyl β-d-thiogalactopyranoside (IPTG) in the presence of 100 μM ZnSO4. All cultures were grown at 25 °C for an additional 5 h and then harvested at 6,000 × g for 20 min at 4 °C. The cell pellets were resuspended in 25 mL of 25 mM HEPES, pH 7.0, containing 2 mM β-mercaptoethanol, 300 mM NaCl and 20% glycerol. The cells were broken by three passages through a cold French pressure cell at 18,000 psi, and debris was removed by centrifugation at 8,000 × g for 20 min at 4 °C. The supernatant was loaded onto a 5-mL Ni-NTA agarose column, equilibrated with 5 mM HEPES, pH 7.0, containing 2 mM β-mercaptoethanol, 300 mM NaCl and 20% glycerol, and the protein was eluted with the 200 mM imidazole. The fractions containing LpxC were pooled, concentrated, and dialyzed into 25 mM HEPES, pH 7.0, containing 2 mM DTT, 150 mM NaCl and 10% glycerol. Protein concentrations were determined using the bicinchoninic assay (30).
Overexpression and Purification of E. coli LpxC and P. aeruginosa LpxC Lacking His Tags
E. coli LpxC was prepared as previously described (18) using lpxC expressed in pET11a (31). For P. aeruginosa LpxC, a fresh colony of BLR (DE3)/pLysS harboring P. aeruginosa lpxC cloned into pET21b was used to inoculate 5 mL of LB broth for an overnight culture. P. aeruginosa LpxC was cloned without the His-tag present on pET21b. A 1.5 L portion of M9 medium (29), containing 100 μg/mL ampicillin and 25 μg/mL chloramphenicol, was inoculated with the overnight culture. Cells were grown at 37 °C to an A600 of 0.5, and then IPTG was added to 1 mM. The bacterial cells were subsequently grown at 30 °C for 3 h, harvested, and lysed as described above. Next, 13.5 g of ammonium sulfate was added to 43 mL of cytosol, and the mixture was incubated on ice for 1.5 h. Protein precipitate was recovered by centrifugation at 8,000 × g for 20 min, and it was resuspended in 30 mL of 25 mM HEPES, pH 7.0, containing 10% glycerol and 2 mM DTT. Remaining ammonium sulfate was removed by overnight dialysis against the same buffer.
The sample was applied to a 30-mL Q-Sepharose (Amersham-Pharmacia) column equilibrated with the same buffer. The column was eluted with a linear gradient of 0 to 1 M KCl dissolved in 25 mM HEPES, pH 7.0, and 2 mM DTT. Fractions (5 mL) containing LpxC activity were pooled and concentrated to 1 mL. The protein was further purified using a 2 cm × 46 cm Sephadex G75 column (Amersham-Pharmacia) equilibrated in the same buffer. Fractions (2 mL) containing LpxC activity were pooled, and purity was assessed by SDS-PAGE. The protein was stored at −80 °C prior to being assayed. Electrospray ionization mass spectrometry (11) was used to confirm the exact mass predicted from the P. aeruginosa LpxC sequence. SDS-PAGE analysis of each of the above LpxC preparations showed no more than 10% contaminating proteins, as judged by densitometry.
Assay of LpxC Activity
UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine and [α-32P]UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine were prepared enzymatically as previously described (31). Assays of LpxC activity were performed with 5 μM substrate, except where noted (11); additionally, 10% DMSO was added to the assay mixtures and held constant at that level when inhibitor (dissolved in DMSO) was added. Except where noted, the concentration of the enzyme was at least 10-fold less than the concentration of either the inhibitor or the substrate. When pre-incubated with or without inhibitor prior to being assayed, the enzyme was diluted in 25 mM sodium phosphate, pH 7.4, containing 1 mg/mL BSA and 10% DMSO. The pre-incubation mixture was held on ice for 15 min before the reaction was initiated by means of a 1:4 dilution of the enzyme into the assay cocktail. Initial velocities were calculated from the linear portion of reaction progress curves (<10% conversion of substrate to product).
Determination of the Mechanism of CHIR-090 binding to E. coli LpxC
In order to assess the slow, tight-binding phenomenon, 0.2 nM E. coli LpxC was first assayed in the presence of 0, 0.5, 1, 2, 3, 4, 6 or 8 nM inhibitor without any pre-incubation. In these assays, 2.2 × 106 DPM of [α-32P]UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine were included per reaction. Equation 1, describing time-dependent inhibition, was fitted to the data as previously described (Scheme 1) (11). The rate of EI* formation (kobs) was determined from curve fitting with:
Scheme 1.

Two step mechanism for slow, tight-binding inhibition
| (Eq. 1) |
where [P] is the concentration of product at time t, vi is the initial velocity, vs is the steady state velocity and kobs is the first order exponential term for the formation of the EI* complex.
A modified form of Equation 1 was used to fit the rate of formation of free E (k′obs) by measuring product accumulation after a 100-fold rapid dilution following a 60 min preincubation (32). The time-dependent inhibition equation was modified by setting initial velocity (vi) to 0 and was subsequently rearranged:
| (Eq. 2) |
The microscopic rate constants defining the rate of the formation and dissociation of the EI* complex in Scheme 1 were computed from:
| (Eq. 3) |
where k5 and k6 are defined as described by Morrison (11, 32, 33), IC50 was determined by fitting the IC50 equation to the initial velocities, and [I] is the concentration of CHIR-090 in the assay. The Ki* describing the potency of CHIR-090 after the onset of time-dependent inhibition was derived using:
| (Eq. 4) |
Ki* for E. coli LpxC was determined by independently varying CHIR-090 from 50 pM - 4 nM, keeping the concentration of enzyme constant at 0.2 nM. The Morrison equation was then fitted to the data (32):
| (Eq. 5) |
where vi is the initial velocity of the reaction, vo is the velocity of the uninhibited reaction, [E]T is the empirically determined total enzyme concentration, and [I]T is the total concentration of CHIR-090.
Determination of Kinetic Parameters for E. coli and R. leguminosarum LpxC
KM and Vmax were determined by varying substrate from 0.5 to 50 μM for E. coli LpxC, and from 1 to 100 μM for R. leguminosarum LpxC. Data were analyzed using an Eadie-Hofstee plot (34) and by a non-linear curve-fitting program (KaleidaGraph, Synergy Software); the resultant values were nearly identical and within the error of the experiment. To determine a Ki for CHIR-090 against R. leguminosarum LpxC, CHIR-090 concentrations were varied from 0.1 to 50 μM, and an equation describing a binding isotherm was fit to the data (32). A Ki value was calculated using:
| (Eq. 6) |
Construction of E. coli W3110RL
In this strain, R. leguminosarum lpxC was used to replace E. coli lpxC. A PCR product containing the R. leguminosarum ORF was amplified from R. leguminosarum genomic DNA. PCR was used to construct a linear piece of DNA containing R. leguminosarum lpxC (primers shown in Table S1), flanked on the 5′ end by 40 bp of DNA complementary to the 5′ region upstream of E. coli lpxC, and on the 3′ end by 40 bp of DNA complementary to the 3′ region downstream of E. coli lpxC. This fragment was extracted from an agarose gel, purified using a Qiagen PCR cleanup kit, and eluted with distilled water. The fragment was then electroporated into E. coli W3110 cells, containing the temperature-sensitive pDK46 recombination plasmid (35), using a BioRad Gene Pulser II set to 2.5 kV, 25 μF, and 400 Ω. Because no CHIR-090 resistant E. coli W3110 colonies were observed on LB plates supplemented with 1 μg / mL CHIR-090 (data not shown), transformants could be selected directly using CHIR-090 without introducing a closely linked resistance cassette for a different antibiotic marker. Recombinant colonies containing R. leguminosarum lpxC were selected on LB agar plates, containing 1 μg/mL CHIR-090. Genomic DNA from resistant colonies was isolated, and the region around lpxC was sequenced. One clone in which R. leguminosarum lpxC had replaced E. coli lpxC was selected and grown at 37 °C to eliminate the pKD46 plasmid. This strain was subsequently named E. coli W3110RL.
P1vir Transduction Experiments
E. coli W3110RL was transduced to kanamycin resistance using a P1vir lysate prepared on E. coli ΔyacF-21 (strain EDCM371 obtained from the E. coli Genetic Resource Center at Yale University, which contains a kanamycin resistance cassestte near LpxC around minute 2 on the E. coli chromosome). More than 100 kanamycin-resistant colonies were recovered, and 12 were restreaked onto fresh LB plates containing kanamycin (25 μg/ml); these were then tested on LB plates containing 1 μg/mL CHIR-090 to verify the genetic linkage of kanamycin and CHIR-090 resistance in W3110RL.
LpxC Activity in Cell-Free Lysates
E. coli W3110 and W3110RL were each grown to an A600 of 0.6 and harvested at 4,000 × g for 15 min at 4 °C. Cell pellets were resuspended in 2 mL of 25 mM HEPES, pH 7.0, containing 2 mM DTT, and lysed by three passages through a cold French pressure cell at 18,000 psi. Lysates were cleared by centrifugation at 8,000 × g for 20 min at 4 °C. LpxC activity was determined as described above, except that 0.5 mM AMP was included to inhibit CDP-diglyceride hydrolase-catalyzed cleavage of the substrate (16).
Disk Diffusion Assays of Antibiotic Sensitivity and Bacterial Growth Tests
Disk diffusion was conducted as previously described (11), except that 10 μg of each antibiotic compound was used per filter. Growth in liquid medium in the presence of CHIR-090 was evaluated as follows: cells from overnight cultures were inoculated into 50 mL portions of LB broth at an A600 of 0.02 and grown with shaking at 30 °C. When the A600 reached 0.15, parallel cultures were treated with either 6 μl of 500 μg/mL CHIR-090 in DMSO or 6 μl of DMSO. To assess cumulative growth, cultures were maintained in log phase growth by 10-fold dilution into pre-warmed medium, containing the same concentrations of DMSO or DMSO/CHIR-090, whenever the A600 reached 0.4.
Antibiotic Minimal Inhibitory Concentrations
The minimal inhibitory concentration was defined as the lowest antibiotic concentration at which no measurable bacterial growth was observed in LB medium containing 1% DMSO (v/v), when inoculated at a starting density of A600 = 0.01. Cultures were incubated with shaking for 24 h at 30 °C in the presence of CHIR-090. Experiments were performed in triplicate.
Results
CHIR-090 is a two-step, slow, tight-binding inhibitor of E. coli LpxC
CHIR-090 is a competitive, two-step, slow, tight-binding inhibitor of A. aeolicus LpxC (Scheme 1) (11). Although CHIR-090 also inhibits E. coli LpxC in the low nM range and has potent antibiotic activity against this organism (11), the mechanism of inhibition of the E. coli enzyme was not previously determined. We therefore investigated the question of whether or not CHIR-090 is a time-dependent inhibitor of E. coli LpxC. Equation 1 was fitted to the data from reaction progress curves (Figure 3A), which clearly show slow tight-binding behavior. Analysis of the data from the first minute of the reaction (Figure 3B) demonstrates that CHIR-090 reduces the initial rate of deacetylation, consistent with a two-step mechanism (Scheme 1).
Figure 3. Slow, tight-binding inhibition of E. coli LpxC by CHIR-090.
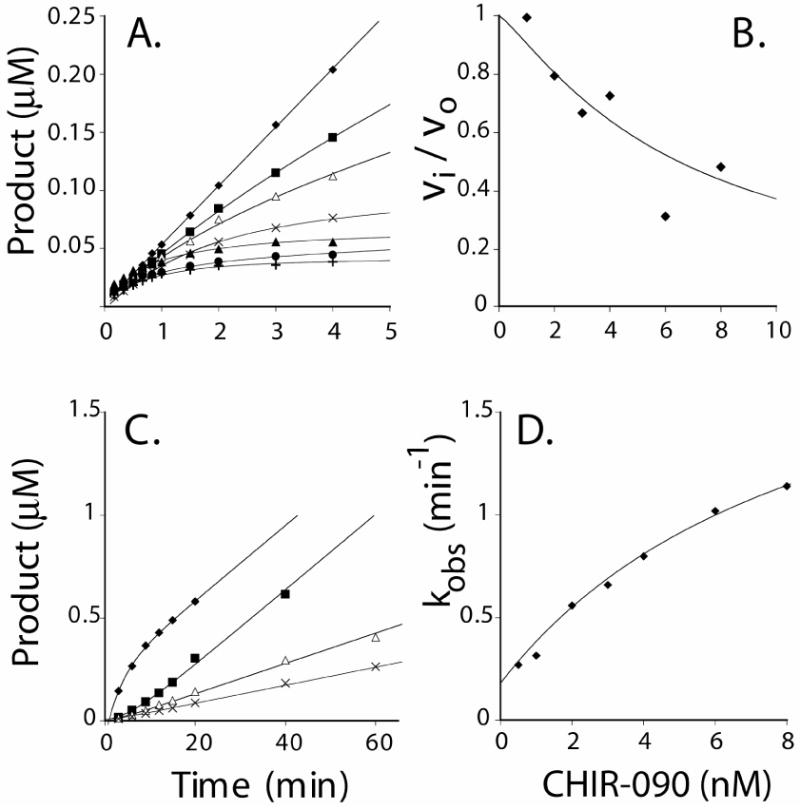
Panel A. Progress curves for product formation when reactions are started by addition of enzyme in the presence 0 nM (υ), 1 nM (ν), 2 nM (Δ), 3 nM (5), 4 nM (σ), 6 nM (λ), or 8 nM (:) CHIR-090. Panel B. Plot of fractional velocities extracted from the results in Panel A as a function of CHIR-090 concentration. A standard IC50 equation was fit to the data with an IC50 of 9 nM. Panel C. Progress curves obtained after rapid dilution of E. coli LpxC pre-incubated with CHIR-090. (υ) Control reaction in which 0.25 nM LpxC was used to start the reaction in the presence of 0.5 nM CHIR-090; (ν) pre-incubation of 50 nM CHIR-090 with 25 nM LpxC diluted 100 times into the assay mixture; (Δ) pre-incubation of 100 nM CHIR-090 with 25 nM LpxC diluted 100 times into the assay mixture; and (5) pre-incubation of 200 nM CHIR-090 with 25 nM LpxC diluted 100 times into the assay mixture. Preincubations were done at 30 °C. Panel D. A plot of the observed first order rate constant for the formation of the EI* complex as fitted with Equation 3. The Y-intercept (0.18 min-1) was determined independently from the dilution experiment shown in Panel C. A repeat experiment (not shown) gave identical results (within 10 %).
The first order rate constant describing the formation of the EI* complex (kobs) was plotted as a function of inhibitor concentration (Figure 3D). The hyperbolic nature of this plot is likewise indicative of a two-step mechanism (Scheme 1) (11, 33). An additional numeric constraint for k6, which describes the reversal of the EI* complex to the EI complex (Scheme 1), was determined independently using a rapid dilution method (32). E. coli LpxC (25 nM) was pre-incubated with 50, 100 or 200 nM CHIR-090, and then diluted 100-fold into a reaction mixture containing 5 μM substrate. The progress curves show that enzyme activity slowly returns (Figure 3C), consistent with the formation of free enzyme. Equation 2 was fit to the data shown in Figure 3C, and a value for k′obs, representative of the microscopic rate constant k6, of 0.18 min-1 was determined. This value was similar to the value determined when CHIR-090 concentration was extrapolated to zero in Figure 3D. The k′obs for this transition provides a lower bound for the true k6.
Using the estimated value of k6, Equation 3 was fit to the data shown in Figure 3D, yielding a value of 1.9 ± 0.3 min-1 for k5 (32, 33). Further analysis, using Equations 3 and 4, yielded a Ki of 4.0 ± 1.0 nM (describing k4/k3) (33), and an overall inhibition constant (Ki*) of 0.4 ± 0.1 nM. Analysis of the microscopic rate constants predicts a half-life of 20 s for the formation of the EI* complex, not dissimilar to the value of 56 s determined for A. aeolicus LpxC (11). In the latter case, however, EI* formation is irreversible.
To confirm the Ki* determined from the microscopic rate constants, we measured the residual activity of E. coli LpxC after the slow-binding step had reached equilibrium. In order to use the Morrison Equation (Eq. 5) to derive Ki*, it was necessary to know the active enzyme concentration. This value was determined by finding the concentration at which LpxC activity became measurable when the protein was titrated into a reaction mixture containing 100 nM CHIR-090 (32). By this criterion the active enzyme concentration was within 10% of the estimated total protein concentration, as determined by A280 as previously described (Figure 4A) (31). E. coli LpxC (0.2 nM) was next assayed in the presence of 0.05 nM to 4 nM inhibitor (Figure 4B). In this case, the reaction rates were determined from the linear portions of the progress curves after the full onset of slow, tight-binding inhibition (or 5 min into the reaction). The Ki* value determined using Eq. 5 (0.5 ± 0.1 nM) was similar to that determined by the curve fitting method described above (0.4 ± 0.1 nM).
Figure 4. Determination of Ki* under conditions of similar enzyme and CHIR-090 concentration.

A plot of initial velocity measurements with different amounts of E. coli LpxC in the presence of 100 nM CHIR-090 is shown in Panel A. The active enzyme concentration was determined by extrapolating to zero a line representing the linear fit of reactions with measurable activity. For these data, this line intersects the x-axis at 90 nM, suggesting the active enzyme concentration is within 10% of the enzyme concentration estimated by UV absorption. In Panel B, fractional activity (vi/vo) was measured over a range of CHIR-090 concentrations, where the amount of enzyme was held constant at 0.2 nM. Using the active enzyme concentration calculated from the data shown in Panel A, Equation 5 was fitted to the data in Panel B, yielding a Ki* value of 0.5 ± 0.1 nM. A repeat experiment (not shown) gave identical results (within 10 %).
CHIR-090 is a potent inhibitor of diverse LpxC orthologues
We next determined whether or not CHIR-090 inhibits LpxCs from other Gram-negative organisms. Four diverse LpxC orthologues (P. aeruginosa, H. pylori, N. meningiditis, and R. leguminosarum), which share 57 %, 43 %, 49 % and 43 % sequence identity to E. coli LpxC respectively, were chosen. To measure the potency of CHIR-090 inhibition against these orthologues, we compared the initial reaction velocities under standard assay conditions using purified enzymes in the presence or absence of CHIR-090. As with E. coli LpxC (Figure 5A), H. pylori LpxC (Figure 5B), P. aeruginosa LpxC (Figure 5C) and N. meningitidis LpxC (data not shown) were inhibited 75 % or more by 4 nM CHIR-090 when enzyme was used to start the reactions. The specific activity of the N. meningitidis protein was at least 20-fold less than that of the others, suggesting that the expression or assay conditions for N. meningitidis LpxC may not be optimal (Table S1). To investigate possible time-dependent inhibition by CHIR-090, we pre-incubated each enzyme with 16 nM CHIR-090 for 15 min before diluting four-fold into the assay mixture containing the substrate. We then compared the rates and extents of product formation to those obtained without pre-incubation (Figure 5A, B and C). For the assays done without pre-incubation, there was considerably more product accumulation by the first time point, consistent with slow-binding inhibition in each case (Figure 5A, B and C). A residual slow rate of product formation was observed with each of these enzymes (Figure 5A, B and C), suggesting that EI* formation is reversible, in contrast to A. aeolicus LpxC (11).
Figure 5. The effect of CHIR-090 on diverse LpxC orthologues.

In each panel the symbols indicate the following order of addition: (Δ) the CHIR-090 concentration was 16 nM during a 15 min pre-incubation with enzyme and 4 nM following dilution into the assay system; (ν) the CHIR-090 concentration was 0 nM during the pre-incubation but 4 nM in the assay mixture; (◆) the CHIR-090 concentration was 0 nM during the pre-incubation and 0 nM in the assay mixture. Panel A. E. coli LpxC. Panel B. H. pylori LpxC. Panel C. P. aeruginosa LpxC. Panel D. R. leguminosarum LpxC. R. leguminosarum LpxC was assayed with ten-fold higher CHIR-090 concentrations (160 nM during the pre-incubation and 40 nM in the assay mixture). Preincubations were done at 4 °C and reactions at 30 °C. The points are connected for ease of viewing. A repeat experiment (not shown) gave identical results (within 10 %).
CHIR-090 is a weak competitive inhibitor of R. leguminosarum LpxC
Surprisingly, R. leguminosarum LpxC was not inhibited by 40 nM CHIR-090 (Figure 5D) under the same conditions that showed > 75 % inhibition of the other orthologues by only 4 nM CHIR-090 (Figure 5A, B and C). There was no evidence for time-dependent inhibition with the R. leguminosarum enzyme (Figure 5D) at any concentration tested. Furthermore, CHIR-090 did not inhibit the growth of R. leguminosarum, Rhizobium etli, Sinorhizobium meliloti or Agrobacterium tumefaciens, as judged by standard disk diffusion assays (data not shown) (11). These CHIR-090 resistant bacteria all belong to the Rhizobiaceae family, a group of soil-borne Gram-negative organisms that infect plants (36).
The KM and kcat of R. leguminosarum LpxC were determined (Table I) as described in the Methods section. The R. leguminosarum values were similar to those for E. coli and were consistent with previous studies (31). There was no evidence for allosteric effects or product inhibition (data not shown).
Table 1.
Kinetics and Inhibition of E. coli versus R. leguminosarum LpxC
| kcat /KM | CHIR-090 | CHIR-090 | |||
|---|---|---|---|---|---|
| LpxC source | KM (μM) | kcat (s-1) | (M-1 s-1) | Ki (nM) | Ki* (nM) |
| E. colia | 4.0 ± 0.5 | 4.2 ± 1.3 | 11 × 105 | 4.0 ± 1.0c | 0.4 ± 0.1c |
| 0.5 ± 0.1d | |||||
| E. colib | 2.1 ± 0.5 | 3.3 ± 0.2 | 15 × 105 | n.d. | n.d. |
| R. leguminosaruma | 4.8 ± 0.4 | 1.7 ± 0.5 | 3.5 × 105 | 340 ± 60 | n.a. |
n.a. – not applicable
n.d. – not determined
this work
Jackman et al. (31)
determined by curve fitting
determined by linear fitting
To determine the Ki of CHIR-090, the R. leguminosarum enzyme was assayed with 5 μM substrate in the presence of 4 nM to 50 μM inhibitor (Figure 6). Using the kinetic parameters established above and the measured IC50 in conjunction with Equation 6 for a simple competitive inhibitor, the Ki for CHIR-090 was calculated to be 340 ± 60 nM (Table 1). There was no evidence for slow, tight-binding inhibition.
Figure 6. Inhibition of R. leguminosarum LpxC by of CHIR-090.

An IC50 for CHIR-090 inhibition of R. leguminosarum LpxC activity at 5 μM substrate was determined from a plot of fractional activity (vi/vo) versus CHIR-090 concentration. The IC50 value was calculated using the equation vi / vo = 1 / (1 + [I] / IC50), yielding an IC50 value of 0.69 μM. A repeat experiment (not shown) gave identical results (within 10 %).
CHIR-090 primarily targets E. coli LpxC in vivo
CHIR-090 is a potent antibiotic against E. coli and inhibits E. coli LpxC activity in vitro in the low nM range (Figure 3). E. coli W3110 colonies resistant to 1 μg/mL CHIR-090 are not observed without prior chemical mutagenesis (data not shown). To determine whether or not CHIR-090 targets other enzymes besides LpxC, we constructed a strain of E. coli W3110 (Figure 7), designated W3110RL, in which R. leguminosarum lpxC replaces the chromosomal copy of E. coli lpxC. This strain is able to grow on LB agar plates containing 1 to 10 μg/mL CHIR-090, which is 4 to 40 times above the MIC of 0.25 μg/mL under our conditions for wild-type E. coli W3110 (data not shown). The doubling time of W3110RL was 40 min in the presence of 1 μg/mL CHIR-090, which is exactly the same rate as wild-type in the absence of inhibitor (Figure 8A). Wild-type cells stopped growing after about 2 h in the presence of 1 μg/mL CHIR-090 (Figure 8A).
Figure 7. E. coli W3110RL is much less sensitive to CHIR-090.

Disk diffusion tests were used to test for antibiotic sensitivity. Panel A. CHIR-090 inhibits the growth of the parental strain E. coli W3110 with potency that is intermediate between tobramycin and ciprofloxacin. Panel B. E. coli W3110RL is completely resistant to L-161,240 and much less sensitive to CHIR-090 than the parental strain. The compounds were tested at 10 μg per disk: tobramycin (1), L,161-240 (2), CHIR-090 (3) and ciprofloxacin (4). Abbreviations: Ec, E. coli; gDNA, genomic DNA; ORF, open reading frame.
Figure 8. Growth of E. coli W3110RL in the presence of CHIR-090 and resistance of LpxC to CHIR-090 in W3110RL extracts.
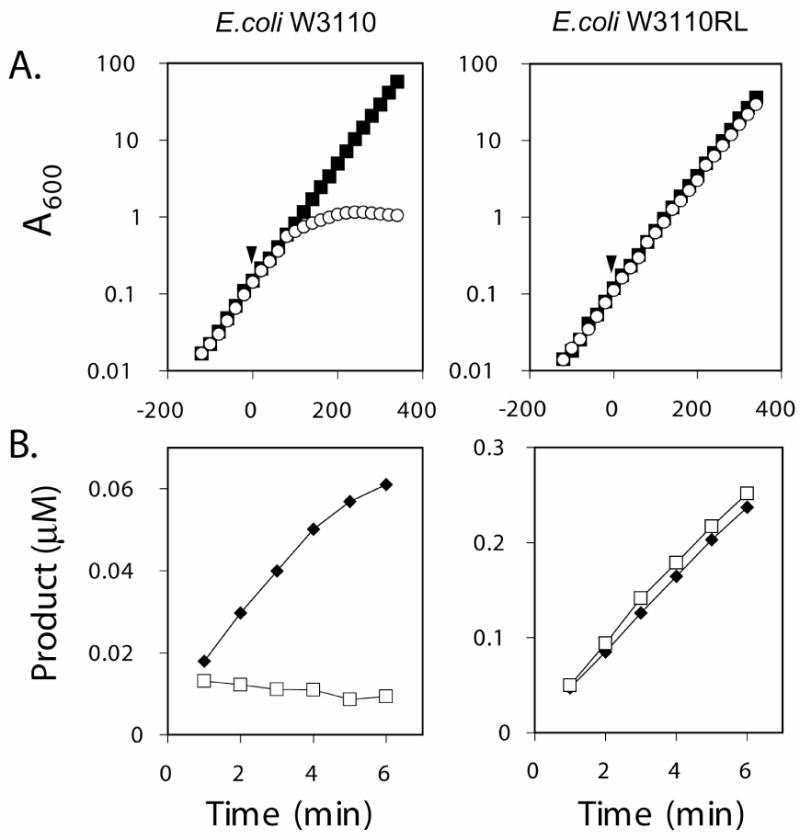
Panel A. The cumulative growth yield of the parental strain E. coli W3110 (left graph) was compared to that of E. coli W3110RL (right graph) when DMSO (ν) or CHIR-090 dissolved in DMSO ( ) was added at time 0, as indicated by the arrow. The final concentration of CHIR-090 and DMSO in the culture medium was 1 μg/mL and 0.012%, respectively. Panel B. LpxC activity in cell-free extracts of E. coli W3110 (left graph) or W3110RL (right graph) was measured in the presence of 10% DMSO (♥) or 10% DMSO plus 50 nM CHIR-090 (
) was added at time 0, as indicated by the arrow. The final concentration of CHIR-090 and DMSO in the culture medium was 1 μg/mL and 0.012%, respectively. Panel B. LpxC activity in cell-free extracts of E. coli W3110 (left graph) or W3110RL (right graph) was measured in the presence of 10% DMSO (♥) or 10% DMSO plus 50 nM CHIR-090 ( ). The concentration of cell free extract in the assay system was 0.83 mg/mL for E. coli W3110 and 0.99 mg/mL for E. coli W3110RL. A repeat experiment (not shown) gave identical results (within 10 %).
). The concentration of cell free extract in the assay system was 0.83 mg/mL for E. coli W3110 and 0.99 mg/mL for E. coli W3110RL. A repeat experiment (not shown) gave identical results (within 10 %).
We next tested whether or not the CHIR-090 resistance of E. coli W3110RL is due to the presence of R. leguminosarum lpxC (located at minute 2.3 on the E. coli chromosome) or is caused by a spontaneous mutation not linked to this locus. Transduction of E. coli W3110RL with a P1vir lysate prepared on E. coli ΔyacF-21, a strain containing a kanamycin-resistance marker at minute 2.4, generated kanamycin resistant colonies with the expected frequency. Of 12 randomly selected kanamycin resistant colonies, all were sensitive to 1 μg / mL CHIR-090. Therefore, the CHIR-090 resistance of E. coli W3110RL shows the expected genetic linkage to the lpxC locus.
The W3110RL phenotype is stable, as demonstrated by removing CHIR-090 for several generations without loss of resistance. DNA sequencing confirmed that the R. leguminosarum lpxC gene replaced E. coli lpxC without altering the native chromosomal DNA sequence upstream or downstream of the insert.
As shown by diffusion assays with 10 μg antibiotic per disk (Figures 7A and 7B), W3110 and W3110RL were equally susceptible to tobramycin and ciprofloxacin. However, W3110RL was much less sensitive to CHIR-090 than was W3110, and W3110RL was completely resistant to L-161,240 (Figures 7A and 7B). The MIC of CHIR-090 against W3110RL in liquid medium is 100 μg/mL, compared to 0.25 μg/ mL for W3110 (data not shown). This ∼ 400-fold reduction in sensitivity of W3110RL to CHIR-090 is consistent with the ∼ 600-fold increase in the Ki of R. leguminosarum LpxC versus the Ki* of E. coli LpxC for CHIR-090 (Table 1).
To confirm the presence of R. leguminosarum LpxC activity in E. coli W3110RL, we assayed cell-free extract of this strain in the presence and absence of 50 nM CHIR-090 (Figure 8B). There was no inhibition of LpxC activity in extracts of this construct, whereas LpxC activity was inhibited completely in extracts of E. coli W3110 (Fig. 8B). R. leguminosarum lpxC expressed from an appropriate promoter may therefore be useful as a selectable antibiotic resistance marker in the transformation or transduction of CHIR-090-sensitive bacteria.
Based on these data, we conclude that LpxC is indeed the primary intracellular target for CHIR-090 in wild-type E. coli at concentrations up to at least 50 μg/mL.
Discussion
In the present study we have demonstrated that CHIR-090 is a potent slow, tight-binding inhibitor of LpxC orthologues from several important Gram-negative pathogens. CHIR-090 is therefore an excellent lead compound for the further development of lipid A biosynthesis inhibitors as clinical antibiotics. Because its mode of action is distinct from those of all commercial antibiotics, CHIR-090 should be effective against multi-drug resistant Gram-negative bacteria, as are often encountered in cystic fibrosis patients or debilitated individuals (37, 38). As yet, the pharmacokinetics, efficacy and safety of CHIR-090 in animal models have not been reported.
Our kinetic evaluation of E. coli LpxC showed that, similar to A. aeolicus LpxC, CHIR-090 inhibition occurs by a two-step, time-dependent mechanism with a low nM Ki (1.0 - 1.7 nM for A. aeolicus LpxC versus 4.0 nM for E. coli LpxC) (11). Likewise, the H. pylori and P. aeruginosa LpxC orthologues are inhibited by low nM levels of CHIR-090 in a time-dependent manner (Figure 5). A thorough analysis of inhibition kinetics is essential when evaluating therapeutic leads. Time-dependent inhibitors are highly desirable because they mitigate the effects of substrate accumulation (37, 38). Slow, tight-binding effects are therefore expected to enhance the relative potency of a compound, reducing the necessary dosage and avoiding potential off-target toxic effects.
The observation that R. leguminosarum and related Gram-negative bacteria are resistant to CHIR-090 afforded an opportunity to validate the specificity and mechanism of this compound as an antibiotic. We found that CHIR-090 inhibits R. leguminosarum LpxC 600-fold less effectively than E. coli LpxC (Table I). Furthermore, CHIR-090 does not display slow, tight-binding inhibition with R. leguminosarum LpxC, and it does not inhibit the growth of R. leguminosarum cells, as judged by disk diffusion assays (data not shown). The small halo of growth inhibition seen with E. coli W3110RL (Figure 7) in which R. leguminosarum lpxC replaces E. coli lpxC does suggest that the insensitivity of R. leguminosarum LpxC to CHIR-090 is not the only reason for the resistance of R. leguminosarum cells. This discrepancy could be explained in several ways. E. coli K-12 and R. leguminosarum (biovar viciae strain 3841) differ in that R. leguminosarum synthesizes LPS-containing O-antigen repeats, which are necessary for the establishment of symbiosis (39-41). The R. leguminosarum outer membrane may therefore be less permeable to CHIR-090 than that of E. coli W3110, which lacks O-antigen (42). Alternatively, CHIR-090 may be pumped out of the R. leguminosarum cytosol or be detoxified by a unique modification pathway. Furthermore, we cannot exclude the possibility that a secondary target for CHIR-090 accounts for the residual antibiotic activity seen with E. coli W3110RL (Figure 7). Given the relative resistance of W3110RL towards CHIR-090 (MIC of 100 μg/mL versus 0.25 μg/mL for W3110), we can conclude that CHIR-090 is indeed highly selective for LpxC in E. coli K-12 (Figure 8).
Unlike the situation with L-161,240 (9, 10), spontaneous resistant colonies are not seen in disk diffusion assays with CHIR-090. Furthermore, no spontaneous resistant colonies are observed when 109 W3110 cells are plated onto agar containing 1 or 10 μg/mL CHIR-090. Prior to CHIR-090, the most effective LpxC inhibitor was BB-78485 (Figure 2) and the related compound BB-78484. As with L-161,240, BB-78484-resistant colonies could be isolated easily from cultures grown with BB-78484 present at 8-times its MIC (12). Most of the BB-78484-resistant bacteria contained single amino acid substitutions in either FabZ (12), the 3-hydroxyacyl-ACP dehydratase of fatty acid biosynthesis (43), or in LpxC (12). Although not seen with wild-type E. coli W3110, CHIR-090-resistant colonies were observed in the disk diffusion assay of E. coli W3110RL (not visible in Figure 7B). These colonies have not yet been characterized. Taken together, the data suggest that multiple mutations in one or more genes of wild-type E. coli may be required to generate CHIR-090 resistance. This feature should limit the development of resistance to CHIR-090 in clinical settings.
We are currently exploring the structural basis for the insensitivity of R. leguminosarum LpxC to CHIR-090. Several amino acid residues that line the fatty acid binding tunnel of LpxC are not conserved in R. leguminosarum and related soil organisms, when compared to sensitive strains. These differences might account for the resistance of R. leguminosarum LpxC to CHIR-090. The NMR (18, 20) and crystal (21, 22) structures of the CHIR-090-sensitive A. aeolicus LpxC are available and should facilitate the structural analysis of R. leguminosarum LpxC by molecular modeling. It will also be very informative to determine a high-resolution structure of a CHIR-090-sensitive LpxC orthologue with bound CHIR-090, and to determine whether or not the biphenyl acetylene unit of CHIR-090 (Figure 2) is positioned within the fatty acid binding tunnel of LpxC. Structure-based methods were not used to design any of the available LpxC inhibitors. When the results of ongoing structural studies are incorporated into the design of new compounds, it is likely that LpxC inhibitors with greater antibiotic potency, alternative zinc binding motifs, and/or enhanced pharmacokinetics will be identified.
Supplementary Material
Table S1 (Primers Used in this Study) and Table S2 (Purification of LpxC orthologues)
Acknowledgments
We would like to thank Dr. Johannes Rudolph for helpful discussions, assistance with curve fitting and a critical reading of the manuscript, and Dr. Z. Guan for his help with electrospray ionization mass spectrometry.
This research was supported by NIH grants GM-51310 to C. R. H. Raetz and AI-055588 to P. Zhou. A. Barb was supported by Cellular and Molecular Biology training grant GM-07184 to Duke University.
List of Abbreviations
- ACP
acyl carrier protein
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- Kdo
3-deoxy-d-manno-octulosonic acid
- IC50
concentration of half maximal inhibition
- IPTG
isopropyl β- d -thiogalactopyranoside
- LPS
lipopolysaccharide
- MIC
minimal inhibitory concentration
- NMR
nuclear magnetic resonance
- ORF
open reading frame
- PCR
polymerase chain reaction
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- UDP-3-O-acyl-GlcNAc
UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine
- UDP-GlcNAc
UDP-N-acetylglucosamine
Footnotes
Supporting Information Available: This material is available free of charge via the internet at: http://pubs.acs.org
References
- 1.Levy SB. Antibiotic resistance-the problem intensifies. Adv Drug Deliv Rev. 2005;57:1446–1450. doi: 10.1016/j.addr.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004;10:S122–129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 3.Wright MO. Multi-resistant gram-negative organisms in Maryland: a statewide survey of resistant Acinetobacter baumannii. Am J Infect Control. 2005;33:419–421. doi: 10.1016/j.ajic.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 4.Walsh CT. Where will new antibiotics come from? Nat Rev Microbiol. 2003;1:65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- 5.Projan SJ. New (and not so new) antibacterial targets - from where and when will the novel drugs come? Curr Opin Pharmacol. 2002;2:513–522. doi: 10.1016/s1471-4892(02)00197-2. [DOI] [PubMed] [Google Scholar]
- 6.Gerdes SY, Scholle MD, Campbell JW, Balazsi G, Ravasz E, Daugherty MD, Somera AL, Kyrpides NC, Anderson I, Gelfand MS, Bhattacharya A, Kapatral V, D'Souza M, Baev MV, Grechkin Y, Mseeh F, Fonstein MY, Overbeek R, Barabasi AL, Oltvai ZN, Osterman AL. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J Bacteriol. 2003;185:5673–5684. doi: 10.1128/JB.185.19.5673-5684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raetz CRH. Biochemistry of endotoxins. Annu Rev Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 8.Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen MH, Patchett AA, Galloway SM, Hyland SA, Anderson MS, Raetz CRH. Antibacterial agents that inhibit lipid A biosynthesis. Science. 1996;274:980–982. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 10.Jackman JE, Fierke CA, Tumey LN, Pirrung M, Uchiyama T, Tahir SH, Hindsgaul O, Raetz CRH. Antibacterial agents that target lipid A biosynthesis in gram-negative bacteria. Inhibition of diverse UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylases by substrate analogs containing zinc binding motifs. J Biol Chem. 2000;275:11002–11009. doi: 10.1074/jbc.275.15.11002. [DOI] [PubMed] [Google Scholar]
- 11.McClerren AL, Endsley S, Bowman JL, Andersen NH, Guan Z, Rudolph J, Raetz CRH. A slow, tight-binding inhibitor of the zinc-dependent deacetylase LpxC of lipid A biosynthesis with antibiotic activity comparable to ciprofloxacin. Biochemistry. 2005;44:16574–16583. doi: 10.1021/bi0518186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clements JM, Coignard F, Johnson I, Chandler S, Palan S, Waller A, Wijkmans J, Hunter MG. Antibacterial activities and characterization of novel inhibitors of LpxC. Antimicrob Agents Chemother. 2002;46:1793–1799. doi: 10.1128/AAC.46.6.1793-1799.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson MS, Bulawa CE, Raetz CRH. The biosynthesis of gram-negative endotoxin: formation of lipid A precursors from UDP-GlcNAc in extracts of Escherichia coli. J Biol Chem. 1985;260:15536–15541. [PubMed] [Google Scholar]
- 14.Anderson MS, Robertson AD, Macher I, Raetz CRH. Biosynthesis of lipid A in Escherichia coli: identification of UDP-3-O-(R-3-hydroxymyristoyl)-α-D-glucosamine as a precursor of UDP-N2-O3-bis-(R-3-hydroxymyristoyl)-α-D-glucosamine. Biochemistry. 1988;27:1908–1917. doi: 10.1021/bi00406a017. [DOI] [PubMed] [Google Scholar]
- 15.Anderson MS, Bull HS, Galloway SM, Kelly TM, Mohan S, Radika K, Raetz CRH. UDP-N-acetylglucosamine acyltransferase of Escherichia coli: the first step of endotoxin biosynthesis is thermodynamically unfavorable. J Biol Chem. 1993;268:19858–19865. [PubMed] [Google Scholar]
- 16.Sorensen PG, Lutkenhaus J, Young K, Eveland SS, Anderson MS, Raetz CRH. Regulation of UDP-3-O-[R-3-hydroxymyristoyl]-N-acetyl-glucosamine deacetylase in Escherichia coli. The second enzymatic step of lipid A biosynthesis. J Biol Chem. 1996;271:25898–25905. doi: 10.1074/jbc.271.42.25898. [DOI] [PubMed] [Google Scholar]
- 17.McClerren AL, Zhou P, Guan Z, Raetz CRH, Rudolph J. Kinetic analysis of the zinc-dependent deacetylase in the lipid A biosynthetic pathway. Biochemistry. 2005;44:1106–1113. doi: 10.1021/bi048001h. [DOI] [PubMed] [Google Scholar]
- 18.Coggins BE, McClerren AL, Jiang L, Li X, Rudolph J, Hindsgaul O, Raetz CRH, Zhou P. Refined solution structure of the LpxC-TU-514 complex and pKa analysis of an active site histidine: insights into the mechanism and inhibitor design. Biochemistry. 2005;44:1114–11126. doi: 10.1021/bi047820z. [DOI] [PubMed] [Google Scholar]
- 19.Hernick M, Gennadios HA, Whittington DA, Rusche KM, Christianson DW, Fierke CA. UDP-3-O-((R)-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase functions through a general acid-base catalyst pair mechanism. J Biol Chem. 2005;280:16969–16978. doi: 10.1074/jbc.M413560200. [DOI] [PubMed] [Google Scholar]
- 20.Coggins BE, Li X, McClerren AL, Hindsgaul O, Raetz CRH, Zhou P. Structure of the LpxC deacetylase with a bound substrate-analog inhibitor. Nat Struct Biol. 2003;10:645–651. doi: 10.1038/nsb948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whittington DA, Rusche KM, Shin H, Fierke CA, Christianson DW. Crystal structure of LpxC, a zinc-dependent deacetylase essential for endotoxin biosynthesis. Proc Natl Acad Sci U S A. 2003;100:8146–8150. doi: 10.1073/pnas.1432990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gennadios HA, Whittington DA, Li X, Fierke CA, Christianson DW. Mechanistic inferences from the binding of ligands to LpxC, a metal-dependent deacetylase. Biochemistry. 2006;45:7940–7948. doi: 10.1021/bi060823m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kline T, Andersen NH, Harwood EA, Bowman J, Malanda A, Endsley S, Erwin AL, Doyle M, Fong S, Harris AL, Mendelsohn B, Mdluli K, Raetz CRH, Stover CK, Witte PR, Yabannavar A, Zhu S. Potent, novel in vitro inhibitors of the Pseudomonas aeruginosa deacetylase LpxC. J Med Chem. 2002;45:3112–29. doi: 10.1021/jm010579r. [DOI] [PubMed] [Google Scholar]
- 24.Pirrung MC, Tumey LN, McClerren AL, Raetz CRH. High-throughput catch-and-release synthesis of oxazoline hydroxamates. Structure-activity relationships in novel inhibitors of Escherichia coli LpxC: in vitro enzyme inhibition and antibacterial properties. J Am Chem Soc. 2003;125:1575–86. doi: 10.1021/ja0209114. [DOI] [PubMed] [Google Scholar]
- 25.Mdluli KE, Witte PR, Kline T, Barb AW, Erwin AL, Mansfield BE, McClerren AL, Pirrung MC, Tumey LN, Warrener P, Raetz CRH, Stover CK. Molecular validation of LpxC as an antibacterial drug target in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2006;50:2178–2184. doi: 10.1128/AAC.00140-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanjilal-Kolar S, Basu SS, Kanipes MI, Guan Z, Garrett TA, Raetz CRH. Expression cloning of three Rhizobium leguminosarum lipopolysaccharide core galacturonosyltransferases. J Biol Chem. 2006;281:12865–12878. doi: 10.1074/jbc.M513864200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hyland SA, Eveland SS, Anderson MS. Cloning, Expression, and Purification of UDP-3-O-Acyl-GlcNAc deacetylase from Pseudomonas aeruginosa: a metalloamidase of the lipid A biosynthesis pathway. J Bacteriol. 1997;179:2029–2037. doi: 10.1128/jb.179.6.2029-2037.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen NH, Bowman J, Erwin A, Harwood E, Kline T, Mdluli K, Pfister KB, Shawar R, Wagman A, Yabannavar A. Patent WO 2004/062601 A2. Chiron; Emeryville California: 2004. Antibacterial Agents. World Intellectual Property Organization; p. 324. [Google Scholar]
- 29.Miller JR. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 30.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 31.Jackman JE, Raetz CRH, Fierke CA. UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase of Escherichia coli is a zinc metalloenzyme. Biochemistry. 1999;38:1902–1911. doi: 10.1021/bi982339s. [DOI] [PubMed] [Google Scholar]
- 32.Copeland RA. Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists. Wiley-Interscience; Hoboken, NJ: 2005. [PubMed] [Google Scholar]
- 33.Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 34.Dowd JE, Riggs DS. A Comparison Of Estimates Of Michaelis-Menten Kinetic Constants From Various Linear Transformations. J Biol Chem. 1965;240:863–869. [PubMed] [Google Scholar]
- 35.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stacey G, Burris RH, Evans HJ. Biological Nitrogen Fixation. Routledge, Chapman and Hall, Inc.; New York: 1992. p. 943. [Google Scholar]
- 37.Conway SP, Brownlee KG, Denton M, Peckham DG. Antibiotic treatment of multidrug-resistant organisms in cystic fibrosis. Am J Respir Med. 2003;2:321–332. doi: 10.1007/BF03256660. [DOI] [PubMed] [Google Scholar]
- 38.Garau J, Gomez L. Pseudomonas aeruginosa pneumonia. Curr Opin Infect Dis. 2003;16:135–143. doi: 10.1097/00001432-200304000-00010. [DOI] [PubMed] [Google Scholar]
- 39.de Maagd RA, Rao AS, Mulders IH, Goosen-de Roo L, van Loosdrecht MC, Wijffelman CA, Lugtenberg BJ. Isolation and characterization of mutants of Rhizobium leguminosarum bv. viciae 248 with altered lipopolysaccharides: possible role of surface charge or hydrophobicity in bacterial release from the infection thread. J Bacteriol. 1989;171:1143–1150. doi: 10.1128/jb.171.2.1143-1150.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carlson RW, Reuhs B, Chen TB, Bhat UR, Noel KD. Lipopolysaccharide core structures in Rhizobium etli and mutants deficient in O-antigen. J Biol Chem. 1995;270:11783–11788. doi: 10.1074/jbc.270.20.11783. [DOI] [PubMed] [Google Scholar]
- 41.Noel KD, Forsberg LS, Carlson RW. Varying the abundance of O antigen in Rhizobium etli and its effect on symbiosis with Phaseolus vulgaris. J Bacteriol. 2000;182:5317–5324. doi: 10.1128/jb.182.19.5317-5324.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stevenson G, Neal B, Liu D, Hobbs M, Packer NH, Batley M, Redmond JW, Lindquist L, Reeves P. Structure of the O-antigen of Escherichia coli K-12 and the sequence of its rfb gene cluster. J Bacteriol. 1994;176:4144–4156. doi: 10.1128/jb.176.13.4144-4156.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohan S, Kelly TM, Eveland SS, Raetz CRH, Anderson MS. An Escherichia coli gene (fabZ) encoding R-3-hydroxymyristoyl acyl carrier protein dehydrase. Relation to fabA and suppression of mutations in lipid A biosynthesis. J Biol Chem. 1994;269:32896–32903. [PubMed] [Google Scholar]
- 44.Raetz CRH, Garrett TA, Reynolds CM, Shaw WA, Moore JD, Smith DC, Jr, Ribeiro AA, Murphy RC, Ulevitch RJ, Fearns C, Reichart D, Glass CK, Benner C, Subramaniam S, Harkewicz R, Bowers-Gentry RC, Buczynski MW, Cooper JA, Deems RA, Dennis EA. Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J Lipid Res. 2006;47:1097–1111. doi: 10.1194/jlr.M600027-JLR200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 (Primers Used in this Study) and Table S2 (Purification of LpxC orthologues)