

Abstract
Purpose
Potent endogenous protection from ischemia can be induced in the retina by ischemic preconditioning (IPC). Protein kinase B/Akt is a cellular survival factor. We hypothesized that Akt was integral to IPC based upon differential effects of Akt subtypes.
Methods
Rats were subjected to retinal ischemia after IPC or IPC-mimicking by the opening of mitochondrial KATP (mKATP) channels. The effects of blocking Akt using wortmannin, API-2, or small interfering RNA (siRNA) were examined. Electroretinography assessed functional recovery after ischemia, and TUNEL examined retinal ganglion cell apoptosis. We studied the relationship between Akt activation, and known initiators of IPC, including adenosine receptor stimulation and the opening of mKATP channels.
Results
The PI-3 kinase inhibitor wortmannin 1 or 4 mg/kg (i.p.), the specific Akt inhibitor API-2, 5-500 μM in the vitreous, or intravitreal siRNA directed against Akt2 or -3, but not Akt1, significantly attenuated the neuroprotective effect of IPC. Interfering RNA against any of the three Akt subtypes significantly but time-dependently attenuated mKATP channel opening to mimic IPC. Adenosine A1 receptor blockade (DPCPX), A2a blockade (CSC), or the mKATP channel blocker 5-hydroxydecanoic acid significantly attenuated Akt activation after IPC. Interfering RNA directed against Akt subtypes prevented the ameliorative effect of IPC on post-ischemic apoptosis.
Conclusions
All three Akt subtypes are involved in functional retinal neuroprotection by IPC or IPC-mimicking. Akt is downstream of adenosine A1 and A2a receptors and mKATP channel opening. The results indicate the presence in the retina of robust and redundant endogenous neuroprotection based upon subtypes of Akt.
Keywords: Akt, electroretinography, ischemic preconditioning, protein kinase B, retinal ischemia
Retinal ischemia is associated with vascular diseases that cause visual loss. The retina's blood and oxygen supply may be decreased in diabetic retinopathy, central retinal artery occlusion, carotid artery stenosis, and sickle cell retinopathy. Previously we demonstrated that a profound endogenous protective capacity in the rat retina can be activated (ischemic preconditioning, IPC) to attenuate ischemic injury (Roth et al. 1998; Zhang et al. 2002; Roth et al. 2006). Such protection offers a model for uncovering pathways and agents to treat retinal ischemia. Ischemic preconditioning (IPC) protects the rat retina against the deleterious effects of a severe ischemic event (Roth et al. 1998; Li and Roth 1999; Li et al. 2000; Li et al. 2003).
The cytoplasmic proto-oncogene 57 kD serine/threonine protein kinase Akt (protein kinase B, PKB), is one of the most important kinases, and at the core of numerous and diverse physiological functions. It is activated by receptor tyrosine kinases, integrins, G-protein coupled receptors, and other stimuli, with over 100 reported non-redundant Akt substrates (Manning and Cantley 2007). The Akt pathway has been identified as a cellular survival signal in diverse neuronal cell types and in protection from oxidative stress (Chong et al. 2005). Akt phosphorylates Ser136 on Bad, thus interfering with mitochondrial cytochrome C release (Datta et al. 2000), and it inhibits caspase-9 by phosphorylating Ser196 (Cardone et al. 1998). Via phosphorylation of transcription factors including FOXO and p53, FOXO-mediated transcription of apoptosis-promoting proteins is inhibited (Greer and Brunet 2005). Inhibition of Akt has received increasing attention as a potential anti-cancer treatment (Robey and Hay 2006).
The three mammalian enzymatic isoforms, Akt1, 2, and 3, have extensive homology to protein kinases A, G, and C. Isotypes are similar in structure, size, and substrate specificity, but differ in organ distribution (Manning and Cantley 2007). Knockouts differentially result in developmental defects (Akt1), insulin-resistance (Akt2), and defective brain development (Akt3) (Fayard et al. 2005). These distinct phenotypes suggest cellular differences in functionality. Few data are available, however, on the role of Akt subtypes in disease states.
Upregulation of Akt has ameliorative effects upon ischemic organs (Mullonkal and Toledo-Pereyra 2007). Recent evidence supports a role for Akt in survival of retinal cells under diverse conditions impacting retinal survival, including chronic ocular hypertension, (Huang et al. 2008) diabetic retinopathy, (Liu et al. 2008) photoreceptor light damage, (Li et al. 2007; Ueki et al. 2008), angiogenesis (Kitamura et al. 2008), retinal development, (Chavarria et al. 2007) and growth hormone action. (Sanders et al. 2006) Vascular endothelial growth factor protected axotomized retinal ganglion cells (RGCs) by activation of Akt. (Kilic et al. 2006) Moreover, neurotrophin signaling complex LINGO-1 antagonists increased Akt activation, and rescued RGCs from cell death after optic nerve transaction or ocular hypertension, an effect eliminated by PI-3 kinase blockade.(Fu et al. 2008)
Because of its involvement in cell survival, Akt is a logical candidate as a critical mediator in the ischemic tolerance induced by IPC. Indeed, pharmacological blockade of Akt attenuated hypoxic preconditioning in human brain endothelial cells (Zhang et al. 2007) and in the brains of post-natal rats (Yin et al. 2007). Results were conflicting when PC12 cells were exposed to oxygen and glucose deprivation (Hillion et al. 2006). The findings suggest that Akt may be an essential mediator in the endogenous protection provided by IPC, and also that enhancing Akt-mediated mechanisms of survival may decrease neuronal ischemic injury. We hypothesized differential actions for Akt subtypes in retinal IPC. In this study we examined the effects of the Akt isoforms in vivo, using a well described model of retinal IPC. We also examined the relationship between Akt's role as a mediator in IPC and some of the significant IPC initiators we previously uncovered including adenosine receptors and mitochondrial KATP channels.
Methods
Ischemia methodology
Procedures (Roth et al. 2003; Roth et al. 2006) conformed to the Association for Research in Vision and Ophthalmology Resolution on the Use of Animals in Research and were approved by our Animal Care Committee. Sprague–Dawley rats (200–250 gm) from Harlan (Indianapolis, IN) were maintained on a 12 h on/12 h off light cycle. Before ischemia, rats were anesthetized with chloral hydrate, 450 mg/kg i.p. For baseline and post–ischemic follow–up electroretinograms, rats were injected i.p. with ketamine (Parke–Davis, Morris Plains, NJ) 35 mg/kg, and xylazine (Miles, Shawnee Mission, KS) 5 mg/kg. Corneal analgesia was achieved with 1–2 drops of 0.5% proparacaine (Allergan, Puerto Rico). Pupils were dilated with 0.5% tropicamide (Alcon, Humacao, Puerto Rico) and cyclomydril (0.2% cyclopentolate HCl and 1% phenylephrine HCl, Alcon, Fort Worth, TX). Body temperature was maintained at 36.5–37.0 C with a servo–controlled heating blanket (Harvard Apparatus, Natick, MA).
For preconditioning, the intraocular pressure (IOP) was increased to 160 mm Hg for 8 min using a pressurized 1000–ml plastic sterile normal saline bag (Baxter, North Chicago, IL) connected to a 27-g needle in the anterior chamber of the eye. For ischemia, performed 24 h after preconditioning, IOP was increased to 110 mm Hg by elevating the saline reservoir above the eye for 45 min, as we previously described (Roth et al. 2003; Roth et al. 2006; Dreixler et al. 2008).
Electroretinography
Our procedures used have been described in detail previously (Roth et al. 2003; Roth et al. 2006; Dreixler et al. 2008). In brief, the scotopic ERG was recorded from rats dark-adapted for at least 1 h before experiments by platinum needle EEG electrodes (Grass, Providence, RI) in contact with the corneal surfaces of both eyes and a reference electrode on the tongue. Responses to 10-μs white light flashes from a Nicolet Ganzfeld stimulator (Madison, WI) with the rat's head centered 6 in from it were recorded on a Nicolet Spirit 486 System. The intensity of the unattenuated light flash was 0.75 log cd.s/m2, and the high/low pass filter settings were 1 kHz/1 Hz. Baseline b-wave amplitudes in both eyes were 800–1100 μV. Data are the average of three flashes delivered at least 2 min apart. ERG wave amplitudes 7 d after ischemia were measured and reported as a percentage of the baseline, non–ischemic wave amplitude.
RNA interference
Target sequences for interfering RNA (siRNA; Qiagen, Valencia, CA) are in Table 1. siRNAs were designed using neural-network technology as described previously (Huesken et al. 2005; Dreixler et al. 2008). Confirmed by BLAST, siRNAs were 100% homologous to the mRNA sequence of the respective rat Akt subtypes. Design was checked for homology to all other sequences of the genome, 3′ UTR/seed analysis, single nucleotide polymorphisms, and interferon motif avoidance (Farh et al. 2005; Hornung et al. 2005; Judge et al. 2005). A 2-μl mixture of four different sequences of siRNA to the Akt subtypes, or a single non-silencing, negative control sequence (Qiagen) in RPMI media (Invitrogen, Carlsbad, CA) and RNAiFect transfection reagent (Qiagen), at a final concentration of 3 μM was injected into the mid-vitreous of both eyes with a microsyringe (Hamilton, Reno, NV) as previously described (Roth et al. 2006).
Table 1. Percentage (± s.e.m.) of TUNEL positive cells in the retinal ganglion cell layer in siRNA-treated retinas.
| Normal Retinas | Ischemic Retinas | P-value | siRNA Sequence(s) | |
|---|---|---|---|---|
| Non-silencing siRNA/Sham IPC/ischemia | 4.3 ± 1.3 | 24.3 ± 3.8 | 0.0004 | ATT TCT CCG AAC GTG TCA CGT |
| Non-silencing siRNA/IPC/ischemia | 7.7 ± 1.4 | 4.6 ± 1.5 | 0.1627 | ATT TCT CCG AAC GTG TCA CGT |
| Akt1 siRNA/IPC/ischemia | 5.3 ± 3.2 | 17.2 ± 4.5 | 0.0443 | ATG GAG TGT GTG GAC AGT GAA ACG CTA CTT CCT CCT CAA GAA TGG GAA GGT GAT CCT GGT GAA CCG CCT CTG CTT TGT CAT GGA |
| Akt2 siRNA/IPC/ischemia | 4.0 ± 2.5 | 24.4 ± 3.3 | 0.0001 | AAC GAC TTC GAT TAT CTC AAA TCG GTT CTT CCT CAG CAT CAA ACA AGG TAC TTT GAT GAT GAA AAC AAT TTC TCT GTA GCA GAA |
| Akt3 siRNA/IPC/ischemia | 1.5 ± 0.9 | 9.1 ± 2.0 | 0.0027 | AGC GAT GTT ACC ATC GTT AAA CAG GAT CAT GAG AAA CTC TTT CGG CTC ATT CAT AGG CTA TAA CAG GAA GAC TTG CAT TAT CTT |
Western blotting (Junk et al. 2002; Zhang et al. 2002; Roth et al. 2003)
Retinas were rapidly dissected, frozen in liquid nitrogen, crushed with a tissue pulverizer (Beckman, Fullerton, CA) on dry ice, and solubilized in 9 M urea, 4% Nonidet P-40 and 2% 2-mercaptoethanol (pH 9.5). Protease inhibitor cocktail (P8340; Sigma) consisting of 4-(2-aminoethyl) benzenesulfonyl fluoride, pepstatin A, bestatin, leupeptin, and E-64 prevented protease activity. Samples were centrifuged 10 min at 10,000g, the supernatant used for SDS-PAGE and the pellet discarded. Protein concentration was determined by modified Bradford assay (Bio-Rad, Hercules, CA).
Equal amounts of protein per lane (40 μg) were diluted with SDS sample buffer and loaded onto gels (4%-20% or 16%; Invitrogen). Proteins were electroblotted to polyvinylidene difluoride (PVDF) membranes (Immobilon-P; Millipore, Bedford, MA) with the efficiency of transfer confirmed by Ponceau S red (Sigma). Non-specific binding was blocked with 5% nonfat dry milk in Tween-Tris-buffered saline. Membranes were incubated overnight at 4°C with rabbit polyclonal anti-phospho-Akt (ser 473, 1:300, Cell Signaling), mouse monoclonal anti-Akt1 (1:250, Cell Signaling), mouse monoclonal anti-Akt2 (1:250, Invitrogen), and rabbit polyclonal anti-Akt3 (1:250; Cell Signaling) primary antibodies.
Anti-rabbit horseradish peroxidase (HRP)-conjugated (goat IgG; Jackson ImmunoResearch) or anti-mouse HRP-conjugated (sheep IgG; Amersham, Buckinghamshire, England) secondary antibodies were applied at 1:20,000. Chemiluminescence was developed with a kit (Super Signal West Pico; Pierce, Rockford, IL). Protein bands were digitally imaged with a CCDBIO 16SC Imaging System (Hitachi Genetic Systems/MiraiBio, Alameda, CA) and quantified by densitometry (Gene Snap and Gene Tools; Syngene, Frederick, MD). Equal protein loading was checked by Ponceau S red gel staining and by immunoblotting with mouse monoclonal rhodopsin (clone Rho4D2 at 1:1500; a gift from Robert Molday, University of British Columbia, Victoria, British Columbia, Canada), rabbit polyclonal anti-Akt (Cell Signaling; 1:500), mouse monoclonal anti-β-actin (Sigma, 1:500), or mouse monoclonal anti-β-tubulin (Sigma; 1:500).
Fluorescent TUNEL
Fluorescent TUNEL used a Fluorescein FragEL DNA Fragmentation Detection Kit (Calbiochem, La Jolla, CA) on 10-μm thick retinal cryosections (Singh et al. 2001; Zhang et al. 2002). Briefly, frozen tissue was fixed and hydrated in 4% formaldehyde then immersed in TBS. After permeation with proteinase K in 10 mM Tris pH = 8 (1:100), tissue was labeled by TdT enzymatic reaction.
Immunohistochemistry (Junk et al. 2002; Roth et al. 2006)
Enucleated eyes were fixed in 4% paraformaldehyde for 3 h at room temperature. After removal of the anterior segment, the posterior eye was post-fixed in the same fixative overnight at 4°C, then placed in 25% sucrose overnight again at 4°C for cryoprotection. Eyecups were embedded in OCT (Sakura Finetec, Torrance, CA) and cut into 10-μm thick cryosections.
Primary antibodies (1:50 concentration) were rabbit polyclonal anti-Akt1 (Calbiochem, La Jolla, CA), polyclonal anti-Akt2 (Cell Signaling, Beverly, MA), and polyclonal anti-Akt3 (Cell Signaling). Control sections were incubated with non-immune serum. After sections were exposed to goat anti-rabbit IgG fluorescein-conjugate (1:500, Invitrogen), antifade mounting media containing DAPI (EMC Biosciences, La Jolla, CA) was applied and sections cover-slipped. Antibody processing was standardized by utilizing standard antibody concentrations and antibody exposure times, of both the primary and secondary antibodies to allow for quantification of fluorescent intensities.
Imaging
For imaging of the fluorescently stained frozen retinal sections (immunohistochemistry and TUNEL), we utilized a fluorescence microscope (Olympus IX81 inverted microscope), a Fast firewire Retiga EXi chilled CCD camera, and a 40X oil lens. Excitation/dichroic/emission settings were 530-550 nm – 570DM-590LP for greens (fluorescein). TUNEL positive cells were identified as previously reported (Singh et al. 2001; Zhang et al. 2002).
Image analysis
Immunohistochemical fluorescent intensities were measured with NIH ImageJ v.1.33, adapted from our previous methods (Roth et al. 2003). We measured the mean fluorescent intensity for the retinal ganglion cell and inner plexiform layers, the inner nuclear, and photoreceptor layers. Three 40X images, 200 μm apart in the same region of the retina, were measured and found to be repetitive. These measurements were thus averaged for the quantification. All measurements were obtained at the same laser intensity and time exposure for all paired experiments. For studying the effects of siRNA, the mean intensities were normalized to the non-silencing control siRNA's fluorescent exposure levels. TUNEL was quantitated by averaging the percentage of TUNEL positive cells, via manual cell counts, in the RGC layer in three different images of the same retinal cryosections, as described for the fluorescent intensity measurements.
Studies
To examine the role of Akt in retinal IPC, a dose-response study used the highly specific Akt inhibitor API-2 (Calbiochem) (Yang et al. 2004), injected intravitreally (5, 50, and 500 μM), 15 min before IPC, followed by 45 min of ischemia 24 h later in one eye. Doses of all agents injected into the vitreous were estimated assuming an approximately 30- μl vitreous volume as determined from earlier experiments; thus the concentration after a 2- μl injection was assumed to be diluted by a factor of 15 (Roth et al. 2003). A control group was injected with 200 μM HCl (in PBS) vehicle diluent. We also tested the impact of blocking the immediately upstream Akt activator phosphoinositol-kinase (PI-3K) using wortmanin (Sidhu et al. 2001) (Sigma), injected 15 min before IPC at doses of 1 or 4 mg/kg i.p., followed by ischemia for 45 min in one eye 24 h later. Interfering RNA to Akt1, 2, and 3, or a non-silencing sequence (Table 1) was injected into the vitreous 6 h before IPC, followed by 45 min of ischemia in one eye 24 h later. In these experiments, the non-ischemic eye underwent sham IPC and ischemia and was injected with the same material and volume as the paired ischemic eye. Functional outcome following ischemia was examined 7 d later using electroretinography
To examine the relationship between Akt and mKATP channels, we injected the mKATP channel opener diazoxide (Sigma) 40 mg/kg i.p. 24 h before 45 min of ischemia in one eye; at this dose, diazoxide mimicked IPC in the retina (Roth et al. 2006). To assess the time course of siRNA effects, either 6 or 24 h before the injection of diazoxide, siRNA for one of the three Akt subtypes or non-silencing siRNA was injected into the vitreous of both eyes. Functional outcome following ischemia was examined 7 d later using electroretinography.
Levels of activated (phosphorylated) Akt were measured in whole retinal homogenates 1, 6, or 24 h after IPC with or without the addition of inhibitors. To test the effectiveness of API-2 to decrease activation of Akt as indicated by its phosphorylation, levels of phospho-Akt were measured after injection of API-2. To test the impact of blocking mKATP channels and assess the hypothesis that opening of the mKATP channel was a predecessor of Akt activation, we injected 5-hydroxydecanoic acid (5-HD) 200 mg/kg (Roth et al. 2006) i.p. 15 min before IPC. To examine the relationship between adenosine receptors, previously shown as essential mediators in initiating IPC and Akt activation, we injected DPCPX 4.5 mg/kg (Sigma) to block adenosine A1 receptors or CSC 1 mg/kg (Sigma) to antagonize adenosine A2a receptors, 15 min before IPC. These doses effectively blocked IPC or IPC-mimicking by adenosine receptor agonists (Li et al. 1999; Li and Roth 1999; Li et al. 2000).
Statistical analysis
Data (mean ± SEM) were analyzed as previously described, with ANOVA and post hoc t–testing using Stata version 6.0 (College Station, TX) (Roth et al. 2003; Roth et al. 2006). Results between paired eyes were compared using paired t tests, and between time-matched groups from different animals using an unpaired t test. P < 0.05 was used for statistical significance.
Results
Inhibition of Akt by API-2 and wortmannin
Recovery of the ERG a- and b-waves of the rat retina after IPC then ischemia was significantly diminished in a dose-dependent manner by intravitreal injection of the Akt activation inhibitor API-2 (Fig. 1A-B). Recovery after ischemia was a-wave 55 ± 7% and b-wave 39 ± 5% (both p < 0.03 vs vehicle control injected; n = 6) for 5 μM API-2; a-wave 45 ± 9% and b-wave 32 ± 7% (both p < 0.003 vs vehicle control injected; n = 10) for 50 μM API-2; and a-wave 28 ± 14% (p < 0.03 vs vehicle control injected) and b-wave 20 ± 9% (p < 0.003 vs vehicle control injected; n = 6) for 500 μM API-2. Vehicle control was 200 μM HCl (in PBS; a-wave 80 ± 6%, b-wave 61 ± 5%; n = 9). There was no effect of API-2 injection on the opposite, non-ischemic control retinas. Western blot quantification of phosphorylated Akt (Fig. 1C-D) showed that 500 μM API-2 partially but significantly inhibited the Akt activation as reflected by Akt phosphorylation 1 h after its intravitreal injection (69 ± 4%, p < 0.0002 versus vehicle control; n = 4). Additionally, levels of total Akt did not change in the paired samples.
Fig. 1.

Inhibition of Akt activation by API-2 attenuated recovery of the electroretinogram (ERG) a- and b-waves after preconditioning (IPC) followed by ischemia. Eyes were subjected to IPC, and ischemia followed 24 h later. A. Both a- and b-wave recovery after ischemia in the preconditioned rat retina were significantly diminished in a dose-dependent manner by intravitreal injection of the Akt phosphorylation inhibitor API-2. Vehicle control was 200 μM HCl (in PBS). B. Representative ERG traces for the baseline and 7 days after ischemia. C. Western blot quantification of phosphorylated Akt showed that API-2 significantly inhibited Akt activation 1 h after API-2 intravitreal injection. D. Representative Western blot bands of the 60 kD phosphorylated Akt, with no change in levels of total Akt, indicating equal loading of protein.
Systemic injection (i.p.) of the PI-3K inhibitor wortmannin, 1 mg/kg (a-wave 88 ± 5% and b-wave 70 ± 4%, p < 0.01 vs control; n = 5) or 4 mg/kg (a-wave 80 ± 5%, p < 0.01 vs control, b-wave 66 ± 5%, p < 0.01 vs control; n = 6) partially attenuated the neuroprotective effect of IPC (a-wave 101 ± 3% and b-wave 90 ± 6%, n = 10 in controls; Fig. 2A-B).
Fig. 2.

PI-3K inhibition, by wortmannin, blocked neuroprotection with ischemic preconditioning (IPC). Eyes were subjected to IPC, and ischemia followed 24 h later. A. Representative traces of the electroretinogram (ERG) for baseline and 7 days after ischemia. B. IPC recovery shown on ERG (both a- and b-wave) in the rat retina was significantly diminished by wortmannin.
Akt subtype siRNA specificity
The specificity of the siRNA for the Akt subtypes is demonstrated in Fig. 3-4. By Western blot, Akt3 levels were significantly attenuated by Akt3 siRNA 24 h after injection (Fig 4A, 31 ± 11% of non-silencing siRNA control, p = 0.0007; n = 4). We also examined the levels of Akt1 and Akt2 after injection of siRNA. Whole retinal protein lysates examined 54 h after injection of siRNA (Fig 3B) showed that Akt1 protein levels decreased 82% versus non-silencing siRNA-injected eyes from an intensity of 4,230,753 ± 126,883 arbitrary units to 765,128 ± 298,707 (n = 4, p < 0.0005).
Fig. 3.

Akt subtype siRNA specificity. A. Akt3 protein levels were significantly reduced 24 h after intravitreal injection as compared to the non-silencing siRNA control. Representative Western blots are shown (right) as well as rhodopsin protein levels that serve as a control. B. Western blot analysis of Akt1 siRNA show decreases in their respective Akt subtype protein levels 54 h after injection. Actin served as a control.
Fig. 4.

Immunostained cryosections of retina injected with Akt2 siRNA showed significant reduction of Akt2 levels in the retinal ganglion cell (RGC) and inner nuclear layers (INL). Akt2 levels were unchanged in the photoreceptor (PR) layer (Fig. 5A). Representative immunostaining of Akt2 is shown (Fig. 5B).Retinal cryosections from paired eyes injected with siRNA to Akt2 or non-silencing siRNA and harvested 24 h later were reacted with anti-Akt2 antibody and fluorescein-conjugated secondary antibody. Green color indicates staining for Akt2 protein (shown in the middle panels). DAPI (far left images) are for orientation (cell layers shown in white letters), where cell nuclei stain in blue. The right panels show double labeling for DAPI and Akt2. White arrows indicate some of the cells positive for Akt2 protein in the GCL and INL; also shown in the far right panel are cells double labeling for DAPI and Akt2, indicating cellular localization of Akt2 protein in cells in the RGC and INL layers. It is evident that Akt2 positivity was diminished by siRNA directed against it. Fig. 5C is a section reacted with non-immune serum to indicate levels of background staining.
Akt2 protein decreased significantly at 24 h in immunostained cryosections from injected eyes (Fig 4A-B). The Akt2 protein levels were diminished to 69 ± 11% of the non-silencing siRNA control in the RGC/inner plexiform layers (p < 0.05; n = 4), to 73 ± 4% in the inner nuclear layer (p < 0.001; n = 4), and to 84 ± 7% in the photoreceptor layer (NS; n = 4). The Akt3 protein levels had a decreasing trend to 74 ± 11% of the non-silencing siRNA 24 h after Akt3 siRNA injection in the RGC layer (p = 0.06, not shown). Protein levels of Akt1, measured by immunohistochemistry, were not significantly changed by Akt1 siRNA administration at this time point.
siRNA to Akt subtypes attenuate IPC
Injection of 2 μl of 3 μM siRNA into the vitreous for Akt2 (a-wave 40 ± 13%, p < 0.02 vs non-silencing siRNA and b-wave 25 ± 8%, p < 0.004 vs non-silencing siRNA; n = 6) and for Akt3 (a-wave 50 ± 7%, p < 0.004 vs non-silencing siRNA and b-wave 39 ± 6%, p < 0.03 vs non-silencing siRNA; n = 13) significantly attenuated post-ischemic recovery after IPC as compared to a non-silencing siRNA control (a-wave 87 ± 6% and b-wave 60 ± 5%; n = 11; Fig. 5A-B). Akt1 siRNA was without effect (a-wave 80 ± 7% and b-wave 56 ± 5%; n = 6). Injection of siRNA to Akt2 before sham IPC, followed by ischemia 24 h later, did not alter the recovery compared to Akt2 siRNA injection and IPC and ischemia (Fig. 5C).
Fig. 5.

Akt subtype specific siRNA blocked neuroprotection with ischemic preconditioning (IPC). Eyes were subjected to IPC and ischemia followed 24 h later. A. Representative traces of the electroretinogram (ERG) at baseline and 7 days after ischemia. B. Injection of 2 μl of 3 μM siRNA to Akt2 or Akt3 into the vitreous 6 h before IPC significantly attenuated post-ischemic recovery of both the a- and b-waves on ERG. Akt1 siRNA was ineffective. Intravitreal of injections of a non-silencing siRNA served as the control. C. Akt2 siRNA intravitreal injection before sham IPC, followed by ischemia 24 h later, did not alter the recovery compared to Akt2 siRNA with IPC and ischemia.
siRNA to Akt subtypes inhibits IPC-mimicking by diazoxide
Injection of siRNA to the three Akt subtypes altered IPC-mimicking by diazoxide in a time-dependent manner. Intravitreal injection of 2 μl of 3 μM subtype-specific Akt2 (46 ± 5%; n = 10) and Akt3 (48 ± 5%; n = 18) siRNA 6 h before diazoxide injection (40 mg/kg i.p.) significantly decreased IPC-mimicking for the ERG a-wave only (p < 0.03 for both) as compared to a non-silencing siRNA control (63 ± 4%, n = 13; Fig. 6A). The Akt1 siRNA (63 ± 12%; n = 10) was ineffective. However, Akt1 siRNA significantly reduced the ERG recovery (a-wave 20 ± 9%, p < 0.002 and b-wave 10 ± 4%, p < 0.002; n = 5) when injected 24 h before diazoxide as compared to the non-silencing siRNA control (a-wave 74 ± 8% and b-wave 53 ± 6%; n = 7; Fig. 6C). Akt2 siRNA significantly attenuated only the ERG b-wave recovery (a-wave 55 ± 7% and b-wave 35 ± 6%, p < 0.05; n = 10). Akt3 siRNA was not effective (a-wave 64 ± 9% and b-wave 52 ± 11%; n = 5).
Fig. 6.
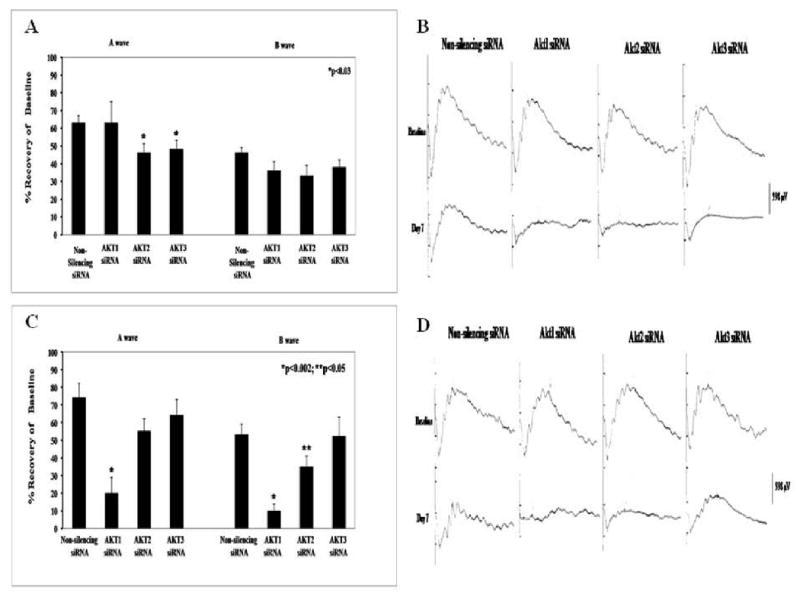
Akt subtype specific siRNA attenuated ischemic preconditioning (IPC)-mimicking by diazoxide in a time-dependent manner. Rats were injected with diazoxide, and ischemia followed 24 h later. A. Intravitreal injection of 2 μl of 3 μM subtype-specific Akt2 and Akt3 siRNA 6 h before diazoxide injection (i.p.) significantly decreased IPC-mimicking by the mKATP channel activator for the ERG a-wave. Akt1 siRNA was ineffective. A non-silencing siRNA served as control. B. Representative traces of electroretinogram (ERG) at baseline and 7 days after ischemia for the 6 h prior to diazoxide-injected groups. C. Akt1 siRNA significantly reduced the post-ischemic recovery (for both a- and b-waves) when injected 24 h before diazoxide. Akt2 siRNA significantly attenuated the recovery of b-wave on ERG. Akt3 siRNA was without effect. D. Representative ERG traces for the baseline and 7 days after ischemia for the 24 h experiments.
Akt involvement in IPC: upstream mechanisms
Western blot analysis (Fig. 7) of phosphorylated Akt protein levels (224 ± 31% vs paired sham IPC eye, n = 4) showed a complete and significant reduction, at 6 h after IPC, by the specific mKATP channel blocker 5-HD, 200 mg/kg (99 ± 23%, p < 0.05; n = 4). Akt phosphorylation was significantly attenuated by the adenosine A1 receptor antagonist DPCPX (128 ± 22%, p < 0.05 vs control; n = 3) and the adenosine A2a receptor blocker CSC (116 ± 7%, p < 0.05 vs control; n = 3).
Fig. 7.

Akt activation was reduced by mKATP channel and adenosine receptor antagonists. A. Western blot analysis of phosphorylated Akt protein levels were significantly reduced compared to levels at 6 h after ischemic preconditioning (IPC), by the specific mKATP channel blocker 5-HD at a concentration of 200 mg/kg, the adenosine A1 receptor antagonist DPCPX and the adenosine A2 receptor inhibitor CSC. These results suggest that Akt activation is downstream from both the mKATP channel and adenosine receptor pathways. B. Representative Western blot bands of the 60 kD phosphorylated Akt, with no change in the beta tubulin control in the paired eyes.
Akt involvement in IPC: anti-apoptosis effects
The percentage of TUNEL-positive cells in the RGC layer was significantly increased compared to the non-ischemic control eyes in the non-silencing siRNA sham IPC with ischemia group (i.e., ischemia only) as well as for each Akt subtype siRNA group with IPC and ischemia (Fig. 8; Table 1). Moreover, the non-silencing siRNA with IPC and ischemia showed clearly the protective effect of IPC on apoptosis. Because there was only sparse TUNEL staining in the inner and outer nuclear layers, we did not count the number of positive cells in those regions. TUNEL positive staining was not enhanced in any of the paired normal control eyes that were injected with Akt subtype siRNA.
Fig. 8.

Fluorescent TUNEL staining and Akt subtype involvement in apoptosis. Shows representative images of the fluorescent TUNEL staining for the normal RGC layer (left) versus the ischemic (right). Images are shown with altered/false colors where the arrows denote co-localization of the fluorescent TUNEL stain (red) and the DAPI (green).
Discussion
Our results showed the role of the three Akt subtypes in the neuroprotective pathways of retinal IPC. We demonstrated that: (1) activation of Akt with ischemic preconditioning, as indicated by its phosphorylation, was downstream of the adenosine A1 and A2a receptors and mKATP channel opening; (2) the specific inhibitor of Akt activation, API-2, the PI-3 kinase inhibitor wortmannin, and siRNA directed against Akt2 and -3 attenuated the protective effects of IPC on retinal function after ischemia; (3) functional neuroprotection by IPC or IPC-mimicking by diazoxide was diminished by specific siRNA targeted against the three Akt subtypes; and (4) inhibition of the Akt subtypes by siRNA increased apoptotic retinal cell death. Akt inhibition itself by siRNA (Akt2) that attenuated the protective effects of IPC on ischemic injury did not alter outcome after sham IPC and ischemia, indicating that siRNA was not working via a toxic or direct effect upon ischemia itself.
It has been shown that several other signaling pathways and/or cellular survival proteins underlie the neuroprotective effects of IPC. We have previously demonstrated that binding of adenosine to its receptors is a trigger for IPC (Li and Roth 1999) and that downstream signal transduction factors, including mitochondrial KATP channels (mKATP), protein kinase C (PKC), reactive O2 species (ROS), nitric oxide synthase (NOS), PKC, and erythropoietin, are components of this neuroprotection(Junk et al. 2002; Roth et al. 2006; Dreixler et al. 2008). More recently, microarray analysis of different paradigms of retinal preconditioning show a number of retinal genes undergoing changes in gene expression (Kamphuis et al. 2007; Thiersch et al. 2008). Activation of the Akt pathway may be one of the mechanisms involved in IPC protection.
A previous study using in situ hybridization showed localization of Akt1, -2, and -3 to the retinal ganglion cell layer in rat retina (Reiter et al. 2003). All subtypes were present in photoreceptor cells in mice (Li et al. 2007). Akt subtype retinal localization correlates with and may explain some of the protective actions of all three Akt subtypes in the present work. In our study, a- and b-wave amplitudes were decreased by API-2 and wortmannin with IPC, the a-wave changes after ischemia indicating photoreceptor damage, and b-wave decreases, damage to inner retinal layer cells. In vivo gene knockdown of the Akt subtypes by siRNA attenuated functional IPC recovery or IPC-mimicking by diazoxide via photoreceptor damage as indicated by a-wave amplitude reduction. Studies in knock-out mice (Li et al. 2007) showed that Akt2 was involved in the protection of photoreceptor cells from light-induced apoptotic cell death, findings attributable to abnormal photoreceptor development. In our transient in vivo siRNA Akt subtype knockdown, followed by IPC and ischemia, photoreceptor injury may be attributed to diminished protein levels of Akt subtypes in the outer retinal layers. Involvement of the Akt subtypes in the inner retinal layers, including in RGCs and amacrine, and horizontal cells, is seen by decreases in the ERG b-wave amplitude after ischemia in our studies of the attenuation of IPC by API-2, wortmannin, and siRNA to Akt2 and Akt3, as well as b-wave reduction by siRNA application of all three Akt subtypes with IPC-mimicking by mKATP channel activation by diazoxide.
Early events in the triggering of IPC include the activation of A1 and A2a adenosine receptors (Li et al. 1999; Li and Roth 1999; Li et al. 2000) and opening of mKATP channels (Roth et al. 2006). In the present study, Akt was functioning downstream of adenosine A1 and A2a receptors in the signal transduction of IPC, and thereby provides a link to previous pathways of retinal IPC that we have demonstrated. Others have noted a relationship between Akt activation and the adenosine A1, but not A2a receptor in myocardium (Krieg et al. 2002) but no previous data have shown a link in CNS between adenosine and Akt. The inhibition of Akt activation by blockade of two different adenosine receptor subtypes suggests that the activation of Akt in the retina via adenosine receptors may be achieved by two divergent pathways that provide redundant means to induce ischemic tolerance by IPC. Our study only addressed a few of the mechanisms activating Akt, including adenosine receptors, mitochondrial KATP channel, and PI3-kinase, but other growth factor pathways and oxygen free radicals and interaction with phosphatases are other known mechanisms in other systems. (Parcellier et al. 2008)
Akt activation, as indicated by its phosphorylation (Alessi et al. 1996), is neuroprotective by preventing apoptotic injury (Datta et al. 1997; Cardone et al. 1998; Fukunaga and Kawano 2003; Nakazawa et al. 2003; Hashiguchi et al. 2004; Saito et al. 2004; Chong et al. 2005; Zhao et al. 2005). The present results showed that IPC protects via blockade of ischemia-induced apoptosis, and that all three Akt subtypes are involved in this anti-apoptosis effect of IPC. Conversely, inactivation of the Akt pathway was associated with photoreceptor cell death in rd mice (Jomary et al. 2006), and enhanced cell death in retinal pigment epithelial cells in vitro (Yang et al. 2006). Moreover, acute IOP elevation activated PI3K/akt pathway in the inner nuclear layer and in RGCs, and cell death was enhanced by blockade of PI3K (Huang et al. 2008). The protective effects of the opening of mKATP channels have been shown previously in retina (Roth et al. 2006). Here we showed that all three Akt subtypes are effective in retinal IPC protection and IPC-mimicking when mKATP channels are activated by diazoxide, thereby linking earlier findings to provide another step in the IPC pathway. Our results indicate that the Akt subtypes are essential for IPC to protect against ischemic injury, and function via a redundant set of pathways via Akt subtypes.
Akt subtypes appear to have different time courses for their actions. The protective effects of IPC on recovery after ischemia of the a- and b-waves were attenuated by both Akt2 and Akt3 siRNA intravitreal injection 6 h prior to IPC, while Akt1 siRNA was without effect. In experiments with siRNA directed against the specific Akt subtypes in our model of IPC-mimicking by diazoxide, Akt2 and Akt3 were effective in blocking mimicking when injected 6 h before diazoxide administration with effects only on the a wave, and Akt1 was without effect. But with injection 24 h before diazoxide, Akt1 and Akt2 were effective and Akt3 was not. These time-dependent responses may be explainable by the siRNA mediated reductions in Akt protein subtypes. While Akt 2 and 3 protein levels were decreased 24 h after injection, Akt3 protein level reduction was not documented until 54 h later.
The time course disparity may be attributed to the different pathways for protective actions, variable protein turnover of Akt subtypes, and other as yet unknown mechanisms or factors that may affect the timing of the Akt subtype activation or the siRNA degradation of the specific subtypes. It appears that the time-dependent differences between the Akt subtype siRNA actions with diazoxide are due to different actions within the mKATP channel activation neuroprotective pathway. Similarly, the different actions of the siRNAs with IPC seem to be IPC pathway-dependent.
There was a disparity between the effects of siRNA directed to the specific Akt subtypes in their attenuation of protection by IPC and that by IPC-mimicking by diazoxide. Akt1 siRNA was effective in blocking only the protection of IPC-mimicking when mKATP channels were opened in a time-dependent manner, while it was without effect with IPC. Akt2 and Akt3 siRNA diminished protection in both IPC and IPC-mimicking by diazoxide. Neuroprotection by IPC and IPC-mimicking by the opening of mKATP channels was accomplished by divergent cellular mechanisms (Roth et al. 2006). The Akt1 subtype may be activated only in the pathway of IPC-mimicking by diazoxide. Apoptotic-related gene levels increased when Akt1 siRNA was injected 6 h before IPC. This shorter time point, sufficient to increase apoptosis, does not seem to affect retinal function.
There was minimal effect of the non-silencing siRNA on the IPC neuroprotection and IPC-mimicking by diazoxide, supporting the notion that our results with siRNA can be attributed to specific effects. Akt knockout mice, an alternative experimental model, introduce systemic effects such growth retardation, insulin resistance, or abnormal brain development, which could potentially confound experimental interpretation (Dummler et al. 2006). Our method is advantageous because it resulted in an in vivo, localized gene “knockdown” for specific investigation of the significance of each Akt subtype in the retina. Moreover, the intravitreal injection may be causing a transient cell dysfunction and not apoptotic cell death. This idea is backed up by the fact that there was little difference in TUNEL positivity of the non-ischemic eyes injected with the siRNAs.
RNA interference specificity was shown in vivo by decreased, but not complete, blockade of protein levels on Western blots of whole retinal homogenates or immunostained retinal sections. This result suggests that only a small proportion of these proteins are functionally active, either because of sequestration or because they are rendered inactive through effects of other proteins.
In conclusion, we showed that specific Akt subtypes function in IPC and IPC-mimicking by diazoxide. Only Akt2 and Akt3 subtypes were involved directly in the neuroprotection induced by IPC in the rat retina. All three Akt subtypes were operative in in vivo signaling downstream and IPC-mimicking from the opening of mitochondrial KATP channels. The activation of Akt is downstream of the activation of adenosine receptors and opening of mKATP channels. All three Akt types exhibit protective mechanisms that block the apoptosis caused by retinal ischemia, indicating an intrinsic redundancy in the Akt pathways in vivo that protects against ischemic damage.
Acknowledgments
This research was supported by National Institutes of Health grants EY10343 (SR), DK058547 (HTL), EY11253 (DMR), the Office of Medical Education of the University of Chicago Pritzker School of Medicine (JCH and YS), and by grants-in-aid from the Illinois Society for the Prevention of Blindness (SR and SKS). Jon W. Hemmert and Yang Shen were recipients of Student Research Fellowships from the American Academy of Neurology, Minneapolis, Minnesota, and Yang Shen was the recipient of a Student Scholarship in Cerebrovascular Disease from the American Heart Association, Dallas, Texas. Immunostained images were generated at the University of Chicago Cancer Center Confocal Microscopy Core Facility.
Footnotes
Proprietary Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Cardone MH, Roy N, Stennicke HR, Salvasen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- Chavarria T, Valenciano AI, Mayordomo R, Egea J, Comella JX, Hallbook F, de Pablo F, de la Rosa EJ. Differential, age-dependent MEK-ERK and PI3K-Akt activation by insulin acting as a survival factor during embryonic retinal development. Dev Neurobiol. 2007;67:1777–1788. doi: 10.1002/dneu.20554. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005;75:207–246. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotah Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;911:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 2000;6:41–51. [PubMed] [Google Scholar]
- Dreixler JC, Shaikh AR, Shenoy SK, Roth S. Protein kinase C subtypes and retinal ischemic preconditioning. Exp Eye Res. 2008;87:300–311. doi: 10.1016/j.exer.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummler B, Tschopp O, Hynx D, Yang ZZ, Dirnhofer S, Hemmings BA. Life with a single isoform of Akt: Mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol. 2006;26:8042–8051. doi: 10.1128/MCB.00722-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- Fayard E, Tintignac LA, Baudry A, Hemmings BA. Protein kinase B/Akt at a glance. J Cell Sci. 2005;118:5675–5678. doi: 10.1242/jcs.02724. [DOI] [PubMed] [Google Scholar]
- Fu QL, Hu B, Wu W, Pepinsky RB, Mi S, So KF. Blocking LINGO-1 function promotes retinal ganglion cell survival following ocular hypertension and optic nerve transection. Invest Ophthalmol Vis Sci. 2008;49:975–985. doi: 10.1167/iovs.07-1199. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Kawano T. Akt is a molecular target for signal transduction therapy in brain ischemic insult. J Pharmacol Sci. 2003;92:317–327. doi: 10.1254/jphs.92.317. [DOI] [PubMed] [Google Scholar]
- Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- Hashiguchi A, Yano S, Morioka M, Hamada J, Ushio Y, Takeuchi Y, Fukunaga K. Up-regulation of endothelial nitric oxide synthase via phosphatidylinositol 3-kinase pathway contributes to ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab. 2004;24:271–279. doi: 10.1097/01.WCB.0000110539.96047.FC. [DOI] [PubMed] [Google Scholar]
- Hillion JA, Li Y, Maric D, Takanohashi A, Klimanis D, Barker JL, Hallenbeck JM. Involvement of Akt in preconditioning-induced tolerance to ischemia in PC12 cells. J Cereb Blood Flow Metabol. 2006;26:1323–1331. doi: 10.1038/sj.jcbfm.9600286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005;11:263–270. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- Huang Y, Cen LP, Luo JM, Wang N, Zhang MZ, van Rooijen N, Pang CP, Cui Q. Differential roles of phosphatidylinositol 3-kinase/akt pathway in retinal ganglion cell survival in rats with or without acute ocular hypertension. Neuroscience. 2008;153:214–225. doi: 10.1016/j.neuroscience.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Huesken D, Lange J, Mickanin C, Weiler J, Asselbergs F, Warner J, Meloon B, Engel S, Rosenberg A, Cohen D, Labow M, Reinhardt M, Natt F, Hall J. Design of a genome-wide siRNA library using an artificial neural network. Nat Biotech. 2005;23:995–1001. doi: 10.1038/nbt1118. [DOI] [PubMed] [Google Scholar]
- Jomary C, Cullen J, Jones SE. Inactivation of the Akt survival pathway during photoreceptor apoptosis in the retinal degeneration mouse. Invest Ophthalmol Vis Sci. 2006;47:1620–1629. doi: 10.1167/iovs.05-1176. [DOI] [PubMed] [Google Scholar]
- Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotech. 2005;23:457–462. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- Junk AK, Mammis A, Savitz SI, Singh M, Roth S, Malhotra S, Rosenbaum PS, Cerami A, Brines M, Rosenbaum DM. Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2002;99:10659–10664. doi: 10.1073/pnas.152321399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Dijk F, Bergen AAB. Ischemic preconditioning alters the pattern of gene expression changes in response to full retinal ischemia. Mol Vis. 2007;13:1892–1901. [PubMed] [Google Scholar]
- Kilic U, Kilic E, Jarve A, Guo Z, Spudich A, Bieber K, Barzena U, Bassetti CL, Marti HH, Hermann DM. Human vascular endothelial growth factor protects axotomized retinal ganglion cells in vivo by activating ERK-1/2 and Akt pathways. J Neurosci. 2006;26:12439–12446. doi: 10.1523/JNEUROSCI.0434-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Asai N, Enomoto A, Maeda K, Kato T, Ishida M, Jiang P, Watanabe T, Usukura J, Kondo T, Costantini F, Murohara T, Takahashi M. Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat Cell Biol. 2008;10:329–337. doi: 10.1038/ncb1695. [DOI] [PubMed] [Google Scholar]
- Krieg T, Qin Q, McIntosh EC, Cohen MV, Downey JM. ACh and adenosine activate PI3-kinase in rabbit hearts through transactivation of receptor tyrosine kinases. Am J Physiol. 2002;283:H2322–2330. doi: 10.1152/ajpheart.00474.2002. [DOI] [PubMed] [Google Scholar]
- Li B, Jennings NM, Rosenbaum PS, Maxwell KM, Roth S. Differential roles of adenosine receptor subtypes in retinal ischemia-reperfusion injury in the rat. Exp Eye Res. 1999;68:9–17. doi: 10.1006/exer.1998.0573. [DOI] [PubMed] [Google Scholar]
- Li B, Roth S. Retinal ischemic preconditioning in the rat: requirement for adenosine and repetitive induction. Invest Ophthalmol Vis Sci. 1999;40:1200–1216. [PubMed] [Google Scholar]
- Li B, Yang C, Rosenbaum DM, Roth S. Signal transduction mechanisms involved in ischemic preconditioning in the rat retina in vivo. Exp Eye Res. 2000;70:755–765. doi: 10.1006/exer.2000.0843. [DOI] [PubMed] [Google Scholar]
- Li G, Anderson RE, Tomita H, Adler R, Liu X, Zack DJ, Rajala RVS. Nonredundant role of Akt2 for neuroprotection of rod photoreceptor cells from light-induced cell death. J Neurosci. 2007;27:203–211. doi: 10.1523/JNEUROSCI.0445-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Roth S, Laser M, Ma JX, Crosson CE. Retinal preconditioning and the induction of heat-shock protein 27. Invest Ophthalmol Vis Science. 2003;44:1299–1304. doi: 10.1167/iovs.02-0235. [DOI] [PubMed] [Google Scholar]
- Liu X, Mameza MG, Lee YS, Eseonu CI, Yu CR, Kang Derwent JJ, Egwuagu CE. Suppressors of cytokine-signaling proteins induce insulin resistance in the retina and promote survival of retinal cells. Diabetes. 2008;57:1651–1658. doi: 10.2337/db07-1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. Akt/Pkb signaling: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullonkal CJ, Toledo-Pereyra LH. Akt in ischemia and reperfusion. J Invest Surg. 2007;20:195–203. doi: 10.1080/08941930701366471. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Shimura M, Tomita H, Akiyama H, Yoshioka Y, Kudou H, Tamai M. Intrinsic activation of PI3K/Akt signaling pathway and its neuroprotective effect against retinal injury. Curr Eye Res. 2003;26:55–63. doi: 10.1076/ceyr.26.1.55.14254. [DOI] [PubMed] [Google Scholar]
- Parcellier A, Tintignac LA, Zhuravleva E, Hemmings BA. PKB and the mitochondria: AKTing on apoptosis. Cell Signal. 2008;20:21–30. doi: 10.1016/j.cellsig.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Reiter CE, Sandirasegarane L, Wolpert EB, Klinger M, Simpson IA, Barber AJ, Antonetti DA, Kester M, Gardner TW. Characterization of insulin signaling in rat retina in vivo and ex vivo. Am J Physiol. 2003;285:E763–774. doi: 10.1152/ajpendo.00507.2002. [DOI] [PubMed] [Google Scholar]
- Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006;25:4683–4696. doi: 10.1038/sj.onc.1209595. [DOI] [PubMed] [Google Scholar]
- Roth S, Dreixler JC, Shaikh AR, Lee KH, Bindokas V. Mitochondrial potassium ATP channels and retinal ischemic preconditioning. Invest Ophthalmol Vis Sci. 2006;47:2114–2124. doi: 10.1167/iovs.05-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S, Li B, Rosenbaum PS, Gupta H, Goldstein IM, Maxwell KM, Gidday JM. Preconditioning provides complete protection against retinal ischemic injury in rats. Invest Ophthalmol Vis Sci. 1998;39:775–785. [PubMed] [Google Scholar]
- Roth S, Shaikh AR, Hennelly MM, Li Q, Bindokas V, Graham CE. Mitogen-activated protein kinases and retinal ischemia. Invest Ophthalmol Vis Sci. 2003;44:5383–5395. doi: 10.1167/iovs.03-0451. [DOI] [PubMed] [Google Scholar]
- Saito A, Narasimhan P, Hayashi T, Okuna S, Ferrand-Drake M, Chan PH. Neuroprotective role of a proline-rich Akt substrate in apoptotic neuronal cell death after stroke: relationships with nerve growth factor. J Neurosci. 2004;24:1584–1593. doi: 10.1523/JNEUROSCI.5209-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders EJ, Parker E, Harvey S. Retinal ganglion cell survival in development: mechanisms of retinal growth hormone action. Exp Eye Res. 2006;83:1205–1214. doi: 10.1016/j.exer.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Sidhu JS, Liu F, Boyle SM, Omiecinski CJ. PI3K inhibitors reverse the suppressive actions of insulin on CYP2E1 expression by activating stress-response pathways in primary rat hepatocytes. Mol Pharmacol. 2001;59:1138–1146. doi: 10.1124/mol.59.5.1138. [DOI] [PubMed] [Google Scholar]
- Singh M, Savitz SI, Hoque R, Rosenbaum PS, Roth S, Rosenbaum DM. Cell-specific caspase expression by different neuronal phenotypes in transient retinal ischemia. J Neurochem. 2001;77:466–475. doi: 10.1046/j.1471-4159.2001.00258.x. [DOI] [PubMed] [Google Scholar]
- Thiersch M, Raffelsberger W, Frigg E, Samardzija M, Blank P, Poch O, Grimm C. The hypoxic transcriptome of the retina: identification of factors with potential neuroprotective activity. Adv Exp Med Biol. 2008;613:75–85. doi: 10.1007/978-0-387-74904-4_8. [DOI] [PubMed] [Google Scholar]
- Ueki Y, Wang J, Chollangi S, Ash JD. STAT3 activation in photoreceptors by leukemia inhibitory factor is associated with protection from light damage. J Neurochem. 2008;105:784–796. doi: 10.1111/j.1471-4159.2007.05180.x. [DOI] [PubMed] [Google Scholar]
- Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, Sebti SM, Cheng JQ. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004;64:4394–4399. doi: 10.1158/0008-5472.CAN-04-0343. [DOI] [PubMed] [Google Scholar]
- Yang P, Peairs JJ, Tano R, Jaffe GJ. Oxidant-mediated Akt activation in human RPE cells. Invest Ophthalmol Vis Sci. 2006;47:4598–4606. doi: 10.1167/iovs.06-0140. [DOI] [PubMed] [Google Scholar]
- Yin W, Signore AP, Iwai M, Cao G, Gao Y, Johnnides MJ, Hickey RW, Chen J. Preconditioning suppresses inflammation in neonatal hypoxic ischemia via Akt activation. Stroke. 2007;38:1017–1024. doi: 10.1161/01.STR.0000258102.18836.ca. [DOI] [PubMed] [Google Scholar]
- Zhang C, Rosenbaum DM, Shaikh AR, L Q, Rosenbaum PS, Pelham DJ, Roth S. Ischemic preconditioning attenuates apoptosis following retinal ischemia in rats. Invest Ophthalmol Vis Sci. 2002;43:3059–3066. [PubMed] [Google Scholar]
- Zhang Y, Park TS, Gidday JM. Hypoxic preconditioning protects human brain endothelium from ischemic apoptosis by Akt-dependent survivin activation. Am J Physiol. 2007;292:H2573–H2581. doi: 10.1152/ajpheart.01098.2006. [DOI] [PubMed] [Google Scholar]
- Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, Steinberg GK. Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci. 2005;25:9794–9806. doi: 10.1523/JNEUROSCI.3163-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]