

Abstract
Background and Purpose
Mutations in the Programmed Cell Death 10 (PDCD10) gene cause autosomal dominant familial cerebral cavernous malformations (CCM3). To date, little is known about the function of this gene and its role in disease pathogenesis.
Methods
We examined the effects of overexpression of wild type and two human disease-causing variants of PDCD10 on cell death using three different methods (TUNEL and MTT assays and caspase-3 activation). We analyzed expression of CCM3, activated caspase-3 and p38 in endothelial cell lines using the serum-deprivation model of apoptosis induction. Finally, we assayed the effects of siRNA-mediated inhibition of endogenous PDCD10 expression on cell death in endothelial cell cultures.
Results
Over-expression of wild-type CCM3, but not disease-linked mutant forms, induced apoptosis as confirmed by TUNEL and increased levels of activated caspase-3. Serum starvation of endothelial cells, an inducer of apoptosis, led to increased expression of CCM3 and activation of p38 and ultimately, activated caspase-3. siRNA-mediated inhibition of CCM3 expression resulted in decreased levels of p38 and activated caspase-3, and decreased cell death.
Conclusions
CCM3 is both necessary and sufficient to induce apoptosis in vitro in well-defined cell culture systems. Even though it is currently unclear whether this effect on apoptosis is direct or indirect through modulation of cell cycle, these results lead to the novel hypothesis that CCM lesions may form as a consequence of aberrant apoptosis, potentially altering the balance between the endothelium and neural cells within the neurovascular unit.
Keywords: Cerebral Cavernous Malformations, CCM3, PDCD10, apoptosis, p38, caspase 3
Introduction
Cerebral cavernous malformations (CCM) are vascular lesions affecting almost exclusively the central nervous system (CNS) and the retina1. Grossly, they are a collection of enlarged sinusoidal vascular channels almost resembling a raspberry1. Ultrastructurally, these channels are lined by a single layer of endothelium, lack normal vessel wall elements such as smooth muscle and are devoid of intervening normal CNS parenchyma2, 3. These lesions can occur sporadically, or as a familial form due to mutations in three different transcripts, CCM1, 2, or 3: Krev1 Interaction Trapped 1 (KRIT1), MGC4607 or malcavernin, and Programmed Cell Death 10 or PDCD10, respectively 4-14. Overall, mutations in these 3 genes account for approximately 96% of familial cases 15. Despite the identification and characterization of these three CCM genes, several questions regarding this complex disorder remain unanswered. These include a sophisticated understanding of the function of the CCM genes and how mutations lead to formation of lesions that almost exclusively affect the CNS and are focal in nature. It has been suggested that the CCM lesions may be the consequence of hemorrhage-induced proliferation, a process by which abnormal vascular beds cause reactive angiogenesis with new vessel formation and coalescence 16, but mechanistic insight into lesion formation is lacking.
Recent molecular biological studies on the CCM1 and 2 proteins shed some light into their functions. CCM1 was shown to bind to microtubules in vitro 17-19 and interact individually with Krev1/Rap1a 19-21, ICAP1a 22, 23, β-catenin 21 and CCM2 24 in various in vitro systems. CCM2 binds CCM1 and MEKK3 in a ternary complex and acts as a scaffolding protein signaling through p38 following extracellular stimulation 24, 25. p38 pathway is known to be important in diverse physiological processes ranging from proliferation to differentiation to apoptosis 26. More recently, CCM3 was shown to be a part of this complex in vitro 18, 27, 28.
Despite this recent progress, little is known about the biological functions of the CCM3 protein. It was first identified through a screen of genes expressed during the induction of apoptosis in a premyeloid cell line 11. Recent evidence suggests that CCM3 binds to and is phosphorylated by serine/threonine kinase 25 (STK25) and is dephosphorylated by binding to the phosphatase domain of Fas-associated phosphatase-127.
Based upon these limited data, we sought to investigate the biological function of CCM3 and specifically focused on its potential involvement in apoptotic pathways. We modulated CCM3 expression using cell culture based assays in vitro and examined its effects on cell death. Our results indicate that CCM3 has a pro-apoptotic function in the cell culture models tested and lead to the novel hypothesis that CCM lesions potentially form as a result of aberrant apoptosis in the vasculature within the neurovascular unit.
Methods
Cell Culture
HeLa cells were plated in high glucose DMEM supplemented with 10% FBS, 1% penicillin/streptomycin and 1% L-glutamine. Primary human umbilical vascular endothelial cells (HUVECs) were obtained from the Vascular Biology and Transplantation Program at Yale University (New Haven, CT). HUVECs were grown in M199 medium with 20% fetal bovine serum (FBS) and 1% endothelial cell growth supplement (ECGS), on gelatin-coated (0.1%) plates (J.T. Baker Inc.). Cells used in this study were obtained between passages 3 and 6. For serum starvation assays, HUVECs were seeded on 0.1% gelatin coated culture slides and after 48 hours of attachment in standard media, cells were washed in 0.2% FBS medium, followed by an additional 24 hour incubation in 0.2% FBS.
Expression Vector Construction
A PCR-amplified human PDCD10/CCM3 (hCCM3) cDNA fragment was subcloned into XhoI and HindIII sites of the pEGFP-C3 vector (Clontech) or a pRFP-C3 vector containing monomeric red fluorescent protein, resulting in a C-terminal GFP- or RFP-tagged CCM3, respectively. Both expression constructs are indistinguishable in terms of transfection efficiency and cytotoxicity in HeLa cells (data not shown). Expression plasmids carrying two human disease causing PDCD10/CCM3 point mutations (c.283C>T and c.192delA) 12 were generated from the pEGFP-C3-hCCM3 plasmid using a standard mutagenesis kit (Stratagene, CA). All constructs were verified by direct sequencing.
Transfections
HeLa cells were seeded (1×105) in six-well plates. The pEGFP-C3-hCCM3 or pRFP-C3-CCM3 and pEGFP-C3 or pRFP-C3 (empty) plasmids were transfected into the cell lines using with lipofectAMINE 2000 (Invitrogen Corp., CA, USA) according to the manufacturer’s protocols. After overnight incubation at 37°C to ensure attachment, the cells were transfected with one of the previously mentioned plasmids encoding either the GFP-tagged wild-type hCCM3, hCCM3 with a point mutation (c.283C>T), or a single base-pair deletion (c.192delA).
Western Blotting
Cells were harvested and total protein from the cells was extracted in a lysis buffer (50mM HEPES, pH:7.4, 150mM NaCl, 10% glycerol, 1 %Triton X-100, 1.5mM MgCl2-6H20, 1mM EGTA, 100mM NaF, 10mM sodium pyrophosphate and protease inhibitors, 1mM Na3V04, 10mg/ml leupeptin, 10mg/ml aprotinin and 4mM PMSF). Cell lysates were centrifuged to collect supernatant, and equal amounts of protein, 50 μg/lane, (Bio-Rad protein assay system; Bio-Rad Laboratories, Hercules, CA) were separated with 10% or 4-20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Bio-Rad Laboratories, CA) and transferred onto a PVDF membrane (Bio-Rad Laboratories, CA). The membranes were incubated with primary antibodies: anti-cleaved caspase-3 (Cell Signaling Technology, Inc., Danvers, MA), anti-phospho-p38 or anti-p38 (Cell Signaling Technology), anti-GAPDH (Santa Cruz, CA), or anti-GFP (Sigma Aldrich, MO) followed by the appropriate fluorescence-conjugated (immunohistochemistry) or HRP-conjugated (Western blot) secondary antibodies.
Apoptosis Assays
Cells were permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2 minutes on ice and the terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) reaction mixture (In Situ Cell Death Detection Kit, TMR-red, Roche) was applied for 1 hour at 37°C. Cell counts were performed at 10 different fields in 5 separate transfection experiments.
Cell Viability Assays (MTT)
Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium (MTT) assay. Briefly, 72 hours following transfection, MTT was added to each well at a final concentration of 0.5mg/ml for 4 hrs; the cells were subsequently lysed in dimethyl sulfoxide (DMSO) and the amount of MTT formazan was quantified by determining the absorbance at 550 nm, using a microplate reader. Cell viability is expressed as a percentage of the control culture value (GFP plasmid transfection group).
PDCD10 siRNA Inhibition
HUVECs were seeded on 6-well plates (5×104 cells/well). After attachment overnight, cells were washed in PBS, and transfected with three sets of PDCD10 Stealth™ select siRNAs (25 base pair) (Unigene ID: Hs.478150), following the manufacturer’s protocol (Invitrogen Corporation, California). As control siRNA, stealth RNAi negative control (Invitrogen) was used. After 72 hours of incubation, HUVECs were washed and exposed to low serum (0.2% FBS). At different time points, HUVECs were harvested for Western blotting. The membranes were incubated with the following antibodies: anti-phospho-p38 (Cell Signaling Technology), anti-CCM3 29, or anti-GAPDH (Santa Cruz).
To quantify cell death occurring in the cultures, terminal deoxynucleotidyltransferase-mediated UTP end labeling (TUNEL) analysis was performed as above. The percentage of apoptotic cells was calculated from the positively labeled cells in each field (400x) from at least 5 fields for each group.
Statistical Analyses
Data are presented as means ± SE of absolute values or percent of control and were analyzed using either Student’s t-test (Figs. 1 and 2) or one-way ANOVA (Figs. 3-5). We initially performed a global ANOVA test to check for significance and if proven, proceeded with pair-wise comparisons to examine which groups were significantly different than the baseline. Bonferroni correction was applied to correct for multiple testing unless specified otherwise and p<0.05 was considered significant. The global ANOVA p-values and Bonferroni corrected p-values for pair-wise ANOVA tests are reported.
Figure 1. CCM3 overexpression induces apoptosis in HeLa cells.
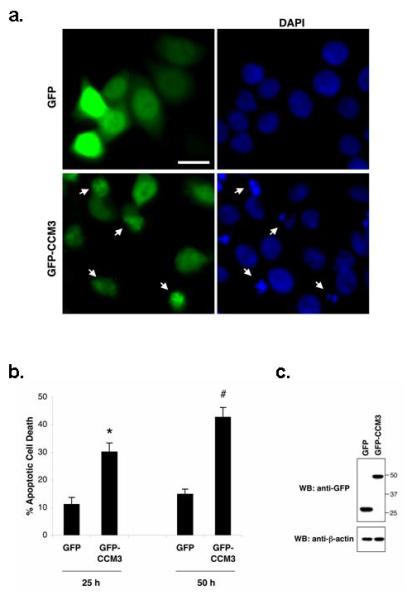
a. HeLa cells were transfected with vector alone or GFP-CCM3 constructs (see Methods). 50 hours (hrs) after transfection, cells were fixed, and nuclei stained with DAPI (blue). Arrows point to the GFP-CCM3-positive cells (green) with condensed or fragmented apoptotic nuclei. Scale bar = 20 μm.
b. GFP-positive cells were quantified for apoptosis by scoring apoptotic nuclear morphology as in (a). The percentage of GFP-positive cells with apoptotic nuclei was represented as mean ± SEM from three and four independent experiments for the 25 and 50 hour (hr) time point, respectively (left panel). *P < 0.002, significant difference from GFP-transfected cells; #P <10-3, significant difference from GFP-transfected cells (by Student’s t-test).
c. Protein expression of GFP and GFP-CCM3 25 hrs after transfection was confirmed by immunoblotting with anti-GFP. Anti-β-actin was used as loading control (bottom panel).
Figure 2. CCM3 activates caspase-3 in HeLa cells.
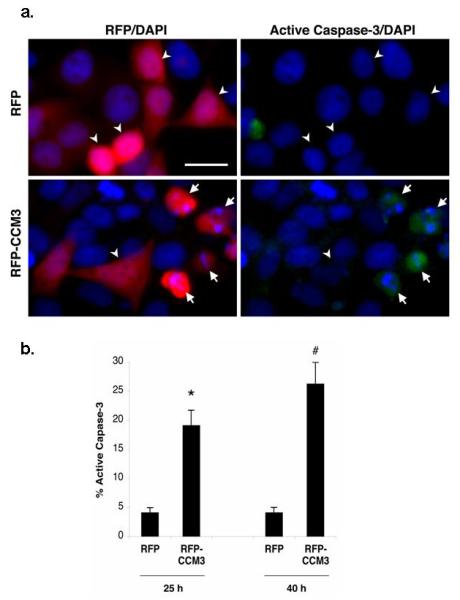
a. HeLa cells were transfected with vector alone or RFP-CCM3 construct. 40 hrs after transfection, cells were fixed and immunostained with FITC-conjugated anti-active caspase-3 (green), and nuclei were labeled with DAPI (blue). Arrows point to RFP-CCM3-positive cells (red) that are immunoreactive for active caspase-3. Arrowheads represent transfected cells that are not active caspase-3 immunoreactive. Scale bar = 20 μm.
b. RFP-positive cells were assessed for caspase-3 activation by scoring active caspase-3-immunoreactivity as indicated in (a). Histogram represents mean ±SEM from three independent experiments for the 25 hr and 40 hr time point, respectively. *P < 10-3, significant difference from RFP-transfected cells; #P <10-3, significant difference from RFP-transfected cells (by Student’s t-test).
Figure 3. Mutant CCM3 fails to induce cell death in HeLa cells.

Transient transfections in HeLa cells with the following expression plasmids: N= untransfected; V= vector alone; CCM3= wild type GFP-CCM3 construct; M1 = GFP-CCM3 construct harboring the 283C>T mutation; M2 = GFP-CCM3 construct harboring the 192delA mutation.
a. Western blotting for anti-GFP demonstrates transfection efficiency. HeLa cells were transiently transfected, cell lysates were prepared and immunoblotted with an antibody against GFP. The anti-CCM3 antibody 29 is downstream to the mutated sequences and thus fails to recognize the truncated protein.
b. Transfection with wild type CCM3 increases cleaved caspase 3 expression levels, while transfection with either mutated CCM3 construct has no effect on cleaved caspase 3 expression. *P < 0.05 compared with untransfected group (n = 3).
c. Transfections with mutated CCM3 fail to elicit significant cell loss in comparison with wild type CCM3. Cell viability was measured using the MTT assay (see Methods) and is graphed as a percentage of the control culture value (GFP vector alone transfection group) as quantified by determining the absorbance of cell cultures at 550 nm. *P < 0.05 vs. GFP plasmid transfection group (n = 3).
Figure 5. siRNA inhibition of CCM3 expression in endothelial cells results in decreased cell death, p38, and cleaved caspase-3 expression.

a. Serum-deprived HUVECs treated with CCM3 siRNA show decreased cell death. Representative images (20x original magnification) at the 3-hour time point are shown for untransfected HUVECs (no siRNA group, left panel) or those transfected either with control siRNA (middle panel) or CCM3 siRNA (right panel). All groups were subjected to serum deprivation (see Methods) and cell death was analyzed at various time points by TUNEL assay. TUNEL-positive cells (green) are scarce in the sample treated with CCM3 siRNA (right panel) compared to those untreated (left panel) or treated with control siRNA (middle panel).
b. The percentage of apoptotic cells was calculated from TUNEL positive cells in each field (400x) and in at least 5 fields for each group (global ANOVA P <10-3 in both control groups across all time points and within the 3 hour group). *corrected P < 0.05 compared with the equivalent groups prior to serum deprivation (0 hour) (n = 3).
c. Serum-deprived HUVECs show decreased cleaved caspase 3 levels after treatment with CCM3 siRNA (global ANOVA P < 10-3 in both control groups across two time points). HUVECs were transfected with control siRNA or CCM3 siRNA, then were cultured under serum deprivation conditions (0.2% FBS). Cells lysates were prepared at various time points and cleaved caspase 3 levels were analyzed using Western blotting. Serum deprivation results in increase of cleaved caspase 3 in both the no siRNA and control siRNA treated groups, but not in the CCM3 siRNA-treated group. *corrected P < 0.05 compared with the equivalent groups prior to serum deprivation (0 hour) (n = 3).
d. HUVECs treated with CCM3 siRNA demonstrate decreased p38 activation (global ANOVA P < 10-3 in p-p38/p38 and CCM3/GSDPH groups across 2 time points). Similar to the procedure in [c], serum deprivation results in increase of p38 activation and CCM3 protein levels in the no siRNA- and control siRNA-treated groups, but not in the CCM3 siRNA-treated group. *corrected P < 0.05 compared with the equivalent groups at each time point (n = 3).
Results
CCM3 Induces Apoptosis in HeLa Cells
We analyzed the effects of CCM3 overexpression in cell culture by transfecting HeLa cells with the expression plasmid pEGFP-C3-hCCM3 (referred as GFP-CCM3 henceforth). GFP-CCM3 (wild type) overexpression resulted in increased levels of apoptosis compared to vector alone (Figure 1). A significantly larger proportion of apoptotic cells with fragmented nuclei was observed in the population of GFP-positive (and, therefore, CCM3 expressing) cells transfected with GFP-CCM3 compared to those from control transfections with empty vector (Figure 1b). Protein expression of GFP and GFP-CCM3 was confirmed by immunoblotting with anti-GFP (Figure 1c).
We then investigated whether CCM3-mediated cell death is associated with caspase-3 activation. Caspase-3 has previously been established as a terminal executioner of the apoptotic cell death pathway 30. Activated caspase-3 was detected in apoptotic cells with fragmented/condensed nuclei expressing CCM3 (Figure 2). These data indicate that CCM3 overexpression in HeLa cells results in caspase-3 activation and cell death.
Disease-causing Mutated CCM3 Fails to Induce Apoptosis
We had previously reported four novel disease-causing PDCD10/CCM3 mutations in CCM families 12. We introduced two of these point mutations (c.283C>T [M1], c.192delA [M2]) into the GFP-CCM3 expression vector. Both mutations lead to production of truncated proteins (Figure 3a). We tested the effects of overexpression of mutated forms of CCM3 in HeLa cells. As expected, overexpression of wild type CCM3 resulted in increased cleaved caspase-3 levels, while overexpression of either mutant form of CCM3 (c.283C>T, c.192delA) had no effect on cleaved caspase-3 expression (Figure 3b). In addition, overexpression of either CCM3 mutant resulted in reduced cell loss by the MTT assay compared to wild type CCM3 (Figure 3c), further suggesting that mutant forms of CCM3 fail to induce apoptosis.
Serum-deprivation of Endothelial Cells Results in Increased CCM3 Levels
Expanding upon a previously established assay for the induction of apoptosis in endothelial cells 30, we cultured human umbilical vein endothelial cells (HUVEC) in low-serum media. Serum-deprived HUVECs demonstrated an increase in cleaved caspase-3 protein levels by Western blotting after 3 hours as expected (Figure 4a). Interestingly, CCM3 expression increased upon serum-deprivation starting at approximately 30 minutes, prior to the increase in cleaved caspase-3 levels (Figure 4b). Following serum starvation, re-incubation of cells in normal media (20% FBS) allows for cell recovery and restores (reduces) CCM3 expression to normal levels (Figure 4c). In order to establish the mechanism of this increase in CCM3 expression, we incubated the cells with different concentrations of actinomycin D, a transcription inhibitor. Actinomycin D treatment inhibits CCM3 expression confirming that serum starvation causes an increase in CCM3 through transcriptional control (Figure 4d).
Figure 4. Serum-deprivation of endothelial cells results in increased CCM3, activated caspase 3 and p38 levels.

a. Serum-deprived HUVECs demonstrate an increase in cleaved caspase 3 levels starting at 3 hrs post serum withdrawal (global ANOVA P<10-3). At different time points following serum withdrawal, cells lysates were prepared, and the cleaved caspase 3 levels were detected with Western blotting. Graphed densitometric analysis of cleaved caspase 3 is normalized to GAPDH. *corrected P < 0.05 compared with the group in 20% FBS at 0 hour (n = 3).
b. CCM3 expression increases upon serum-deprivation starting at approximately 30 minutes, preceding the increase in cleaved caspase-3 levels as above (global ANOVA P <10-5). * pair-wise corrected P < 0.01 compared with the group at 0 hour (n = 3).
c. Addition of 20% FBS to the cell cultures reduces the increase of CCM3 expression elicited by serum deprivation. Cells were cultured under 0.2% FBS for 1 hour, then exposed to 20% FBS or remained in 0.2% FBS, as described in the Figure. The graphed densitometric analysis of cleaved caspase 3 is normalized to GAPDH as demonstrated by the Western blot on top. *P < 0.05 compared between the 0.2% FBS treated group and the 20% FBS-treated group at 8 and 24 hour time points (n = 3).
d. Actinomycin D treatment of serum-deprived HUVECs prevents the increase in CCM3 protein levels, indicating that serum deprivation causes an increase in CCM3 transcription. HUVECs were incubated with various concentrations of actinomycin D, a transcription inhibitor, for 4 hours in 20% FBS, then exposed to 0.2% FBS still containing various concentrations of actinomycin D; cell lysates were prepared after 1 hour, and CCM3 levels were investigated with Western blotting. *corrected P < 0.01 compared with the group at 0 hour (n = 3).
e. Serum deprivation results in increase in phosphorylation of p38, again preceding the increase in cleaved caspase 3 levels [see a.] (global ANOVA P <10-5). p38 phosphorylation increases dramatically upon serum starvation at 5 minutes, followed by a second peak beginning at 3 hours post serum withdrawal. *corrected P < 0.05 compared with non-0.2% FBS-treated group at 0 hour (n = 3).
Because of previous evidence that the CCM1/2 complex signals through p38 24, 25 combined with the possibility that CCM1, 2, and 3 may all interact 18, 27, 28, and signal through either the same or parallel pathways, we hypothesized that CCM3 might also signal through p38. Consistent with this hypothesis, serum deprivation that results in an increase in CCM3 levels also resulted in a bimodal increase in activated phosphorylated p38 (p-p38) and activated caspase-3 levels. Although the levels of p-p38 increase dramatically almost immediately upon serum withdrawal, this early peak may not be related to increased CCM3 expression, as it is occurs early after challenge, and before CCM3 levels significantly change at about 1hour following serum starvation (Figure 4e).
In summary, serum deprivation of endothelial cells results in increases in CCM3 and phosphorylated p38 protein levels followed by caspase-3 activation.
siRNA Inhibition of CCM3 Expression Reduces Endothelial Cell Apoptosis during Serum Deprivation
Using the same serum-starvation model, we then reduced CCM3 protein levels with siRNAs as described in Methods. HUVECs treated with CCM3 siRNA demonstrated decreased levels of cell death, as shown by the TUNEL assay (Figure 5a,b). We then evaluated whether the reduction of cell death correlated with reduction of caspase-3 activation. Serum-starved HUVECs also demonstrated decreased activated caspase-3 levels after treatment with CCM3 siRNA (Figure 5c). This decrease in CCM3 and activated caspase-3 levels correlates with decreased p38 activation after CCM3 siRNA transfection compared to cells treated with siRNA controls (stealth RNAi negative control) (Figure 5d).
Discussion
Because CCM lesions in all three inherited forms of the disease are clinically and pathologically nearly identical, we had hypothesized over 10 years ago that genes causing familial cavernous malformations CCM1, 2 and 3 (KRIT1, MGC4607, and PDCD10, respectively) all participate in a common or parallel pathways 7, 12, 31, and thus disruption of signaling would lead to similar disease manifestations. To test this hypothesis, we previously investigated the expression patterns of CCM1, 2, and 3 and showed that these proteins spatially and temporally show similar expression patterns 29, 31-33. These proteins are specifically expressed throughout the endothelium in many tissues and organs and within the neurovascular unit of the CNS that consists of astrocytes, neurons and vascular endothelium 29, 31-33. Consistent with this observation, recent data from yeast and mammalian systems demonstrate that CCM1, 2, and 3 physically interact 18, 24, 27, 28, confirming findings in zebrafish which demonstrate that CCM1 and 2 act in a common pathway 34 with ccm1 and ccm2 mutants displaying comparable vascular defects35. Similarly, in vivo studies in the mouse based on Ccm1 and 2 knockout models have revealed their non-redundant, vital roles in angiogenesis 36, 37 but failed to reveal any breakthrough insights into their function.
On the other hand, multiple levels of in vitro evidence suggest that the CCM genes might play a role in a variety of biological processes linking extracellular signals, including adhesion (through β1 integrin 22, 23 or β-catenin 21 interactions) or stress (including osmo- or mechano-stress ) to various biological responses, including changes in morphology and proliferation 38. In this manuscript, we present data that link CCM signaling to apoptotic pathways.
Indeed, in vitro work has shown that the CCM1 protein, KRIT1, binds to microtubules 17, 18 in a folded state 19. Both Rap1a and integrin cytoplasmic domain associated protein 1 alpha (ICAP1α) can disrupt KRIT1’s association with microtubules localizing it to endothelial cell—cell junctions 19, 21. Interestingly, only ICAP1α appears to be able to unfold KRIT1 19. After localization to the cell membrane through its FERM domain, KRIT1 binds to β-catenin and plays a role in stabilizing adherens junctions and regulates endothelial permeability 21. Through its NPXY domains, KRIT1 also binds CCM2 24, potentially recruiting CCM2 to these junctions. CCM2 (Malcavernin) has been identified as a human paralog of the mouse osmosensing scaffold protein for MEKK3 (OSM), which, after binding with CCM1 and potentially this complex, might participate in the p38 mitogen-activated protein kinase (p38 MAPK) stress signaling pathway 24, 25. More recent evidence suggests that CCM3, at least in certain biological states, might be part of this complex 18, 27, 28. Furthermore, upon activation of this complex, CCM1 shuttles from the cytoplasm and the cell membrane into the nucleus and therefore might interact with other nuclear molecules, including transcription factors, which might influence basic cellular functions such as proliferation and apoptosis 39. Consistent with this hypothesis is the observation in C. elegans, in which kri-1, a CCM1 ortholog, interacts with DAF-16, a transcription factor, such that mutations in kri-1 lead to an increase in life span 40.
Based on these observations, we investigated the biological functions of CCM3 using over-expression or siRNA mediated inhibition of CCM3 expression. Previously published studies showed CCM3 to be a part of a cohort of proteins up-regulated upon growth factor deprivation and apoptosis induction in the TF-1 premyeloid cell line 11 as well as in a fibroblast cell line 41. Based on these data which suggested that CCM3 might play a role in apoptosis signaling, we initially focused our attention at understanding the capacity of CCM3 to induce apoptosis in the absence of exogenous pro-apoptotic stimuli. Over expression of CCM3 in HeLa cells was in fact sufficient to induce increased levels of cell death as evidenced by both nuclei morphology as well as caspase-3 activation, a well established common end pathway protein in apoptosis 30, 42, 43. This increase in apoptosis and activated caspase-3 levels was abrogated when cells were transfected with biologically inactive forms of CCM3, identified as mutations causing CCM in patients with the familial form of the disease 12.
We then focused on whether CCM3 was induced in the presence of pro-apoptotic stimuli. With the induction of serum starvation in HUVECs, CCM3 levels increased concomitantly with increased levels of activated caspase-3 and p38. Interestingly, this increase in percentage of apoptotic cells and activated caspase-3 and p38 expression levels was diminished when endogenous CCM3 was blocked by siRNA, suggesting that CCM3 might be necessary for induction of apoptosis in this in vitro model. Furthermore, although HUVECs represent a well-characterized model of endothelial cell responses in culture, it is always possible that in the context of the organism, endothelial cells behave differently. Thus, in vivo experiments are needed to extend and confirm these findings. In addition, even though some of the molecules involved in CCM signaling are known 18, many remain anonymous. Identification of these molecules along with in vivo experiments are needed to dissect the biological role of CCM signaling. Our results demonstrating CCM3 to be both necessary and sufficient for programmed cell death in vitro in cell culture models provide a novel insight into this complex pathophysiology.
The effects of CCM3 on apoptosis could be direct or indirect through cell cycle modulation. Previous evidence in the literature suggests that interference with cell cycle progression can lead to apoptosis 44. If true, CCM signaling can affect both proliferation 38 and apoptosis in a cell type dependent manner, potentially explaining observed effects of CCM3 on these biological processes. Further work will be needed to answer these questions. Our results, however, lead to the novel hypothesis of attenuated apoptosis affecting endothelial cells within the neurovascular unit as a molecular mechanism underlying cavernous malformations. If proven to be true in vivo, this might lead to a mechanistic understanding of the role of apoptosis and cell cycle control in CCM pathophysiology, potentially having significant implications in designing new therapies.
Conclusions
CCM3 is both necessary and sufficient to induce apoptosis in vitro in well-defined cell culture systems. These results lead to a novel hypothesis that CCM lesions may form due to aberrant apoptosis, potentially altering the balance between the endothelium and neural cells within the neurovascular unit. Even though it is currently unclear whether this effect on apoptosis is direct or indirect through modulation of cell cycle, the current data provide a testable hypothesis about CCM lesion development and has potentially important implications for a mechanistic understanding of CCM pathophysiology.
Acknowledgements
This study was supported by NIH/NINDS grant RO1-NS046521 (MG) and is part of the Ph.D. thesis of GT, who was partially supported by the Research Fund of Akdeniz University, Antalya, Turkey. We thank Dr. Katsuhito Yasuno for his help with statistical analysis.
Footnotes
The authors disclose no conflicts of interest.
References
- 1.Russell DS, Rubenstein LJ. Pathology of tumors of the nervous system. Williams and Wilkins; Baltimore: 1989. [Google Scholar]
- 2.Tu J, Stoodley MA, Morgan MK, Storer KP. Ultrastructural characteristics of hemorrhagic, nonhemorrhagic, and recurrent cavernous malformations. J Neurosurg. 2005;103:903–909. doi: 10.3171/jns.2005.103.5.0903. [DOI] [PubMed] [Google Scholar]
- 3.Wong JH, Awad IA, Kim JH. Ultrastructural pathological features of cerebrovascular malformations: A preliminary report. Neurosurgery. 2000;46:1454–1459. doi: 10.1097/00006123-200006000-00027. [DOI] [PubMed] [Google Scholar]
- 4.Dubovsky J, Zabramski JM, Kurth J, Spetzler RF, Rich SS, Orr HT, Weber JL. A gene responsible for cavernous malformations of the brain maps to chromosome 7q. Hum Mol Genet. 1995;4:453–458. doi: 10.1093/hmg/4.3.453. [DOI] [PubMed] [Google Scholar]
- 5.Johnson EW, Iyer LM, Rich SS, Orr HT, Gil-Nagel A, Kurth JH, Zabramski JM, Marchuk DA, Weissenbach J, Clericuzio CL, Davis LE, Hart BL, Gusella JF, Kosofsky BE, Louis DN, Morrison LA, Green ED, Weber JL. Refined localization of the cerebral cavernous malformation gene (ccm1) to a 4-cm interval of chromosome 7q contained in a well-defined yac contig. Genome Res. 1995;5:368–380. doi: 10.1101/gr.5.4.368. [DOI] [PubMed] [Google Scholar]
- 6.Gunel M, Awad IA, Finberg K, Anson JA, Steinberg GK, Batjer HH, Kopitnik TA, Morrison L, Giannotta SL, Nelson-Williams C, Lifton RP. A founder mutation as a cause of cerebral cavernous malformation in hispanic americans. N Engl J Med. 1996;334:946–951. doi: 10.1056/NEJM199604113341503. [DOI] [PubMed] [Google Scholar]
- 7.Gunel M, Awad IA, Finberg K, Steinberg GK, Craig HD, Cepeda O, Nelson-Williams C, Lifton RP. Genetic heterogeneity of inherited cerebral cavernous malformation. Neurosurgery. 1996;38:1265–1271. doi: 10.1097/00006123-199606000-00059. [DOI] [PubMed] [Google Scholar]
- 8.Craig HD, Gunel M, Cepeda O, Johnson EW, Ptacek L, Steinberg GK, Ogilvy CS, Berg MJ, Crawford SC, Scott RM, Steichen-Gersdorf E, Sabroe R, Kennedy CT, Mettler G, Beis MJ, Fryer A, Awad IA, Lifton RP. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum Mol Genet. 1998;7:1851–1858. doi: 10.1093/hmg/7.12.1851. [DOI] [PubMed] [Google Scholar]
- 9.Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF, Tournier-Lasserve E. Truncating mutations in ccm1, encoding krit1, cause hereditary cavernous angiomas. Nat Genet. 1999;23:189–193. doi: 10.1038/13815. [DOI] [PubMed] [Google Scholar]
- 10.Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T, Verlaan D, Balogun F, Hughes L, Leedom TP, Plummer NW, Cannella M, Maglione V, Squitieri F, Johnson EW, Rouleau GA, Ptacek L, Marchuk DA. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73:1459–1464. doi: 10.1086/380314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, Coubes P, Echenne B, Ibrahim R, Irthum B, Jacquet G, Lonjon M, Moreau JJ, Neau JP, Parker F, Tremoulet M, Tournier-Lasserve E. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42–51. doi: 10.1086/426952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guclu B, Ozturk AK, Pricola KL, Bilguvar K, Shin D, O’Roak BJ, Gunel M. Mutations in apoptosis-related gene, pdcd10, cause cerebral cavernous malformation 3. Neurosurgery. 2005;57:1008–1013. doi: 10.1227/01.neu.0000180811.56157.e1. [DOI] [PubMed] [Google Scholar]
- 13.Verlaan DJ, Roussel J, Laurent SB, Elger CE, Siegel AM, Rouleau GA. Ccm3 mutations are uncommon in cerebral cavernous malformations. Neurology. 2005;65:1982–1983. doi: 10.1212/01.wnl.0000188903.75144.49. [DOI] [PubMed] [Google Scholar]
- 14.Liquori CL, Berg MJ, Squitieri F, Ottenbacher M, Sorlie M, Leedom TP, Cannella M, Maglione V, Ptacek L, Johnson EW, Marchuk DA. Low frequency of pdcd10 mutations in a panel of ccm3 probands: Potential for a fourth ccm locus. Hum Mutat. 2006;27:118. doi: 10.1002/humu.9389. [DOI] [PubMed] [Google Scholar]
- 15.Labauge P, Denier C, Bergametti F, Tournier-Lasserve E. Genetics of cavernous angiomas. Lancet Neurol. 2007;6:237–244. doi: 10.1016/S1474-4422(07)70053-4. [DOI] [PubMed] [Google Scholar]
- 16.Awad IA, Robinson JR, Jr., Mohanty S, Estes ML. Mixed vascular malformations of the brain: Clinical and pathogenetic considerations. Neurosurgery. 1993;33:179–188. doi: 10.1227/00006123-199308000-00001. discussion 188. [DOI] [PubMed] [Google Scholar]
- 17.Gunel M, Laurans MS, Shin D, DiLuna ML, Voorhees J, Choate K, Nelson-Williams C, Lifton RP. Krit1, a gene mutated in cerebral cavernous malformation, encodes a microtubule-associated protein. Proc Natl Acad Sci U S A. 2002;99:10677–10682. doi: 10.1073/pnas.122354499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hilder TL, Malone MH, Bencharit S, Colicelli J, Haystead TA, Johnson GL, Wu CC. Proteomic identification of the cerebral cavernous malformation signaling complex. J Proteome Res. 2007;6:4343–4355. doi: 10.1021/pr0704276. [DOI] [PubMed] [Google Scholar]
- 19.Beraud-Dufour S, Gautier R, Albiges-Rizo C, Chardin P, Faurobert E. Krit 1 interactions with microtubules and membranes are regulated by rap1 and integrin cytoplasmic domain associated protein-1. FEBS J. 2007;274:5518–5532. doi: 10.1111/j.1742-4658.2007.06068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serebriiskii I, Estojak J, Sonoda G, Testa JR, Golemis EA. Association of krev-1/rap1a with krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21-22. Oncogene. 1997;15:1043–1049. doi: 10.1038/sj.onc.1201268. [DOI] [PubMed] [Google Scholar]
- 21.Glading A, Han J, Stockton RA, Ginsberg MH. Krit-1/ccm1 is a rap1 effector that regulates endothelial cell cell junctions. J Cell Biol. 2007;179:247–254. doi: 10.1083/jcb.200705175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zawistowski JS, Serebriiskii IG, Lee MF, Golemis EA, Marchuk DA. Krit1 association with the integrin-binding protein icap-1: A new direction in the elucidation of cerebral cavernous malformations (ccm1) pathogenesis. Hum Mol Genet. 2002;11:389–396. doi: 10.1093/hmg/11.4.389. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Clatterbuck RE, Rigamonti D, Chang DD, Dietz HC. Interaction between krit1 and icap1alpha infers perturbation of integrin beta1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum Mol Genet. 2001;10:2953–2960. doi: 10.1093/hmg/10.25.2953. [DOI] [PubMed] [Google Scholar]
- 24.Zawistowski JS, Stalheim L, Uhlik MT, Abell AN, Ancrile BB, Johnson GL, Marchuk DA. Ccm1 and ccm2 protein interactions in cell signaling: Implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet. 2005;14:2521–2531. doi: 10.1093/hmg/ddi256. [DOI] [PubMed] [Google Scholar]
- 25.Uhlik MT, Abell AN, Johnson NL, Sun W, Cuevas BD, Lobel-Rice KE, Horne EA, Dell’Acqua ML, Johnson GL. Rac-mekk3-mkk3 scaffolding for p38 mapk activation during hyperosmotic shock. Nat Cell Biol. 2003;5:1104–1110. doi: 10.1038/ncb1071. [DOI] [PubMed] [Google Scholar]
- 26.Zarubin T, Han J. Activation and signaling of the p38 map kinase pathway. Cell Res. 2005;15:11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- 27.Voss K, Stahl S, Schleider E, Ullrich S, Nickel J, Mueller TD, Felbor U. Ccm3 interacts with ccm2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics. 2007;8:249–256. doi: 10.1007/s10048-007-0098-9. [DOI] [PubMed] [Google Scholar]
- 28.Stahl S, Gaetzner S, Voss K, Brackertz B, Schleider E, Surucu O, Kunze E, Netzer C, Korenke C, Finckh U, Habek M, Poljakovic Z, Elbracht M, Rudnik-Schoneborn S, Bertalanffy H, Sure U, Felbor U. Novel ccm1, ccm2, and ccm3 mutations in patients with cerebral cavernous malformations: In-frame deletion in ccm2 prevents formation of a ccm1/ccm2/ccm3 protein complex. Hum Mutat. 2008;29:709–717. doi: 10.1002/humu.20712. [DOI] [PubMed] [Google Scholar]
- 29.Tanriover G, Boylan AJ, Diluna ML, Pricola KL, Louvi A, Gunel M. Pdcd10, the gene mutated in cerebral cavernous malformation 3, is expressed in the neurovascular unit. Neurosurgery. 2008;62:930–938. doi: 10.1227/01.neu.0000318179.02912.ca. discussion 938. [DOI] [PubMed] [Google Scholar]
- 30.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/akt signal transduction pathway. Requirement for flk-1/kdr activation. J Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 31.Seker A, Pricola KL, Guclu B, Ozturk AK, Louvi A, Gunel M. Ccm2 expression parallels that of ccm1. Stroke. 2006;37:518–523. doi: 10.1161/01.STR.0000198835.49387.25. [DOI] [PubMed] [Google Scholar]
- 32.Guzeloglu-Kayisli O, Amankulor NM, Voorhees J, Luleci G, Lifton RP, Gunel M. Krit1/cerebral cavernous malformation 1 protein localizes to vascular endothelium, astrocytes, and pyramidal cells of the adult human cerebral cortex. Neurosurgery. 2004;54:943–949. doi: 10.1227/01.neu.0000114512.59624.a5. discussion 949. [DOI] [PubMed] [Google Scholar]
- 33.Guzeloglu-Kayisli O, Kayisli UA, Amankulor NM, Voorhees JR, Gokce O, DiLuna ML, Laurans MS, Luleci G, Gunel M. Krev1 interaction trapped-1/cerebral cavernous malformation-1 protein expression during early angiogenesis. J Neurosurg. 2004;100:481–487. doi: 10.3171/ped.2004.100.5.0481. [DOI] [PubMed] [Google Scholar]
- 34.Mably JD, Chuang LP, Serluca FC, Mohideen MA, Chen JN, Fishman MC. Santa and valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development. 2006;133:3139–3146. doi: 10.1242/dev.02469. [DOI] [PubMed] [Google Scholar]
- 35.Hogan BM, Bussmann J, Wolburg H, Schulte-Merker S. Ccm1 cell autonomously regulates endothelial cellular morphogenesis and vascular tubulogenesis in zebrafish. Hum Mol Genet. 2008;17:2424–2432. doi: 10.1093/hmg/ddn142. [DOI] [PubMed] [Google Scholar]
- 36.Whitehead KJ, Plummer NW, Adams JA, Marchuk DA, Li DY. Ccm1 is required for arterial morphogenesis: Implications for the etiology of human cavernous malformations. Development. 2004;131:1437–1448. doi: 10.1242/dev.01036. [DOI] [PubMed] [Google Scholar]
- 37.Plummer NW, Squire TL, Srinivasan S, Huang E, Zawistowski JS, Matsunami H, Hale LP, Marchuk DA. Neuronal expression of the ccm2 gene in a new mouse model of cerebral cavernous malformations. Mamm Genome. 2006;17:119–128. doi: 10.1007/s00335-005-0098-8. [DOI] [PubMed] [Google Scholar]
- 38.Ma X, Zhao H, Shan J, Long F, Chen Y, Zhang Y, Han X, Ma D. Pdcd10 interacts with ste20-related kinase mst4 to promote cell growth and transformation via modulation of the erk pathway. Mol Biol Cell. 2007;18:1965–1978. doi: 10.1091/mbc.E06-07-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Rigamonti D, Dietz HC, Clatterbuck RE. Interaction between krit1 and malcavernin: Implications for the pathogenesis of cerebral cavernous malformations. Neurosurgery. 2007;60:353–359. doi: 10.1227/01.NEU.0000249268.11074.83. discussion 359. [DOI] [PubMed] [Google Scholar]
- 40.Berman JR, Kenyon C. Germ-cell loss extends c. Elegans life span through regulation of daf-16 by kri-1 and lipophilic-hormone signaling. Cell. 2006;124:1055–1068. doi: 10.1016/j.cell.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 41.Busch CR, Heath DD, Hubberstey A. Sensitive genetic biomarkers for determining apoptosis in the brown bullhead (ameiurus nebulosus) Gene. 2004;329:1–10. doi: 10.1016/j.gene.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 42.Fernandes-Alnemri T, Litwack G, Alnemri ES. Cpp32, a novel human apoptotic protein with homology to caenorhabditis elegans cell death protein ced-3 and mammalian interleukin-1 beta-converting enzyme. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- 43.Kumar S. The apoptotic cysteine protease cpp32. Int J Biochem Cell Biol. 1997;29:393–396. doi: 10.1016/s1357-2725(96)00146-x. [DOI] [PubMed] [Google Scholar]
- 44.Aylon Y, Oren M. Living with p53, dying of p53. Cell. 2007;130:597–600. doi: 10.1016/j.cell.2007.08.005. [DOI] [PubMed] [Google Scholar]