

Abstract
This article provides a perspective on collisions of ions with surfaces, including surface-induced dissociation (SID) and reactive ion scattering spectrometry (RISS). The content is organized into sections on surface-induced dissociation of small ions, surface characterization of organic thin films by collision of well-characterized ions into surfaces, the use of SID to probe peptide fragmentation, and the dissociation of large non-covalent complexes by SID. Examples are given from the literature with a focus on experiments from the authors’ laboratory. The article is not a comprehensive review but is designed to provide the reader with an overview of the types of results possible by collisions of ions into surfaces.
Introduction
Tandem mass spectrometry (MS/MS) is an essential tool for elucidating ion structure. The MS/MS experiment involves mass selection of a precursor ion followed by ion activation and subsequent dissociation. The ion activation step is commonly accomplished via collision-induced dissociation (CID) in which the initial kinetic energy of a projectile ion is converted into internal energy through inelastic collisions with a neutral gas. Several alternative activation methods have been employed in tandem mass spectrometry, one of which is surface-induced dissociation (SID). SID is analogous to collision-induced dissociation except that a surface replaces the neutral gas as the collision target. A typical ion-surface collision event is illustrated in Figure 1.
Figure 1.

Diagram of surface-induced dissociation where surface collision event deposits energy into the precursor ion. Collisions can generate fragment ions, neutralized precursor molecules, sputtered surface atoms and ion-surface reaction products. Inset shows an all trans configuration alkanethiolate (e.g. CH3(CH2)11S-Au) self-assembled monolayer on gold.
The incorporation of a surface into a mass spectrometer for ion activation was pioneered in the laboratory of R. Graham Cooks in the mid-1970s and early 1980’s [1–3]. Since that time, collisions of low-energy (eV) organic ions with surfaces within the tandem mass spectrometer have been valuable for analyzing surface composition, characterizing reactions between organic projectile ions and surface adsorbates, chemically modifying surfaces, and determining projectile ion structure. A major motivation for development of SID is that energy transfer to ionic projectiles can be improved by increasing the mass of the collision target. The total available energy for transfer into the internal modes of the projectile ion is defined by the center-of-mass energy (ECOM) described by Equation 1:
| (1) |
where ELAB is the laboratory collision energy and MION and MN are the masses of the projectile ion and neutral, respectively. Energy transfer in CID is limited by the mass of the collision partner, typically inert gases such as helium, argon or xenon. In SID, if one assumes that the surface is an infinitely large collision partner, ECOM becomes independent of mass and approaches the laboratory collision energy. The assumption that the entire surface can be viewed as the collision partner is not always valid however, and there are instances where the mass of the terminal groups on the surface influence the amount of energy transfer [4, 5]. Nonetheless, the use of a massive surface target should, in theory, provide greater energy transfer.
Use of a surface has several other attractive features, including no gas load on the instrument, no need to reproduce a specific gas pressure, and the ability to deposit energy in one fast large deposition step rather than “slow heating” of the ions by CID (slow heating may lead to more rearrangement products). Figure 2 is a simple depiction of how these different types of activation methods may affect the observed dissociation pathways as described previously by Wysocki and Kenttamaa for ion traps versus triple quadrupoles [6] and Laskin, Denisov, and Futrell for SID versus multiple collision CID [7]. Surface-induced dissociation has been implemented in many different instrument platforms including BE-surface-EQ, reflectron TOF MS, Q-surface-Q, TOF-surface-TOF, Q-surface-TOF, FTICR, and MALDI-IM-SID-TOF; information on different types of tandem mass spectrometers used to examine SID phenomena are available in a review from 2001 [8]. Many types of surfaces have been used in these experiments including contaminated metals, graphite, diamond, Langmuir-Blodgett films, and the often used hydrocarbon, fluorocarbon, and functionalized alkanethiolate self-assembled monolayer films illustrated in the inset of Figure 1. Because the chemical composition of the surface has a large effect on the nature of the ion-surface collision, it is important to understand the extent of energy deposition, electron transfer, and reactivity of different surfaces, as well as to examine the effects of surface stiffness and roughness.
Figure 2.

Reaction coordinates for SID and CID depicting sudden vs. gradual activation of protein complexes. When internal energy is deposited in a single event, dissociation pathways that do not involve unfolding may be accessible [6,7].
This brief perspective describes the surface-induced dissociation (SID) activation method for fragmenting projectile ions in tandem mass spectrometry. It should be noted that ion-surface reactions can result in a number of chemically interesting phenomena and are not limited to surface-induced dissociation. Chemical sputtering, elastic and reactive ion scattering, and soft landing are all prevalent chemical processes that occur in the hyperthermal (1–100 eV) collision energy regime, and descriptions of each can be found in recent reviews [8, 9]. The purpose of this article is to focus on ion-surface collisions as they pertain to ion activation, offering a perspective on SID and its application to various systems of interest and to present only a brief description of reactive ion scattering spectrometry (RISS). For SID, extensive fragmentation has been accomplished for small molecules, isomeric compounds, salt crystals, fullerenes, peptides, and, recently, non-covalent protein complexes. The content presented here progresses from collisions of small ions with surfaces to collisions of large non-covalent complexes with surfaces. For some systems, CID and SID spectra look very similar; presumably the fragmentation pathways and kinetics of fragmentation control the spectral appearance such that differences in the distribution of energy deposited or the stepwise nature of energy deposition do not lead to differences in spectra. For other systems, such as the large non-covalent complexes, SID achieves fragmentation not observed by CID.
Small Molecule Projectiles: SID and RISS
With the advent of SID as an ion activation method, studies were required to probe the potential of this technique. Low m/z polyatomic radical ions with simple, well characterized fragmentation profiles were used to conduct initial experiments. Low m/z molecular ions were also employed for reactive ion scattering spectrometry experiments that allow characterization of the surface.
Small Projectile Ion Fragmentation by SID
Low m/z polyatomic ions with well known dissociation energetics were employed to study the utility of ion surface collisions for the fragmentation of low m/z molecules. Small molecules such as Fe(CO)5 and (C2H5)4Si were used by Cooks and coworkers to investigate internal energy distributions deposited into projectiles upon surface collisions[8, 10, 11]. These “thermometer ions” were ideally suited for this type of work because they exhibit consecutive fragmentation with no or minimal competitive reactions [10]. For a given fragment ion the ratio of the relative abundance of the fragment ion to the energy range over which it is detected is used to estimate the average internal energy that must have been deposited into the projectile ion [10, 11]. From a series of these single probability values a distribution can be estimated allowing for a measure of the overall average internal energy of the projectile ions [10, 11]. This thermochemical method was used to compare single collision CID to SID. As seen in Figure 3, SID provides variable controlled ion internal energy with increasing collision energy [12]. In addition it was also shown that the average internal energy deposited changes linearly over a broad range of collision energies [13].
Figure 3.

Internal energy distributions of [Fe(CO)5]+• activated by collisions with a stainless steel surface. Ion-surface collisions demonstrate a more narrow energy distribution than gas collisions. Figure adapted from reference 12 with permission of John Wiley & Sons Limited.
While the thermometer method is advantageous in the calculation of energy deposition, it is limited by the number of small molecules with simple consecutive fragmentation pathways for which it can be applied. In order to increase the potential molecular candidates for energy deposition calculations, an extended deconvolution method was implemented by Vekey et al. [14]. This method allowed the incorporation of competitive fragmentation pathways thus enabling the calculation of energy deposition for a broader range of compounds with more complicated fragmentation schemes. While analogous to the thermometer method, the deconvolution method relies on an experimentally determined breakdown curve of the ion rather than threshold appearance energies of fragment ions. The method is limited by the number of molecules with fully characterized breakdown curves [14].
After reports of SID fragmentation of well characterized small molecules, larger simple cluster projectile ions were also characterized by SID. Whetten and coworkers probed the cleavage of sodium fluoride crystals by colliding mass selected clusters on surfaces. The ability to control the collision energies allowed for the determination of single-step low energy cleavages. Magic numbers in the resulting fragments were observed that correlated to stable full crystal lattice structures [15]. The appearance of specific fragments at lower collision energies as opposed to a distribution of products seen at higher energies demonstrated the ability of SID to access a variety of fragmentation pathways that are dependent on the collision energy employed [15].
With the applicability of SID to evaluate the fragmentation profiles of cluster ions, larger covalent molecules such as C60 became of interest. Whetten and coworkers found that unlike smaller ions and clusters, fullerenes did not exhibit fragmentation even at impact energies of 200 eV [16]. These experimental results were bolstered by theoretical calculations. Mowery et al. conducted molecular dynamics studies of C60 collisions on hydrogen-terminated diamond (111) surfaces [17]. It was shown that these large cluster molecules deformed upon collision with the surface but then exhibited resilience by regaining their cage-like structure upon recoil [17, 18]. Figure 4 illustrates the structural changes of a C60 ion during the course of a 250 eV collision with a hydrogen terminated diamond surface [17].
Figure 4.

Atomic positions for a trajectory resulting in non-reactive scattering of C60 with a collision energy of 250 eV a) before ion-surface collision (14 fs), b) during collision event (114 fs), and c) after ion-surface collision (294 fs). Figure adapted from reference 17 with permission of the American Chemical Society.
Busmann and coworkers collided fullerene clusters at higher collision energies with a highly ordered pyrolytic graphite (HOPG) surface and found that unfragmented scattered ion intensity decreased with an increase of energy between 250 and 450 eV [18]. These results also correlate with the molecular dynamics simulations of Mowery from which he suggests that above 250 eV reactive collisions occur where the ions do not maintain enough center-of-mass translational energy to break bonds made with the surface, thus creating “chemisorbed” layers [17]. If there are molecules that gain enough internal energy for fragmentation, these bonds would break on the microsecond time scale which is longer than the time frame of simulations and some of the experimental fragmentation studies undertaken of these fullerenes [17].
Fragmentation of large clusters was also found to be dependent on the surface chosen as the collision partner. Kappes et al. found that graphite contaminated with hydrocarbon species gave low mass sputter ions as well as influenced the overall fragmentation of the fullerene [19]. Busmann found a broader distribution of scattering angles for ions undergoing collisions with roughened diamond surfaces whereas graphite surfaces gave a much more narrow angular distribution [20]. Wysocki and coworkers showed that doubly charged C60 fragments at collision energies as low as 125 eV on a fluorocarbon surface, illustrated in Figure 5a [21], noting that the ions were doubly charged and formed by thermal desorption/electron ionization creating hotter ions as opposed to the laser desorption used by Whetten [22]. When doubly charged fullerenes were collided on a hydrocarbon surface at the same collision energy, however, as shown in Figure 5b, a significant amount of one electron charge reduction of C602+ was observed without the extensive fragmentation seen on the fluorocarbon surface [21]. Because C60 is used as a projectile for secondary ion mass spectrometry (SIMS) there is continued interest in C60 collisions at a variety of surfaces and simulations of collisions at keV collision energies are still ongoing in the Garrison lab [23–26].
Figure 5.

SID of C602+ at 250eV collision energy on a) fluorocarbon and b) hydrocarbon self-assembled monolayers (SAMs). Collisions on the hydrocarbon surface show significant amounts of one electron charge reduction whereas collisions on the fluorinated surface show fragmentation of both the charge reduced ion and the doubly charged precursor. Figure reprinted from reference 21 with permission of John Wiley & Sons Limited.
These experiments involving fullerene dissociation by SID highlighted the significance of surface selection. It may be advantageous to employ a particularly “hard” surface that generates a large fraction of fragment ions or conversely a “softer” surface might be needed in order to retain precursor ion information. Several reports in the literature discuss “hard” versus “soft” surfaces which have been loosely defined by the amount of fragmentation and the resulting scattering angle of the fragments after colliding with the surface. Hard surfaces such as fluorinated self-assembled monolayers (SAMs) generate a population of SID fragments with a narrower angular scattering distribution whereas softer surfaces such as hydrocarbon SAMs create broader distributions [27]. Fluorinated surfaces have been shown to exhibit unique characteristics when used as collision targets, which can be attributed to their terminal mass [27]. The larger terminal mass of a fluorocarbon SAM when compared to a hydrocarbon SAM results in a larger amount of laboratory energy conversion into the vibrational energy modes of a projectile ion when colliding on a fluorinated surface approximately 20%, as opposed to only 13% on a hydrocarbon surface [28]. The amount of energy imparted into projectile ions has also been studied using additional ω-functionalized SAM surfaces in order to investigate the influences of film chain lengths, surface packing density on different substrates, and interfacial hydrogen bonding [29–35].
The neutralization efficiency of the surfaces is also an important determinant of a surface’s utility in SID. Surfaces that exhibit a smaller amount of neutralization, such as fluorinated films, have a higher survival of resulting unfragmented projectile and fragment ions than those films that show a greater amount of electron transfer from the film to the projectile ions. The decrease in performance due to ion neutralization must therefore be taken into account along with the desired amount of fragmentation when selecting a surface partner for collisions.
CID has been used for the distinction of isomers. In order to more fully compare the utility of SID with CID it was important to show that ion activation by ion-surface collisions allowed comparable or better isomeric determinations. SID is attractive for the fragmentation of isomers as it requires only a single collision event which can minimize the effects of rearrangements. Cooks and coworkers demonstrated that isomer discrimination by SID was possible; they investigated a series of C2H4O molecules specifically by utilizing the MH+ precursor ion abundances relative to fragments [36]. This use of the ratio of the precursor ion abundances to fragment abundances as an additional tool for isomeric identification is possible for SID instruments that utilize an orthogonal configuration due to the fact that every precursor ion must collide with the surface. The resulting percentages of fragments to precursor molecules represent a statistical distribution of fragmentation products to non-dissociated precursor molecules in the absence of instrument discrimination effects. This is in contrast to CID where the ambiguity in the number of collisions that each precursor ion undergoes does not allow for direct comparisons of ion abundances between the precursor ions and resulting fragment ions.
Another unique advantage of SID is that some peaks in the MS/MS spectra do not come from simple fragmentation of the precursor molecule. For odd electron precursors, reactions may occur between the projectile ions and surface adsorbates. While the resulting ions are typically discussed as a distinct process of ion-surface reactions, these products are inextricably convoluted into SID spectra and can be an additional asset in studying projectile fragmentation when ion-surface collisions are the ion activation method. Some ion-surface reactions such as radical H and CH3 abstractions from the surface can be used as diagnostic peaks for the distinction of isomers. For the spectra of C6H6 isomers shown in Figure 6, the peaks at 91m/z originate from an abstraction of CH3 followed by a subsequent loss of a neutral H2 molecule [37]. The abstraction product ion abundances relative to that of the remaining intact precursor ions are clearly different and their appearance in SID mass spectra can be an important tool for isomer distinction. Cooks and coworkers used the presence of these reaction products in SID spectra to characterize isomers of furanocoumarins (heterocyclic aromatics). They found that even though product ion spectra acquired by high energy CID could differentiate between linear and cyclic precursors, additional isomers were indistinguishable. Isomers activated by SID, however, reacted differently and generated distinct product/precursor ratios. The use of these reaction peaks allowed the distinction of more isomeric compounds by SID than CID [38]. These reaction products are advantageous for projectile ion distinction and are dependent upon the surface composition. For this reason it is important to characterize the surfaces employed in ion-surface collisions in order to appropriately select surface collision partners used in SID experiments.
Figure 6.

Isomers of [C6H6]+• collided at 35eV on stainless steel surfaces. The 91m/z inset shows reaction peaks used for further differentiating isomers beyond visual fragmentation profiles. Figure reproduced from reference 37 with permission from the American Chemical Society.
Reactive Ion Scattering Spectrometry (RISS)
Reactions can occur between terminal chemical moieties of organic thin film surfaces and the incoming projectile ions at hyperthermal energies (1–100eV) at which bond cleavage and bond formation can occur. The technique of reactive ion scattering spectrometry (RISS) can characterize the surfaces used as collision partners by using well characterized projectile ions. The information gained from these experiments is essential to maximizing the potential control of SID where reaction peaks can be enhanced or diminished depending on the goals of the experiment. Types of reactive projectile ions vary from atomic species such as Cs+ used extensively by Kang and coworkers in a UHV chamber [39–42] to polyatomic ions. Abstractions of CH3• and H• from the surface are particularly useful reactions seen with odd electron polyatomic projectile ions as they can not only be used to characterize hydrocarbon films but can also indicate the cleanliness of thin films containing hydrocarbon as a contaminant. Surfaces employed as collision targets for MS experiments within the pressure range of 10−6 to 10−7 Torr can become contaminated with adventitious hydrocarbons from pump oil [43] or from organics used in experiments [44–46]. Ion-surface collisions of fullerenes on contaminated highly ordered pyrolytic graphite (HOPG) showed this technique to be more sensitive means of detection of sputtered hydrocarbons than low-energy electron scattering and the cleanliness of the surface had a direct impact on the scattering of C60 clusters as mentioned previously [20].
Reaction products containing surface components such as methyl and hydrogen atoms can also be used to monitor orientation of groups at the surface as shown by reactivity results obtained for odd and even chain length films [47]. SAM films of hydrocarbon adsorbates on gold have well defined terminal orientations based on their intrinsic tilt angles upon adsorption as is depicted in Figure 7a [47]. For odd chain length SAMs, the terminal carbon-carbon bond is approximately parallel to the surface. This exposes a more perpendicular H-C bond to the projectile ions and results in an increased observance of H abstraction reaction product ions. An even chain length hydrocarbon SAM has terminal C-C bonds approximately perpendicular to the surface. This creates a more probable reaction between incoming projectile ions and exposed surface carbon atoms leading to increased methyl abstractions as shown in Figure 7b [47].
Figure 7.

(a) Pictorial representation of alkyl SAMs on gold showing terminal carbon-carbon bond orientation with respect to incoming projectile ions and (b) % methyl addition observed on odd and even chain alkyl SAMs with 20eV collisions of pyrazine. Figure adapted from reference 47 with permission of the American Chemical Society.
In addition to odd/even reactivity differences, it has been shown by Wysocki and coworkers that the interaction of the projectile ion is predominantly isolated to the terminal carbon atoms [43]. Terminally labeled Langmuir-Blodgett (LB) film methyl groups (13CH3 and CD3) were interrogated by reactive ion collisions by a variety of both deuterated and non-deuterated projectile ions [43]. Abstraction products showed clear mass shifts for the incorporation of the labeled methyl groups. Additionally, LB films of CD3(CH2)16- and CD3(CD2)16- gave remarkably similar abstraction products showing that the ion beam probes predominantly the uppermost surface at low collision energies.
Peptides and Proteins
Mass spectrometry emerged as a tool to study peptide structure in the early to mid 1980’s with the development of “soft” ionization techniques such as fast atom bombardment (FAB) [48, 49], electrospray ionization (ESI) [50] and matrix-assisted laser desorption ionization (MALDI) [51]. As these methods predominantly produced molecular ions with little fragmentation, few structural details were available from a single stage of mass spectrometry. Consequently tandem mass spectrometry became essential for the structural characterization of biological molecules. The ability to ionize peptides and proteins was met with the realization that traditional methods of activation, namely collision-induced dissociation (CID), did not always provide extensive fragmentation for large biological ions, especially singly charged ions above ~3,000 daltons [52–54]. While the upper mass limit of CID has since been overcome through the increased use of multiple collisions in CID and the ease of fragmenting multiply charged ESI-generated proteins [55], these difficulties in the early days of biological mass spectrometry opened the door for alternative fragmentation methods to be explored.
Advances in sample ionization coincided chronologically with the first routine application of SID in a tandem mass spectrometer by Cooks and coworkers [3]. Early SID studies conducted with small molecules demonstrated efficient conversion of the ion’s translational energy into its internal degrees of freedom (e.g. kinetic to internal energy), suggesting SID could be a promising method for fragmenting larger, biological species. This lead several research groups to investigate the potential of SID for use in peptide and protein activation. Indeed, collaborative work between the McLafferty and Hunt groups and studies from our own laboratory verified that extensive sequence coverage could be achieved for large peptides such as RSBP (MW 3054) and Porcine ATCH (MW 4568) upon surface collision [56, 57]. McLafferty and coworkers also demonstrated the effectiveness of SID as an activation method for multiply protonated proteins, fragmenting 29 kDa carbonic anhydrase [58].
Despite this success, SID has not been incorporated into commercial mass spectrometers, preventing widespread application outside of labs willing to build and develop their own instrumentation. SID has nevertheless proven its worth in fundamental studies of fragmentation mechanisms and energetics, especially in the case of peptides for which accurate activation energies are difficult to estimate. An important feature of SID is the deposition of a well defined internal energy distribution that can be finely varied by changing the laboratory collision energy. This tunability reduces the uncertainty in the amount of internal energy deposited upon activation, providing a method to reliably measure relative energies for fragmentation processes.
The Role of SID in Developing the Mobile Proton Model and Elucidating Peptide Fragmentation Mechanisms
Protein sequencing by mass spectrometry was realized over twenty-five years ago [59–61]. Since that time, improvements in sequencing have been spurred by the development of protein identification algorithms as well as a better understanding of peptide dissociation in the gas-phase. The latter advancement has been achieved by exploring the effects of peptide size, sequence, gas-phase basicity, charge state, and secondary structure on fragmentation [62–70]. Surface-induced dissociation was influential in helping to develop a general framework for peptide fragmentation through the energy-resolved study of systematically varied model peptides. SID results from the Wysocki group contributed to the mobile proton model, a description of peptide dissociation in which fragmentation is initiated by rapid intra-molecular proton transfer among backbone protonation sites.
Evidence for the mobile proton model is illustrated by the relative fragmentation energetics of four penta-peptides (sequence XAAAA, where X = A, P, K, or R) shown in Figure 8a [65]. Each peptide was fragmented by SID on a gold surface coated with a fluorinated SAM film. Their relative fragmentation was compared by plotting the fragmentation efficiency curves (FECs, plots of percent fragmentation vs. laboratory collision energy) for each peptide. The inflection point energy, defined as the laboratory collision energy corresponding to 50% fragmentation of the precursor ion, can be used to provide a single point comparison for relative fragmentation. In this case, it is clear that the inflecton point energies of these four peptides are shifted in accordance with the gas phase basicity of the N-terminal amino acid residue. This is consistent with the idea that when a proton is more tightly sequestered at the site of greatest basicity, more energy is necessary to mobilize the proton and induce fragmentation. Consequently, the energy required to fragment a peptide increases along with the gas-phase basicity of the ion (non-basic peptides < Pro at the N-terminus < Lys-containing < Arg-containing). If this inflection point energy is corrected for the number of degrees of freedom (DOF) in each peptide, fragmentation energetics can be compared between peptides of various size, revealing differences due solely to chemical properties such as gas-phase basicity. It then becomes clear that there is a linear correlation between the DOF corrected energies and the gas-phase basicity of the peptides (Fig. 8b) [65, 71–74].
Figure 8.

(a) Fragmentation efficiency curves of the [M+H]+ ion for four alanine containing peptides: Ala5, Pro-Ala4, Lys-Ala4, and Arg-Ala4. Fragmentation of the electrosprayed ions was performed in a tandem quadrupole instrument via SID. (b) The DOF-corrected inflection point energies from (a) plotted against the gas-phase basicity of each peptide. Figure reproduced from reference 57 with permission from the American Chemical Society.
SID experiments have also played an essential role in characterizing residue-specific peptide cleavages. As the number of tandem MS peptide studies increased, several researchers began to notice enhanced fragmentation at particular amino acid residues that prevented complete backbone cleavage, and resulted in incomplete sequencing information [59, 62, 64, 68, 70, 75–78]. Protein identification algorithms may benefit from the incorporation of selective cleavage rules if residue-specific influences are more completely understood. Studies in our research group focused on characterizing why certain residues cause enhanced cleavages and studying the prevalence of those cleavages over a variety of instrument time frames and activation methods, including SID. For example, the presence of aspartic or glutamic acid in a peptide commonly causes cleavage on the C-terminal side of the acidic residue, particularly when arginine is also present within the peptide and the number of added protons is less than or equal to the number of arginine residues. SID studies of peptides containing acidic residues illustrated that if the number of ionizing protons were equal to the number of Arg residues, then the arginine(s) could sequester the proton(s) allowing the acidic hydrogen on the aspartic or glutamic acid side chain to initiate backbone cleavage [79, 80]. Figure 9a shows SID spectra of singly protonated LDIFSDFR (9a) and LDIFSDF (9b). The former contains one Arg and two Asp residues, and shows selective cleavage at the C-terminus of aspartic acid to preferentially yield y2 and y6 ions. The latter peptide has the same sequence at residues 1–6 but contains no arginine, and non-selective cleavage occurs along the peptide backbone. When the peptide analyzed in Figure 9b is derivatized to add a fixed charge at the N-terminus, selective cleavage is once again observed at Asp because the charge can no longer be mobilized (Figure 9c).
Figure 9.
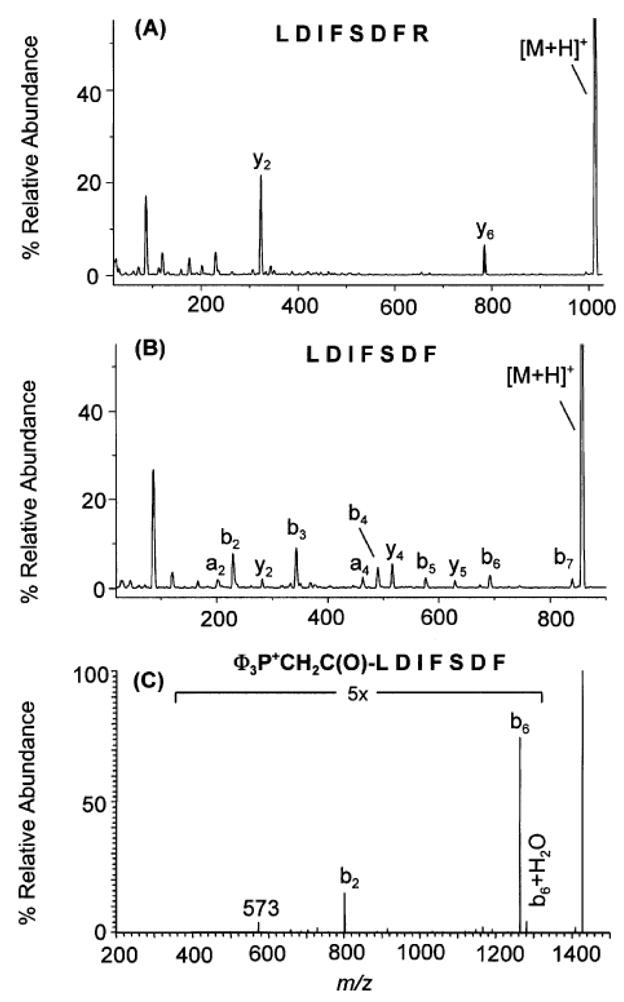
SID spectra of singly protonated LIDFSDFR (a), LDIFSDF (b), and the fixed charge derivative Φ3P+CH2C(O)-LDIFSDF acquired on a quadrupole ion trap (Thermo LCQ). Figure adapted from reference 79 with permission of the American Chemical Society.
A statistical analysis of over 28,000 tryptic peptide ion trap tandem mass spectra of singly, doubly, and triply-charged peptides confirmed the common trends observed in model peptide studies [70], and agrees with a related study of doubly charged peptides [81]. The factors with the greatest influence on peptide dissociation are the mobility of the proton, the position and basicity of the most basic residue, and the presence or absence of proline. Doubly charged peptides are far more likely to contain a mobile proton and follow the random cleavage model upon which sequencing algorithms are based, whereas singly charged peptides demonstrate a greater dependence on the basicity of the C-terminal arginine or lysine. In the case of singly charged tryptic peptides, Lys-ending peptides are far more likely to have a heterogeneous population of protonated forms, or a “partially mobile proton” whereas Arg-ending peptides can more completely sequester the proton at the guanidinium group. When the proton is mobile or partially mobile, cleavage N-terminal to proline, if present, dominates the product ion spectra. When proline is not present, enhanced cleavage is observed at other residues that have atypical Ramachandran Φ, Ψ plots, indicating a steric influence on fragmentation [68]. If the proton is sequestered at a basic residue, the trend of enhanced cleavage C-terminal to acidic residue dominates, as observed for the model peptide in Figure 9a. In this case, enhanced cleavage at Asp, Glu was found to be more dominant in Arg-ending tryptic peptides than in Lys-ending tryptic peptides, a statistical correlation consistent with the notion that protons are more tightly sequestered by the more basic side chain of arginine. While current computer algorithms ignore much of the chemically meaningful relative intensity information available in a tandem mass spectrum, the success rate of protein identification might be improved by taking into account the chemical properties of individual amino acid residues.
Additional efforts to improve protein identification and characterization have promoted novel developments in SID instrumentation spearheaded by Russell’s group. Surface-induced dissociation has proven to be a simple, yet effective method of activating ions in ion mobility-orthogonal time-of-flight instruments [82–84]. The advantage of these instruments is that complex proteomic samples can be separated in the gas-phase by ion mobility followed by comparatively faster MS and tandem MS analysis as species exit the mobility drift cell. This allows simultaneous acquisition of peptide molecular masses and sequence information in a single experiment. The incorporation of SID as the activation method is cost-effective and relieves the need for bath gas, simplifying experiments conducted with high resolution TOF mass analyzers. It also provides finer, more reproducible control over the fragmentation chemistry of peptide samples than CID.
Implementation of SID in an FTICR instrument has been shown to improve high-throughput of peptide dissociation [85]. Unlike SORI-CID, the gas-free nature of SID allows LC/MS/MS studies to be conducted without the need to pump down the ICR cell, increasing the rate of spectral acquisition. This allows a greater number of precursor ions to be selected for MS/MS within a single chromatographic peak. High-resolution SID is particularly amenable to patchwork efforts that have been used to improve protein identification. The concept of the patchwork approach is to utilize accurately identified fragment ions in the low m/z region of tandem mass spectra as compositional qualifiers in peptide sequencing [86]. These qualifiers, consisting mainly of a2, b2, y1, y2, internal and immonium type ions, provide additional restraints during database searching that help ensure the correct peptide is identified. Fernandez et al. demonstrated that SID in an FTICR yielded a large number of these qualifier ions, improving the statistical significance of database searches [87]. In addition, SID conducted on a diamond surface provided improved sequence coverage over SORI-CID owing to the accessibility of both fast and slow dissociation pathways not readily accessible by CID. The wide energy distribution reported was attributed to the energy deposition properties of the diamond collision target itself. This highlights the importance of understanding the role that the surface plays in the study of peptide dissociation. While fundamental aspects of ion-surface collisions involving small molecules were discussed above, a number of studies have focused on the effects of extending SID to ions of increasing mass and the influence of surface composition on the dissociation of biological molecules.
Characterization of Energy Deposition in Peptide Ion-Surface Collisions
The view of the surface as a nearly infinite collision partner implies that ion activation should essentially be independent of mass, making SID potentially even more attractive for the study of large projectile ions such as peptides and proteins [88, 89]. As mentioned earlier however, this assumption is not always valid and several attempts have been made to understand energy conversion in SID as it pertains to biological molecules. Energy transfer in SID has also been addressed in terms of the distribution of internal energy deposited into the ion population. An understanding of these characteristics and how they are affected by ion mass is essential for comparing SID with other activation methods or evaluating different types of surfaces.
While thermometer ions were used to characterize energy deposition in SID of small molecules, the dissociation pathways of peptides are far more complicated. The deconvolution method of Vekey et al. mentioned in the small molecule section has also been employed to estimate activation energies for the dominant fragmentation channels of several peptides [90]. By estimating the internal energy of a peptide ion based on the extent of fragmentation, the kinetic to internal energy conversion upon collision with a given surface can be estimated. Laskin, Denisov, and Futrell [7, 91] have shown that SID collision energy can be converted to an “effective” center of mass energy scale by superimposing the SID and multiple-collision CID fragmentation efficiency curves and using an arbitrary value of MN to represent the surface collision partner. When comparisons between the SID and CID fragmentation efficiency curves for a series of small alanine-containing peptide ions were made in the center of mass reference frame, the best overlap was observed using an effective surface mass between 39 and 46 for all peptides colliding with a fluorinated SAM [7]. This implied that the energy transfer in SID is not independent of projectile ion mass, but that kinetic to internal energy conversion decreases with increasing molecular size. Though the arbitrary value of MN given to the surface in these experiments has no physical significance, it indicates that perhaps the surface is more appropriately defined in terms of an “effective” mass than as a bulk collision partner of essentially infinite mass, and that the mass of the terminal chemical moieties on the surface (i.e. the CF3 group of a fluorinated SAM) dictate how much internal energy is deposited [4]. As mentioned above the Wysocki group has also found the terminal groups of SAM films to have the most effect on the extent of kinetic to internal energy conversion, but the underlying chemical groups also appear to play a secondary role [5].
Laskin, Futrell, and coworkers [91–94] have developed a method for defining the energy deposition function of an activation process that combines RRKM modeling with experimental MS/MS data to define the distribution of internal energy deposited into a peptide ion population. In this approach, the energy deposition function is determined by simulating a breakdown curve based on the microcanonical rate constants for each dissociation channel. The simulated curves are compared to experimentally obtained fragmentation efficiency curves until the best fit is achieved. The energy deposition function has been used to compare SID with multiple-collision CID and to explore the utility of different surfaces in peptide SID. While SID has been shown to offer a narrow internal energy distribution relative to single collision CID [95, 96], Laskin, Futrell and coworkers have found that internal energy distributions for SID and multiple-collision SORI-CID in an FTICR are similar [91, 97]. They have also demonstrated that the width of the energy deposition function in SID is greatly affected by the stiffness of the surface. The modeled energy deposition function of a hydrocarbon SAM film, which is approximately equivalent to a thermal distribution of energies, was found to be 1.6 and 2.3 times narrower than the energy deposition functions of stiffer LiF and diamond surfaces, respectively [4].
Experimental studies of peptide SID have been supported by increasing efforts to characterize energy transfer through simulations of peptide ion-surface collisions [98–102]. Hase and coworkers have shown that several factors affect kinetic to internal energy conversion in the SID process including collision energy, peptide size and conformation, incident collision angle and surface target. Classical trajectory simulations of protonated diglycine and dialanine demonstrated that internal energy transfer in normal-incidence SID (collision perpendicular to the surface) decreases slightly with increasing collision energy [101, 102]. The influence of peptide size has been explored by examining collisions of polyglycine (glyn-H+, n=1–5) with a diamond surface. The percentage of energy transfer into the peptide’s internal modes was found to be slightly greater for larger peptides due to the greater number of vibrational modes [98, 102]. The angular dependence of SID has been explored by varying the incident angle from 0 to 45 degrees (with zero representing a collision perpendicular to the surface) which resulted in a decrease in energy transfer to internal modes of diglycine. Simulations from the Hase group have also helped to define the role of the surface in SID by studying not only internal energy deposited into the projectile ion, but transfer into vibrational modes of the surface as well. In a comparison of diamond and n-hexyl thiolate SAM surfaces, the SAM film was found to absorb a far greater percentage of the projectile ion’s initial kinetic energy [98]. SID of folded (Gly)3 resulted in transfer of 63 % of the kinetic energy into the SAM film as opposed to only 9% into the stiffer diamond surface. The majority of the energy in the collision with diamond remained as translational energy of the fragment ions. The diamond surface was found to be more efficient for activation, transferring approximately twice the amount of internal energy into the projectile ion as the n-hexyl thiolate SAM film. What becomes clear from these theoretical studies is that experimental work over a wide range of surface compositions, collision angles, and ion types is necessary to characterize energy deposition in SID.
Kinetics of Peptide Fragmentation
Collisional activation, whether by gas or surface, is generally considered to occur via two steps: (1) activation via collision with the target and (2) unimolecular dissociation some time after the activation step once the ion has traveled away from the collision target. The observation of fragment ions in an MS/MS experiment is dependent on the rate of the dissociation of the precursor ion and the timescale of the mass spectrometer. Typically, excess energy above the activation barrier must be added for fragmentation to be observed on the timescale of the mass spectrometer, a phenomenon known as the kinetic shift. As the observation time frame in a mass spectrometer can range from nanoseconds to several seconds or longer, the amount of internal energy that must be added to overcome the kinetic shift varies from instrument to instrument. Consequently, dissociation products can differ substantially when produced on the microsecond time-scale of tandem quadrupole SID instruments versus those generated in trapping mass spectrometers. Recently, there has been evidence to suggest that some peptides may fragment by an alternative dissociation process known as shattering, in which fragmentation occurs nearly instantaneously while ions are still in direct contact with the surface. Full understanding of the SID process requires knowledge of the timescale on which dissociation occurs and must be studied using instruments with different observation time windows.
Time-resolved SID studies have been conducted in an FTICR by varying the trapping time following peptide ion-surface collision [103–106]. These studies revealed the time-dependence of peptide fragmentation when the observation time frame ranged from one millisecond to several seconds. The fragmentation efficiency curves for some fragment ions however, proved to be independent of the timescale. The time-dependent fragments were formed through statistical RRKM-type unimolecular decay at low collision energy, whereas the formation of time-independent fragments was attributed to an instantaneous shattering of peptides at the surface under high energy conditions. It should be noted however, that in this case time-independent means only that the protonated peptides had fragmented faster than the msec minimum detection time in the FTMS instrument.
Simulations performed by Hase and coworkers have supported evidence of a shattering fragmentation mechanism given the appropriate laboratory energies, collision angles, and surfaces [99–101, 107]. Simulations of glycine colliding with a diamond surface revealed that as much as 55% of the total fragmentation observed could be attributed to shattering at the surface [99]. For simulations on diglycine, the shattering mechanism resulted in an increase in the number of fragmentation channels available from 6 at 30 eV to 59 at 100 eV and was strongly influenced by the orientation of the diglycine molecule as it collided with the surface [101].
Typically, unimolecular dissociation alone has been reported to dictate the SID product ion spectrum with no evidence of shattering. Hanley and coworkers, who used an axial energy analyzer to measure the kinetic energy distributions of fragment ions, determined that peptide dissociation occurs away from the surface rather than during direct surface contact [108]. In contrast to small ion SID, in which evidence of shattering was provided by the scattering of products with different velocities but similar energies [109], peptides fragments were shown to possess a common velocity. This indicates, according to the conservation of momentum, that larger peptide ions must decompose only after leaving the surface.
Gamage et al. [110] investigated peptide fragmentation kinetics by performing SID in a MALDI-TOF specifically designed for observing sub-microsecond fragmentation products. SIMION trajectory simulations of precursor and product ions were used to estimate fragmentation time based on the experimentally observed surface-to-detector flight times of peptide fragments. Gamage et al. proposed that these fast fragmentation processes occur on the nanosecond timescale, corresponding to a fragmentation distance of 1–3 mm away from the surface and a log k of 7 based on peak shape analysis. No experimental evidence of instantaneous dissociation was observed.
Protein Complexes
In recent years the mass spectrometric analysis of non-covalent macromolecular complexes has rapidly grown into an exciting area of research. Through the contributions of a number of different research groups, numerous non-covalent assemblies have now been investigated in the gas phase [111–117]. The wide array of non-covalent systems that have been studied include complexes as simple as small protein-protein dimers and assemblies as daunting as the intact 70S ribosome, a heterogeneous 2 MDa complex composed of several RNA and over 50 different proteins [113, 116]. Regardless of the complex, analysis by mass spectrometry has typically required nanospray ionization of the analyte from “near-physiological” (i.e. neutral pH) conditions. Furthermore, the efficient transmission of these large ions hinges on the use of elevated source pressures to provide collisional focusing in the source region [118]. In many of these studies the mass-selected complexes have been fragmented using collision induced dissociation (CID), typically with argon as the target gas. It has been shown in numerous studies with a variety of protein-protein complexes that CID causes highly asymmetric dissociation with respect to both mass and charge [112, 114, 119]. Typically, the predominant dissociation products observed are a monomer and (n-1)mer (where n = the oligomeric state of the precursor ion) with each ion retaining a nearly equal number of charges despite the large difference between their molecular weights. From a practical standpoint, this dissociation pathway limits the amount of structural information that can be derived from MS/MS spectra. Furthermore, this unusual phenomenon has sparked a flurry of research seeking to better understand how large non-covalent assemblies dissociate in the gas phase. Through the work of a number of groups the general hypothesis that has emerged explains asymmetric dissociation by a pathway that must involve gas phase proton transfers and significant unfolding of the ejected monomer [112, 114, 120]. Although the precise details of what drives this process are still a matter of debate, it is generally agreed upon that protein unfolding must occur to increase surface area and relieve the Coulomb repulsion induced by the disproportionate number of charges (protons) that are typically carried away by the ejected subunit [112, 114, 120].
In light of the recent interest in the activation and dissociation of gas-phase complexes, our group has begun probing these phenomena via surface collisions. Unlike the common ion activation methods that have been employed previously, by which internal energy is deposited gradually in multiple steps [121], SID takes place following a short time-frame collision event with a surface as depicted in Figure 2. If a protein subunit unfolds during the multiple gas-phase collisions provided by CID then it is expected that the transition state structure of this dissociation reaction would be quite different from that of a folded monomer being ejected. Furthermore, an unfolded monomer is likely to be bound less tightly due to the disruption of its native binding interfaces. Therefore, once unfolded, it is likely that expulsion of this subunit would have a lower activation barrier than expulsion of a folded protein subunit where the integrity of its binding interfaces remains relatively intact. This is not to say that the entire pathway involving monomer unfolding is energetically less demanding than a direct dissociation pathway that does not require disruption of tertiary structure. In fact, monomer unfolding is likely to be quite energetically demanding. The reason why this pathway may be favored in CID is due to the small steps in which the ion is activated, each of which is not sufficient to cause dissociation, but may affect the structures prior to dissociation. We have hypothesized that if stepwise unfolding occurs through a series of incremental increases in energy then it should be possible to access the direct dissociation pathway (i.e. without significant unfolding) by SID. In the latter case, sufficient internal energy can be deposited in a single collision event such that dissociation occurs prior to significant unfolding. Both the unfolding and direct (no unfolding) pathways could contribute to the overall spectrum, because the internal energy accessed may be higher than either of the barriers to dissociation.
This hypothesis was initially explored using non-covalent dimers of cytochrome c. The dimer of cytochrome c was chosen for its relative simplicity and because it was previously studied [114, 122]. In previous research, Williams and coworkers used a combination of sustained off-resonance irradiation CID (SORI-CID) and chemical cross-linking to demonstrate that upon activation and dissociation of the dimer only one of the subunits was likely unfolding [114]. For our investigation a quadrupole time of flight mass spectrometer (QTOF II, Waters) was modified to include an electrostatic device for SID [123]. This device was installed between the mass selection quadrupole and hexapole collision cell of the instrument. Ions could be either deflected toward the surface (“SID mode”) or transmitted without a surface collision (“CID mode”). Therefore, the instrument is capable of both CID and SID experiments. In our initial experiments with this protein-protein dimer, the 11+ precursor ion was mass selected and fragmented by CID in a QTOF II (Waters), as can be seen in Figure 10a [124]. It is evident from this spectrum that the dimer favors dissociation into monomers possessing unequal charge states, with the formation of 8+ and 3+ fragment ions being the predominant pathway. This pathway was favored over a wide range of collision energies, spanning from 550 eV to 990 eV in the laboratory reference frame. These results are quite similar to those of the published SORI-CID experiments [114], and also suggest that one of the subunits is unfolding (i.e. the 8+ ion). It is not clear whether the unfolding is driven by Coulomb repulsion, or if proton transfers are enhanced due to the increased surface area of the monomer. In contrast to CID, a surface collision produces a different spectrum. The SID spectrum in Figure 10b demonstrates this effect, as the predominant pathway is now the formation of the 6+ and 5+ fragment ions, the most symmetric pathway possible from an 11+ ion. This symmetric dissociation pathway was observed from 330 eV to 990 eV laboratory collision energies. If the highly asymmetric behavior induced by CID is indicative of extensive subunit unfolding, then the symmetrical dissociation observed by SID may suggest a more compact protein dimer structure in the dissociative transition state.
Figure 10.

Comparison of CID (a) and SID (b) tandem mass spectra of the 11+ dimer of cytochrome C. The voltage difference between the source hexapole and collision cell or surface is shown as ΔV. The laboratory collision energy (CE), calculated as the product of the collision voltage and precursor ion charge state, is also provided with each spectrum. Spectrum adapted from reference 124 with permission from the American Chemical Society.
The dramatic differences observed between CID and SID of cytochrome C dimers led to the comparison of the dissociation behaviors of larger complexes. As described above, it is believed that SID of the dimer occurs with minimal subunit unfolding, as evidenced by the even distribution of charges. To study the partitioning of both mass and charge, we have compared the CID and SID of various tetramers, a pentamer, and several dodecameric small heat shock proteins. The dissociation of one of these tetramers, concanavalin A from jack bean, is shown in Figure 11. In solution, concanavalin A exists primarily as a dimer below pH 5.8 and as a tetramer between pH 5.8 and 7.0. Both the dimeric and tetrameric forms of the complex have been studied previously by Light-Wahl, et al. including dissociation of both gas-phase species in the capillary-skimmer region [125]. Figure 11a shows the CID MS/MS spectrum of the 22+ tetramer ion (Q22+). In general, CID has been shown to induce highly asymmetric mass and charge partitioning upon dissociation of nearly all protein complexes studied thus far. This is indeed the case here as the 22+ tetramer yields primarily monomer (M) and trimer (T) product ions with the monomer retaining approximately half the charge. The SID spectrum of this complex is shown in Figure 11b and is strikingly different from the CID spectrum. Collision of the 22+ tetramer with a surface produces solely monomeric product ions at a collision energy of 2310 eV. While the monomers ejected by CID retain approximately half of the overall charge of the precursor ion, the monomers produced via SID have an average charge state of 5.8. This corresponds to approximately 26% of the overall charge of the initial precursor, indicating the charge is nearly evenly distributed over the four dissociating subunits. An in-depth study of additional tetrameric and pentameric complexes is currently in preparation and will be presented separately.
Figure 11.

Comparison of CID (a) and SID (b) tandem mass spectra of the 22+ tetramer (Q22+) of concanavalin A from jack bean. The voltage difference and collision energy are defined as in Figure 10. Each protein species is listed as monomers (M), trimers (T), or tetramers (Q) followed by the charge state.
To address large complexes we have compared the CID and SID of the ~200 kDa dodecameric small heat shock protein 18.1 complex (sHSP18.1) from garden pea. This and numerous other sHSPs have been studied quite extensively in the gas phase by Robinson and coworkers. It has been shown that CID induces highly asymmetric mass and charge partitioning upon dissociation of all sHSP studied thus far, with monomer and (n-1)mer of nearly equal charge being the predominant product ions. Furthermore, evidence of a sequential dissociation pathway, in which monomers continue to be ejected from multimeric product ions at higher collision energies, has also been recently described [120]. Figure 12a shows the CID of sHSP18.1 performed in the University of Arizona QTOF instrument modified with the SID device. In this experiment ions were transmitted through this device without a surface collision and were activated in the collision cell using argon as the target gas. The results are quite typical of what can be found in the literature with a monomer being ejected and removing approximately half of the charge from a 33+ precursor ion. The SID spectrum of this complex was acquired by deflecting the mass selected 33+ ion into the surface and transmitting the product ions through the collision cell without the presence of a target gas. This spectrum is displayed in Figure 12b. The first observation that can be made by comparison of the CID and SID data is that the ejected monomer gains less net charge during SID than it does from CID. By CID the most intense monomer charge state was a 16+ ion, whereas SID yielded a 14+ ion as its most abundant species. Furthermore, the charge state distribution from SID is slightly broader than that of CID with peaks from 18+ to 5+ being observed compared to only 19+ to 8+ from CID. Since the higher monomer charge states are the most intense it seems likely that the predominant dissociation pathway remains asymmetric by SID and likely involves some degree of subunit unfolding. However, since the most abundant ions are less protonated it is feasible that the monomer may be slightly more compact in the transition state. Also, we can not rule out that partial proton loss to the surface contributes to the observation of lower charge states by SID. Due to the broadness of the charge state distribution observed by SID, it also appears that many more dissociation pathways may also occur, involving varying degrees of subunit unfolding. For instance, the 5+ ion may result from ejection of a monomer subunit that remains quite compact. At this time it is not entirely clear how to explain the wide distribution of observed charge states. One possibility to consider is that a broad distribution of internal energies and structures exist after the surface collision. Yet another explanation could be that at a given internal energy there exists a number of dissociation pathways that are competitive with the unfolding pathway that is typically dominant (e.g. see Figure 2). Future research will be directed at answering these types of questions.
Figure 12.

SID/CID comparison for the dodecameric protein complex PsHSP18.1, 33+ charge state (DD33). (a) CID at ΔV = 130 V, collision energy (CE) = 4290 eV. On the left is an enhancement of the low m/z region containing monomer fragments centered about the 16+ charge state (M16). (b) SID at ΔV = 70 V, collision energy (CE) = 2310 eV. The left-hand side is the enhancement of the monomer region centered about the 14+ charge state (M14).
Future Directions
In this short perspective we have presented a wide range of applications, from small molecules to large protein complexes, for which SID has been employed over the years. The recent development of an in-line SID device in a QTOF mass spectrometer demonstrates the feasibility of this activation method being implemented in commercial instruments, and is a promising step toward even more widespread use of this technology.
As discussed above, an exciting new area where SID is now being applied is to the study of protein complex dissociation pathways and energetics. Given the rich chemistry of large multimeric protein complexes and the fact that SID has only recently been employed to study these types of systems, there remains a great deal of work to be done in this area. Our laboratory is currently developing methods involving SID and subsequent gas phase reactions that are designed to further our understanding of the dissociation pathways of protein complexes. The difference in charge states of monomer product ions formed from either CID or SID has previously been interpreted to indicate a difference in the extent of unfolding in the transition state structure of the dissociating complex. To study this hypothesis we are in the process of implementing either H/D exchange or ion/ion reactions in the collision cell following the SID device of the QTOF/SID instrument. The reactions with product ions formed either by in-source CID or by SID will be compared. We anticipate that product ions possessing a more unfolded structure will react more extensively than relatively folded counterparts.
Another promising tool for the study of activated ion structures is ion mobility spectrometry (IMS). Robinson and coworkers have demonstrated the feasibility of such an experiment for a protein complex by studying the gas phase structure of the TRAP complex, a multimeric assembly known to possess a ring structure in solution [126]. Additionally, Russell and coworkers recently reported an IMS-SID-TOF instrument in which peptide ions were studied [84]. In this instrument the IMS preceded the target surface and product ions were measured by a TOF analyzer. To measure the collision cross-sections of SID product ions and activated precursors an instrument would require the IMS region to follow SID. Another intriguing possibility would be to use IMS prior to the target surface to select specific structures of a given protein complex and investigate how these structures affect dissociation. We believe that the development of a SID-IMS or IMS-SID instrument for protein complexes is feasible and could be an important advance towards the understanding of the structural transitions of complexes and product ions induced by different activation methods. Furthermore, the utility of such an instrument would not be limited to only protein complexes, and could also be employed to study the structures of peptide fragment ions following a surface collision.
As mentioned above the role of the surface composition and the projectile ion incidence angle in the efficiency of internal energy deposition remains somewhat ambiguous. Future studies involving a wide range of molecules and surface compositions will be necessary to more precisely define the roles of these experimental variables.
In conclusion, SID has been utilized to probe the chemistry of a wide variety of molecules and non-covalent assemblies in numerous different instrument configurations. The relative simplicity of ion activation via a short time-frame collision event with a neutral surface has made SID an attractive tool for the study of many types of gas phase ion chemistries, and is likely to continue providing valuable insights.
Acknowledgments
The work described was funded by grants to V.H.W. NSF grant CHE 0416338 for ion-surface reactions and small molecule dissociation, NSF BIO U54 AI065359 for QTOF instrument development, NIH R01 GM051387 for the dissociation of peptides and proteins, and an NIH NRSA Fellowship F32 GM 75622 to R.L.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cooks RG, Ast T, Beynon JH. Anomalous metastable peaks. International Journal of Mass Spectrometry and Ion Physics. 1975;16:348–352. [Google Scholar]
- 2.Cooks RG, Terwilliger DT, Ast T, Beynon JH, Keough T. Surface modified mass spectrometry. J Am Chem Soc. 1975;97:1583–1585. [Google Scholar]
- 3.Mabud MDA, Dekrey MJ, Cooks RG. Surface-Induced Dissociation of Molecular-Ions. Int J Mass Spectrom. 1985;67:285–294. [Google Scholar]
- 4.Laskin J, Futrell JH. Energy transfer in collisions of peptide ions with surfaces. Journal of Chemical Physics. 2003;119:3413–3420. [Google Scholar]
- 5.Smith DL, Wysocki VH, Colorado R, Shmakova OE, Graupe M, Lee TR. Low-energy ion-surface collisions characterize alkyland fluoroalkyl-terminated self-assembled monolayers on gold. Langmuir. 2002;18:3895–3902. [Google Scholar]
- 6.Wysocki VH, Kenttamaa HI. Collisional activation of distonic radical cations and their conventional isomers in quadrupole tandem mass spectrometry. J Am Chem Soc. 1990;112:5110–5116. [Google Scholar]
- 7.Laskin J, Denisov E, Futrell J. Comparative study of collision-induced and surface-induced dissociation. 2. Fragmentation of small alanine-containing peptides in FT-ICR MS. Journal of Physical Chemistry B. 2001;105:1895–1900. [Google Scholar]
- 8.Grill V, Shen J, Evans C, Cooks RG. Collisions of ions with surfaces at chemically relevant energies: Instrumentation and phenomena. Review of Scientific Instruments. 2001;72:3149–3179. [Google Scholar]
- 9.Gologan B, Green JR, Alvarez J, Laskin J, Cooks RG. Ion/surface reactions and ion soft-landing. Physical Chemistry Chemical Physics. 2005;7:1490–1500. doi: 10.1039/b418056a. [DOI] [PubMed] [Google Scholar]
- 10.Wysocki VH, Kenttamaa HI, Cooks RG. Internal energy distributions of isolated ions after activation by various methods. Int J Mass Spectrom. 1987;75:181–208. [Google Scholar]
- 11.Cooks RG, Ast T, Mabud MA. Collisions of polyatomic ions with surfaces. Int J Mass Spectrom. 1990;100:209–265. [Google Scholar]
- 12.DeKrey MJ, Kenttamaa HI, Wysocki VH, Cooks RG. Energy deposition in iron pentacarbonyl cation radical upon collision with a metal surface. Org Mass Spectrom. 1986;21:193–195. [Google Scholar]
- 13.Bier ME, Amy JW, Cooks RG, Syka JEP, Ceja P, Stafford G. A tandem quadrupole mass spectrometer for the study of surface-induced dissociation. Int J Mass Spectrom. 1987;77:31–47. [Google Scholar]
- 14.Vekey K, Somogyi A, Wysocki VH. Internal Energy-Distribution of Benzene Molecular-Ions in Surface-Induced Dissociation. J Mass Spectrom. 1995;30:212–217. [Google Scholar]
- 15.Beck RD, St John P, Homer ML, Whetten RL. Impact-induced cleaving and melting of alkali-halide nanocrystals. Science. 1991;253:879–883. doi: 10.1126/science.253.5022.879. [DOI] [PubMed] [Google Scholar]
- 16.Beck RD, Warth C, May K, Kappes MM. Surface impact induced shattering of C60. Detection of small Cm fragments by negative surface ionization. Chem Phys Lett. 1996;257:557–562. [Google Scholar]
- 17.Mowrey RC, Brenner DW, Dunlap BI, Mintmire JW, White CT. Simulations of buckminsterfullerene (C60) collisions with a hydrogen-terminated diamond {111} surface. J Phys Chem. 1991;95:7138–7142. [Google Scholar]
- 18.Busmann HG, Lill T, Hertel IV. Near specular reflection of carbon sixty-atom molecular ions in collisions with an HOPG graphite surface. Chem Phys Lett. 1991;187:459–465. [Google Scholar]
- 19.Beck RD, Rockenberger J, Weis P, Kappes MM. Fragmentation of C60+ and higher fullerenes by surface impact. J Chem Phys. 1996;104:3638–3650. [Google Scholar]
- 20.Busmann HG, Lill T, Reif B, Hertel IV, Maguire HG. Energy partition in collisions of C60+ ions with diamond(111) and graphite(0001) surfaces. J Chem Phys. 1993;98:7574–7580. [Google Scholar]
- 21.Callahan JH, Somogyi A, Wysocki VH. Collisions of fullerene C60+. bul. and C602+ at fluorinated and non-fluorinated self-assembled monolayer films. Rapid Commun Mass Spectrom. 1993;7:693–699. [Google Scholar]
- 22.Beck RD, St John P, Alvarez MM, Diederich F, Whetten RL. Resilience of all-carbon molecules C60, C70, and C84: a surface-scattering time-of-flight investigation. J Phys Chem. 1991;95:8402–8409. [Google Scholar]
- 23.Garrison BJ, Ryan KE, Russo MF, Jr, Smiley EJ, Postawa Z. Quadratic Friction Model for Cluster Bombardment of Molecular Solids. Journal of Physical Chemistry C. 2007;111:10135–10137. [Google Scholar]
- 24.Krantzman KD, Kingsbury DB, Garrison BJ. Cluster induced chemistry at solid surfaces: Molecular dynamics simulations of keV C60 bombardment of Si. Nuclear Instruments & Methods in Physics Research, Section B: Beam Interactions with Materials and Atoms. 2007;255:238–241. [Google Scholar]
- 25.Delcorte A, Garrison BJ. keV fullerene interaction with hydrocarbon targets: Projectile penetration, damage creation and removal. Nuclear Instruments & Methods in Physics Research, Section B: Beam Interactions with Materials and Atoms. 2007;255:223–228. [Google Scholar]
- 26.Russo MF, Jr, Garrison BJ. Mesoscale Energy Deposition Footprint Model for Kiloelectronvolt Cluster Bombardment of Solids. Anal Chem. 2006;78:7206–7210. doi: 10.1021/ac061180j. [DOI] [PubMed] [Google Scholar]
- 27.Morris MR, Riederer DE, Jr, Winger BE, Cooks RG, Ast T, Chidsey CED. Ion/surface collisions at functionalized self-assembled monolayer surfaces. Int J Mass Spectrom. 1992;122:181–217. [Google Scholar]
- 28.Miller SA, Riederer DE, Jr, Cooks RG, Cho WR, Lee HW, Kang H. Energy disposal and target effects in hyperthermal collisions of ferrocene molecular ions at surfaces. J Phys Chem. 1994;98:245–251. [Google Scholar]
- 29.Shuler SF, Davis GM, Morris JR. Energy transfer in rare gas collisions with hydroxyl- and methyl-terminated self-assembled monolayers. J Chem Phys. 2002;116:9147–9150. [Google Scholar]
- 30.Day BS, Shuler SF, Ducre A, Morris JR. The dynamics of gas-surface energy exchange in collisions of Ar atoms with w-functionalized self-assembled monolayers. J Chem Phys. 2003;119:8084–8096. [Google Scholar]
- 31.Scott Day B, Davis GM, Morris JR. The effect of hydrogen-bonding and terminal group structure on the dynamics of Ar collisions with self-assembled monolayers. Analytica Chimica Acta. 2003;496:249–258. [Google Scholar]
- 32.Day BS, Morris JR. Packing density and structure effects on energy-transfer dynamics in argon collisions with organic monolayers. J Chem Phys. 2005;122:234714/234711–234714/234710. doi: 10.1063/1.1924693. [DOI] [PubMed] [Google Scholar]
- 33.Day BS, Morris JR, Troya D. Classical trajectory study of collisions of Ar with alkanethiolate self-assembled monolayers: Potential-energy surface effects on dynamics. J Chem Phys. 2005;122:214712/214711–214712/214712. doi: 10.1063/1.1924543. [DOI] [PubMed] [Google Scholar]
- 34.Day BS, Morris JR, Alexander WA, Troya D. Theoretical Study of the Effect of Surface Density on the Dynamics of Ar + Alkanethiolate Self-Assembled Monolayer Collisions. J Phys Chem A. 2006;110:1319–1326. doi: 10.1021/jp054043j. [DOI] [PubMed] [Google Scholar]
- 35.Lohr JR, Day BS, Morris JR. Dynamics of HCl Collisions with Hydroxyl-and Methyl-Terminated Self-Assembled Monolayers. J Phys Chem A. 2006;110:1645–1649. doi: 10.1021/jp0542625. [DOI] [PubMed] [Google Scholar]
- 36.Mabud MA, Ast T, Verma S, Jiang YX, Cooks RG. Ion/surface interactions as a tool for characterizing isomers: [C2H4O]•+ ions. J Am Chem Soc. 1987;109:7597–7602. [Google Scholar]
- 37.Hayward MJ, Mabud MA, Cooks RG. Ion/surface collisions for distinction of isomeric [C6H6]•+ and [C6H6]2+ ions. J Am Chem Soc. 1988;110:1343–1346. [Google Scholar]
- 38.Horning SR, Bier ME, Cooks RG, Brusini G, Traldi P, Guiotto A, Rodighiero P. Characterization of isomeric dimethylfuranocoumarins by electron impact ionization and surface and gaseous collision activation mass spectrometry. Biomed Environ Mass Spectrom. 1989;18:927–934. [Google Scholar]
- 39.Yang MC, Hwang CH, Kang H. Cs+ reactive scattering from a Si(111) surface adsorbed with water. J Chem Phys. 1997;107:2611–2618. [Google Scholar]
- 40.Kang H, Yang MC, Kim KD, Kim KY. Reactive scattering of Cs+ from chemisorbed molecules on a Ni(100) surface. Secondary neutral mass spectrometry with a hyperthermal ion beam. Int J Mass Spectrom. 1998;174:143–154. [Google Scholar]
- 41.Kim KY, Shin TH, Han SJ, Kang H. Identification of the Precursor State in the Initial Stages of Si(111)-(7*7) Oxidation. Physical Review Letters. 1999;82:1329–1332. [Google Scholar]
- 42.Kim CM, Hwang CH, Lee CW, Kang H. Communications: Real-time observation of the H/D exchange reaction between ethylene and hydrogen on Pt(111) Angewandte Chemie, International Edition. 2002;41:146–148. doi: 10.1002/1521-3773(20020104)41:1<146::aid-anie146>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 43.Gu C, Wysocki VH. Ion-Surface Reactions Involving Isotopically Labeled Langmuir-Blodgett Films. J Am Chem Soc. 1997;119:12010–12011. [Google Scholar]
- 44.Wu Q, Hanley L. Reactive scattering, sputtering, and dissociation of 32-eV pyridine ions colliding with clean and pyridine-covered silver(111) J Phys Chem. 1993;97:2677–2685. [Google Scholar]
- 45.Wu Q, Hanley L. Effect of adsorbate coverage and ion beam exposure time on reactive ion-surface collisions: pyridine cation (C5H5N+) reactions with deuterated pyridine on silver surface (C5D5N/Ag(111)) J Am Chem Soc. 1993;115:1191–1193. [Google Scholar]
- 46.Kane TE, Somogyi A, Wysocki VH. Reactive ion-surface collisions: application of ionized acetone-d6, DMSO-d6 and pyridine-d5 as probes for the characterization of self-assembled monolayer films on gold. Org Mass Spectrom. 1993;28:1665–1673. [Google Scholar]
- 47.Angelico VJ, Mitchell SA, Wysocki VH. Low-Energy Ion-Surface Reactions of Pyrazine with Two Classes of Self-Assembled Monolayers: Influence of Alkyl Chain Orientation. Anal Chem. 2000;72:2603–2608. doi: 10.1021/ac0001028. [DOI] [PubMed] [Google Scholar]
- 48.Barber M, Bordoli RS, Sedgwick RD, Tyler AN. Fast Atom Bombardment of Solids (Fab) - a New Ion-Source for Mass-Spectrometry. J Chem Soc Chem Commun. 1981:325–327. [Google Scholar]
- 49.Surman DJ, Vickerman JC. Fast Atom Bombardment Quadrupole Mass-Spectrometry. J Chem Soc Chem Commun. 1981:324–325. [Google Scholar]
- 50.Yamashita M, Fenn JB. Electrospray Ion Source. Another Variation on the Free-Jet Theme. J Phys Chem. 1984;88:4451–4459. [Google Scholar]
- 51.Karas M, Bachmann D, Bahr U, Hillenkamp F. Matrix-Assisted Ultraviolet-Laser Desorption of Nonvolatile Compounds. International Journal of Mass Spectrometry and Ion Processes. 1987;78:53–68. [Google Scholar]
- 52.Kiplinger JP, Bursey MM. Collisionally Activated Decomposition of Poly(Ethylene Glycol)S - an Investigation of High-Mass Ion Abundances in the Collisional Activation Technique with Large Molecules. Organic Mass Spectrometry. 1988;23:342–349. [Google Scholar]
- 53.Alexander AJ, Boyd RK. Experimental Investigations of Factors Controlling the Collision-Induced Dissociation Spectra of Peptide Ions in a Tandem Hybrid Mass-Spectrometer.1. Leucine Enkephalin. International Journal of Mass Spectrometry and Ion Processes. 1989;90:211–240. [Google Scholar]
- 54.Poulter L, Taylor LCE. A Comparison of Low and High-Energy Collisionally Activated Decomposition Ms-Ms for Peptide Sequencing. International Journal of Mass Spectrometry and Ion Processes. 1989;91:183–197. [Google Scholar]
- 55.Smith RD, Barinaga CJ, Udseth HR. Tandem Mass-Spectrometry of Highly Charged Cytochrome-C Molecular-Ions Produced by Electrospray Ionization. Journal of Physical Chemistry. 1989;93:5019–5022. [Google Scholar]
- 56.Williams ER, Henry KD, Mclafferty FW, Shabanowitz J, Hunt DF. Surface-Induced Dissociation of Peptide Ions in Fourier-Transform Mass-Spectrometry. Journal of the American Society for Mass Spectrometry. 1990;1:413–416. doi: 10.1016/1044-0305(90)85022-E. [DOI] [PubMed] [Google Scholar]
- 57.Dongre AR, Somogyi A, Wysocki VH. Surface-induced dissociation: An effective tool to probe structure, energetics and fragmentation mechanisms of protonated peptides. Journal of Mass Spectrometry. 1996;31:339–350. doi: 10.1002/(SICI)1096-9888(199604)31:4<339::AID-JMS322>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 58.Chorush RA, Little DP, Beu SC, Wood TD, Mclafferty FW. Surface-Induced Dissociation of Multiply Protonated Proteins. Analytical Chemistry. 1995;67:1042–1046. doi: 10.1021/ac00102a004. [DOI] [PubMed] [Google Scholar]
- 59.Hunt DF, Yates JR, Shabanowitz J, Winston S, Hauer CR. Protein Sequencing by Tandem Mass-Spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Biemann K. Mass-Spectrometric Methods for Protein Sequencing. Analytical Chemistry. 1986;58:1288. doi: 10.1021/ac00126a001. [DOI] [PubMed] [Google Scholar]
- 61.Hunt DF, Bone WM, Shabanowitz J, Rhodes J, Ballard JM. Sequence-Analysis of Oligopeptides by Secondary Ion-Collision Activated Dissociation Mass-Spectrometry. Analytical Chemistry. 1981;53:1704–1706. [Google Scholar]
- 62.Bean MF, Carr SA, Thorne GC, Reilly MH, Gaskell SJ. Tandem Mass-Spectrometry of Peptides Using Hybrid and 4-Sector Instruments - a Comparative-Study. Analytical Chemistry. 1991;63:1473–1481. doi: 10.1021/ac00014a024. [DOI] [PubMed] [Google Scholar]
- 63.Tang XJ, Thibault P, Boyd RK. Fragmentation Reactions of Multiply-Protonated Peptides and Implications for Sequencing by Tandem Mass-Spectrometry with Low-Energy Collision-Induced Dissociation. Analytical Chemistry. 1993;65:2824–2834. doi: 10.1021/ac00068a020. [DOI] [PubMed] [Google Scholar]
- 64.Qin J, Chait BT. Preferential Fragmentation of Protonated Gas-Phase Peptide Ions Adjacent to Acidic Amino-Acid-Residues. Journal of the American Chemical Society. 1995;117:5411–5412. [Google Scholar]
- 65.Dongre AR, Jones JL, Somogyi A, Wysocki VH. Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: Evidence for the mobile proton model. Journal of the American Chemical Society. 1996;118:8365–8374. [Google Scholar]
- 66.Summerfield SG, Whiting A, Gaskell SJ. Intra-ionic interactions in electrosprayed peptide ions. International Journal of Mass Spectrometry and Ion Processes. 1997;162:149–161. [Google Scholar]
- 67.Tsaprailis G, Nair H, Somogyi A, Wysocki VH, Zhong W, Futrell JH, Summerfield SG, Gaskell SJ. Influence of Secondary Structure on the Fragmentation of Protonated Peptides. J Am Chem Soc. 1999;121:5142–5154. [Google Scholar]
- 68.Huang YY, Triscari JM, Pasa-Tolic L, Anderson GA, Lipton MS, Smith RD, Wysocki VH. Dissociation behavior of doubly-charged tryptic peptides: Correlation of gas-phase cleavage abundance with Ramachandran plots. Journal of the American Chemical Society. 2004;126:3034–3035. doi: 10.1021/ja038041t. [DOI] [PubMed] [Google Scholar]
- 69.Tsaprailis G, Nair H, Zhong W, Kuppannan K, Futrell JH, Wysocki VH. A mechanistic investigation of the enhanced cleavage at histidine in the gas-phase dissociation of protonated peptides. Analytical Chemistry. 2004;76:2083–2094. doi: 10.1021/ac034971j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang YY, Triscari JM, Tseng GC, Pasa-Tolic L, Lipton MS, Smith RD, Wysocki VH. Statistical characterization of the charge state and residue dependence of low-energy CID peptide dissociation patterns. Analytical Chemistry. 2005;77:5800–5813. doi: 10.1021/ac0480949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gorman GS, Speir JP, Turner CA, Amster IJ. Proton Affinities of the 20 Common Alpha-Amino-Acids. Journal of the American Chemical Society. 1992;114:3986–3988. [Google Scholar]
- 72.Wu ZC, Fenselau C. Proton Affinity of Arginine Measured by the Kinetic Approach. Rapid Communications in Mass Spectrometry. 1992;6:403–405. [Google Scholar]
- 73.Wu ZC, Fenselau C. Gas-Phase Basicities and Proton Affinities of Lysine and Histidine Measured from the Dissociation of Proton-Bound Dimers. Rapid Communications in Mass Spectrometry. 1994;8:777–780. [Google Scholar]
- 74.Lias SG, Liebmann LF, Levine RD. J Phys Chem Ref Data. 1984;13:695–808. [Google Scholar]
- 75.Biemann K. Sequencing of Peptides by Tandem Mass Spectrometry and High-Energy Collision-Induced Dissociation. Methods in Enzymology. 1990;193:455–479. doi: 10.1016/0076-6879(90)93433-l. [DOI] [PubMed] [Google Scholar]
- 76.Alexander AJ, Thibault P, Boyd RK, Curtis JM, Rinehart KL. Collision-Induced Dissociation of Peptide Ions.3. Comparison of Results Obtained Using Sector-Quadrupole Hybrids with Those from Tandem Double-Focusing Instruments. International Journal of Mass Spectrometry and Ion Processes. 1990;98:107–134. [Google Scholar]
- 77.Jones JL, Dongre AR, Somogyi A, Wysocki VH. Sequence Dependence of Peptide Fragmentation Efficiency Curves Determined by Electrospray-Ionization Surface-Induced Dissociation Mass-Spectrometry. Journal of the American Chemical Society. 1994;116:8368–8369. [Google Scholar]
- 78.Cox KA, Gaskell SJ, Morris M, Whiting A. Role of the site of protonation in the low-energy decompositions of gas-phase peptide ions. Journal of the American Society for Mass Spectrometry. 1996;7:522–531. doi: 10.1016/1044-0305(96)00019-0. [DOI] [PubMed] [Google Scholar]
- 79.Gu C, Tsaprailis G, Breci L, Wysocki VH. Selective Gas-Phase Cleavage at the Peptide Bond C-Terminal to Aspartic Acid in Fixed-Charge Derivatives of Asp-Containing Peptides. Analytical Chemistry. 2000;72:5804–5813. doi: 10.1021/ac000555c. [DOI] [PubMed] [Google Scholar]
- 80.Wysocki VH, Tsaprailis G, Smith LL, Breci LA. Special feature: Commentary - Mobile and localized protons: a framework for understanding peptide dissociation. Journal of Mass Spectrometry. 2000;35:1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 81.Kapp EA, Schuetz F, Reid GE, Eddes JS, Moritz RL, O’Hair RAJ, Speed TP, Simpson RJ. Mining a tandem mass spectrometry database to determine the trends and global factors influencing peptide fragmentation. Anal Chem. 2003;75:6251–6264. doi: 10.1021/ac034616t. [DOI] [PubMed] [Google Scholar]
- 82.Stone E, Gillig KJ, Ruotolo B, Fuhrer K, Gonin M, Schultz A, Russell DH. Surface-induced dissociation on a MALDI-ion mobility-orthogonal Time-of-flight mass spectrometer: Sequencing peptides from an \”In-solution\” protein digest. Anal Chem. 2001;73:2233–2238. doi: 10.1021/ac001430a. [DOI] [PubMed] [Google Scholar]
- 83.Stone EG, Gillig KJ, Ruotolo BT, Russell DH. Optimization of a matrix-assisted laser desorption ionization-ion mobility-surface-induced dissociation-orthogonal-time-of-flight mass spectrometer: simultaneous acquisition of multiple correlated MS1 and MS2 spectra. Int J Mass Spectrom. 2001;212:519–533. [Google Scholar]
- 84.Sun W, May JC, Russell DH. A novel surface-induced dissociation instrument for ion mobility-time-of-flight mass spectrometry. Int J Mass Spectrom. 2007;259:79–86. [Google Scholar]
- 85.Laskin J, Denisov EV, Shukla AK, Barlow SE, Futrell JH. Surface-induced dissociation in a Fourier transform ion cyclotron resonance mass spectrometer: Instrument design and evaluation. Analytical Chemistry. 2002;74:3255–3261. doi: 10.1021/ac025514q. [DOI] [PubMed] [Google Scholar]
- 86.Schlosser A, Lehmann WD. Patchwork peptide sequencing: Extraction of sequence information from accurate mass data of peptide tandem mass spectra recorded at high resolution. Proteomics. 2002;2:524–533. doi: 10.1002/1615-9861(200205)2:5<524::AID-PROT524>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 87.Fernandez FM, Wysocki VH, Futrell JH, Laskin J. Protein identification via surface-induced dissociation in an FT-ICR mass spectrometer and a patchwork sequencing approach. Journal of the American Society for Mass Spectrometry. 2006;17:700–709. doi: 10.1016/j.jasms.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 88.Shukla AK, Futrell JH. Tandem mass spectrometry: dissociation of ions by collisional activation. Journal of Mass Spectrometry. 2000;35:1069–1090. doi: 10.1002/1096-9888(200009)35:9<1069::AID-JMS54>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 89.Williams ER, Fang LL, Zare RN. Surface Induced Dissociation for Tandem Time-of-Flight Mass-Spectrometry. International Journal of Mass Spectrometry and Ion Processes. 1993;123:233–241. [Google Scholar]
- 90.Vekey K, Somogyi A, Wysocki VH. Average activation energies of low-energy fragmentation processes of protonated peptides determined by a new approach. Rapid Communications in Mass Spectrometry. 1996;10:911–918. doi: 10.1002/(SICI)1097-0231(19960610)10:8<911::AID-RCM593>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 91.Laskin J, Denisov E, Futrell J. A comparative study of collision-induced and surface-induced dissociation. 1. Fragmentation of protonated dialanine. Journal of the American Chemical Society. 2000;122:9703–9714. [Google Scholar]
- 92.Laskin J, Futrell JH. Collisional activation of peptide ions in FT-ICR mass spectrometry. Mass Spectrometry Reviews. 2003;22:158–181. doi: 10.1002/mas.10041. [DOI] [PubMed] [Google Scholar]
- 93.Laskin J, Futrell J. Internal energy distributions resulting from sustained off-resonance excitation in Fourier transform ion cyclotron resonance mass spectrometry. II. Fragmentation of the 1-bromonaphthalene radical cation. Journal of Physical Chemistry A. 2000;104:5484–5494. [Google Scholar]
- 94.Laskin J, Byrd M, Futrell J. Internal energy distributions resulting from sustained off-resonance excitation in FTMS. I. Fragmentation of the bromobenzene radical cation. International Journal of Mass Spectrometry. 2000;196:285–302. [Google Scholar]
- 95.Dekrey MJ, Kenttamaa HI, Wysocki VH, Cooks RG. Energy Deposition in [Fe(Co)5]+. Upon Collision with a Metal-Surface. Organic Mass Spectrometry. 1986;21:193–195. [Google Scholar]
- 96.Wysocki VH, Ding JM, Jones JL, Callahan JH, King FL. Surface-Induced Dissociation in Tandem Quadrupole Mass Spectrometers - a Comparison of 3 Designs. Journal of the American Society for Mass Spectrometry. 1992;3:27–32. doi: 10.1016/1044-0305(92)85015-C. [DOI] [PubMed] [Google Scholar]
- 97.Laskin J, Futrell JH. Collisional activation of peptide ions in FT-ICR mass spectrometry. Rapid Communications in Mass Spectrometry. 2003;17:2694–2694. doi: 10.1002/mas.10041. [DOI] [PubMed] [Google Scholar]
- 98.Meroueh O, Hase WL. Dynamics of energy transfer in peptide-surface collisions. Journal of the American Chemical Society. 2002;124:1524–1531. doi: 10.1021/ja011987n. [DOI] [PubMed] [Google Scholar]
- 99.Meroueh O, Wang YF, Hase WL. Direct Dynamics Simulations of Collision- and Surface-Induced Dissociation of N-Protonated Glycine. Shattering Fragmentation. J Phys Chem A. 2002;106:9983–9992. [Google Scholar]
- 100.Song K, Meroueh O, Hase WL. Dynamics of Cr(CO)6+ Collisions with Hydrogenated Surfaces. J Chem Phys. 2003;118:2893–2902. [Google Scholar]
- 101.Wang YF, Hase WL. Direct dynamics study of N-protonated diglycine surface-induced dissociation. Influence of collision energy. Journal of the American Society for Mass Spectrometry. 2003;14:1402–1412. doi: 10.1016/j.jasms.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 102.Wang JP, Meroueh SO, Wang YF, Hase WL. Efficiency of energy transfer in protonated diglycine and dialanine SID - Effects of collision angle, peptide ion size, and intramolecular potential. International Journal of Mass Spectrometry. 2003;230:57–64. [Google Scholar]
- 103.Laskin J, Bailey TH, Futrell JH. Shattering of peptide ions on self-assembled monolayer surfaces. Journal of the American Chemical Society. 2003;125:1625–1632. doi: 10.1021/ja027915t. [DOI] [PubMed] [Google Scholar]
- 104.Laskin J, Bailey TH, Futrell JH. Fragmentation energetics for angiotensin II and its analogs from time-and energy-resolved surface-induced dissociation studies. International Journal of Mass Spectrometry. 2004;234:89–99. [Google Scholar]
- 105.Laskin J, Bailey TH, Futrell JH. Mechanisms of peptide fragmentation from time- and energy-resolved surface-induced dissociation studies: Dissociation of angiotensin analogs. International Journal of Mass Spectrometry. 2006;249:462–472. [Google Scholar]
- 106.Laskin J. Energetics and dynamics of fragmentation of protonated leucine enkephalin from time-and energy-resolved surface-induced dissociation studies. Journal of Physical Chemistry A. 2006;110:8554–8562. doi: 10.1021/jp057229r. [DOI] [PubMed] [Google Scholar]
- 107.Park K, Song K, Hase WL. An ab Initio Direct Dynamics Simulation of Protonated Glycine Surface-Induced Dissociation. Int J Mass Spectrom. 2007;265:326–336. [Google Scholar]
- 108.Schultz DG, Lim H, Garbis S, Hanley L. Energy partitioning in the surface-induced dissociation of linear and cyclic protonated peptides at an organic surface. Journal of Mass Spectrometry. 1999;34:217–225. [Google Scholar]
- 109.Schultz DG, Hanley L. Shattering of SiMe3+ during surface-induced dissociation. Journal of Chemical Physics. 1998;109:10976–10983. [Google Scholar]
- 110.Gamage CM, Fernandez FM, Kuppannan K, Wysocki VH. Submicrosecond surface-induced dissociation of peptide ions in a MALDI TOF MS. Analytical Chemistry. 2004;76:5080–5091. doi: 10.1021/ac0493121. [DOI] [PubMed] [Google Scholar]
- 111.Benesch J, Robinson C. Mass spectrometry of macromolecular assemblies: preservation and dissociation. Curr Opinion Struct Biol. 2006;16:245–251. doi: 10.1016/j.sbi.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 112.Felitsyn N, Kitova EN, Klassen JS. Thermal decomposition of a gaseous multiprotein complex studied by blackbody infrared radiative dissociation. Investigating the origin of the asymmetric dissociation behavior. Anal Chem. 2001;73:4647–4661. doi: 10.1021/ac0103975. [DOI] [PubMed] [Google Scholar]
- 113.Hanson CL, Fucini P, Ilag LL, Nierhause KH, Robinson CV. Dissociation of Intact Escherichia coli Ribosomes in a Mass Spectrometer. J Biol Chem. 2003;278:1259–1267. doi: 10.1074/jbc.M208966200. [DOI] [PubMed] [Google Scholar]
- 114.Jurchen JC, Garcia DE, Williams ER. Further studies on the origins of asymmetric charge partitioning in protein homodimers. J Am Soc Mass Spectrom. 2004;15:1408–1415. doi: 10.1016/j.jasms.2004.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.McCammon MG, Hernandez H, Sobott F, Robinson CV. Tandem mass spectrometry defines the stoichiometry and quaternary structural arrangement of tryptophan molecules in the multiprotein complex TRAP. J Am Chem Soc. 2004;126:5950–5951. doi: 10.1021/ja0317170. [DOI] [PubMed] [Google Scholar]
- 116.Rostom AA, Fucini P, Benjamin DR, Juenemann R, Nierhaus KH, Hartl FU, Dobson CM, Robinson CV. Detection and selective dissociation of intact ribosomes in a mass spectrometer. Proc Natl Acad Sci. 2000;97:5185–5190. doi: 10.1073/pnas.97.10.5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sobott F, Robinson CV. Protein complexes gain momentum. Curr Opinion Struct Biol. 2002;12:729–734. doi: 10.1016/s0959-440x(02)00400-1. [DOI] [PubMed] [Google Scholar]
- 118.Sobott F, Hernandez H, McCammon MG, Tito MA, Robinson CV. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- 119.Sobott F, McCammon MG, Robinson CV. Gas-phase dissociation pathways of a tetrameric protein complex. Int J Mass Spectrom. 2003;230:193–200. [Google Scholar]
- 120.Benesch JLP, Aquilina JA, Ruotolo BT, Sobott F, Robinson CV. Tandem Mass Spectrometry Reveals the Quaternary Organization of Macromolecular Assemblies. Chem Biol. 2006;13:597–605. doi: 10.1016/j.chembiol.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 121.McLuckey SA, Goeringer DE. Slow heating methods in tandem mass spectrometry. J Mass Spectrom. 1997;32:461–474. [Google Scholar]
- 122.Versluis C, van der Staaij A, Stokvis E, Heck AJR, de Craene B. Metastable ion formation and disparate charge separation in the gas-phase dissection of protein assemblies studied by orthogonal time-of-flight mass spectrometry. J Am Soc Mass Spectrom. 2001;12:329–336. doi: 10.1016/S1044-0305(00)00227-0. [DOI] [PubMed] [Google Scholar]
- 123.Galhena AS, Dagan S, Wysocki VH. 2007 Manuscript in Preparation. [Google Scholar]
- 124.Jones CM, Beardsley RL, Galhena AS, Dagan S, Wysocki VH. Symmetrical Gas-Phase Dissociation of Noncovalent Protein Complexes via Surface Collisions. J Am Chem Soc. 2006;128:15044–15045. doi: 10.1021/ja064586m. [DOI] [PubMed] [Google Scholar]
- 125.Light-Wahl KJ, Schwartz BL, Smith RD. Observation of the Noncovalent Quaternary Associations of Proteins by Electrospray Ionization Mass Spectrometry. J Am Chem Soc. 1994;116:5271–5278. doi: 10.1016/1044-0305(94)85034-8. [DOI] [PubMed] [Google Scholar]
- 126.Ruotolo BT, Giles K, Campuzano I, Sandercock AM, Bateman RH, Robinson CV. Evidence for Macromolecular Protein Rings in the Absence of Bulk Water. Science. 2005;310:1658–1661. doi: 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]