

Abstract
After an inflammatory stimulus, lymphocyte migration into draining lymph nodes increases dramatically to facilitate the encounter of naïve T cells with antigen-loaded dendritic cells. Here we show that CD73 (ecto-5′-nucleotidase) plays an important role in regulating this process. CD73 produces adenosine from AMP and is expressed on high endothelial venules (HEV) and subsets of lymphocytes. Cd73-/- mice have normal sized lymphoid organs in the steady state, but approximately 1.5-fold larger draining lymph nodes and 2.5-fold increased rates of L-selectin-dependent lymphocyte migration from the blood through HEV compared to wild type mice 24 hours after LPS administration. Migration rates of cd73+/+ and cd73-/- lymphocytes into lymph nodes of wild type mice are equal, suggesting that it is CD73 on HEV that regulates lymphocyte migration into draining lymph nodes. The A2B receptor is a likely target of CD73-generated adenosine, as it is the only adenosine receptor expressed on the HEV-like cell line KOP2.16 and it is up regulated by TNFα. Furthermore, increased lymphocyte migration into draining lymph nodes of cd73-/- mice is largely normalized by pretreatment with the selective A2B receptor agonist BAY 60-6583. Adenosine receptor signaling to restrict lymphocyte migration across HEV may be an important mechanism to control the magnitude of an inflammatory response.
Keywords: Rodent, Cell Trafficking, Spleen and lymph nodes, Inflammation
INTRODUCTION
Lymphocyte circulation from the blood stream to lymph nodes is necessary for immune homeostasis (reconnaissance) under normal physiological conditions and for immune responses against exogenous antigens. This trafficking requires coordinated action of adhesion molecules, chemokines, and chemokine receptors expressed on lymphocytes and high endothelial venules (HEV4) (reviewed in 1 and 2). The interaction of L-selectin with peripheral lymph node addressins (PNAd) initiates lymphocyte tethering and rolling on HEV (3). Chemokine receptor signaling activates the integrin LFA-1 on lymphocytes and induces stable adhesion via binding to ICAM-1 on HEV (4,5) which is followed by transmigration. The importance of each molecule associated with the entrance of lymphocytes into lymph nodes through HEV has been shown by decreases in lymph node cellularity and defective immune responses in gene-targeted mice (6-10).
TLR signaling activates innate immune responses (reviewed in 11) in part by inducing APC maturation and recruitment to lymphoid organs via the afferent lymphatics (12,13). Furthermore, recent reports showed that inflammation induced by a TLR 4 or 9 agonist controlled naïve lymphocyte recirculation in an antigen-independent manner, resulting in an increase in the number of naïve lymphocytes in the draining lymph node and an increase in the efficiency of lymphocyte-APC encounters (14). TLR-dependent lymph node hypertrophy was proposed to require vascular growth and arteriole thickening. Although these changes needed at least a few days before they were detectible (14,15), lymph node growth began within 24 h after a stimulus, implying the existence of other mechanisms that contribute to lymph node swelling. Here we present data to show that adenosine (Ado) receptor signaling, mediated by CD73-generated Ado, plays an important role in regulating early migration of lymphocytes to draining lymph nodes.
CD73 is a 70-kDa glycosyl phosphatidylinositol (GPI)-anchored protein with ecto-5′-nucleotidase enzyme activity that catalyzes the dephosphorylation of extracellular nucleoside monophosphates such as AMP to nucleosides such as Ado (16). Extracellular Ado can engage four subtypes of ubiquitously expressed Ado receptors (A1AR, A2AAR, A2BAR and A3AR) to modulate a wide array of physiological responses including vascular tone, neurotransmission, cytokine production, heart rate, and adaptation to hypoxia (reviewed in 17). In addition to being generated by CD73, Ado can also be generated intracellularly through the action of cytoplasmic nucleotidases and then exported via nucleoside transporters. Extracellular Ado has a very short half-life, as it is efficiently taken up into the cytoplasm where it can be phosphorylated to AMP or degraded to inosine by adenosine deaminase (ADA). In humans, ADA can be localized to the cell surface via binding to CD26 (18,19), giving it the potential to inhibit Ado receptor signaling through deamination of extracellular Ado (20). Mice deficient in the expression of each Ado receptor have been engineered and characterized (21-25). Each strain has a variety of interesting phenotypes, revealing the diverse consequences of Ado receptor signaling. However, the mechanism by which extracellular Ado levels are regulated to modulate Ado receptor engagement in vivo is not fully understood.
Ado is a well-known anti-inflammatory mediator (26). Recent studies clearly showed that CD73 makes a major contribution to the generation of extracellular Ado in a number of physiologically relevant experimental models and plays a critical role in host defense systems. For example, CD73 attenuates hypoxia-induced vascular leakage, FMLP-stimulated neutrophil adhesion to endothelial cells, and neutrophil accumulation in tissues (27-29). Furthermore, cd73-deficient mice are susceptible to vascular inflammation and neointima formation due to decreased concentrations of endogenous Ado (30). CD73 deficiency increases VCAM-1 expression on endothelial cells isolated from carotid arteries through NF-κB activation; however, ICAM-1 expression is unchanged. This pro-inflammatory phenotype of cd73-deficient endothelium causes the arrest of monocytes and exacerbates wire-induced injury.
These observations demonstrated a crucial role for CD73-generated Ado in the interaction of myeloid cells with vascular endothelium. However, the in vivo function of this molecule in lymphocyte-endothelium crosstalk remains unclear. In addition to its enzymatic role in the production of extracellular Ado, CD73 has also been characterized as a signaling molecule (31) and an adhesion molecule (32). Engagement of lymphocyte CD73 with anti-CD73 mAbs has been shown to stimulate proliferation, IL-2 secretion and IL-2R expression (33,34). Furthermore, blocking this molecule on lymphocytes with an antibody appears to inhibit adhesion of lymphocytes to cultured endothelial cells (35). Thus, there are multiple mechanisms by which CD73 could impact lymphocyte migration across HEV. We show here that cd73-deficient mice have increased rates of lymphocyte homing to draining lymph nodes and propose that CD73-generated Ado regulates the ability of lymphocytes to migrate across HEV, thus limiting their access to inflamed lymph nodes.
MATERIALS AND METHODS
Mice
Cd73-deficient mice developed in our laboratory (27) were backcrossed onto C57BL/6J for 14 generations. Genotyping by PCR, using primers that differentiate between the wild type cd73 allele and the mutated cd73 allele containing a neomycin resistance cassette, was performed as previously described. A2BAR-/- mice were obtained from Deltagen (San Mateo, CA) and have also been backcrossed onto the C57BL/6 background. All mice were bred and maintained in our animal facility under specific pathogen-free conditions. All protocols were approved by the Oklahoma Medical Research Foundation Institutional Animal Care and Use Committee.
Cell culture
The cell line KOP2.16 was derived from stromal cells taken from pooled mouse lymph nodes and has been described previously (36). It was cultured in DMEM supplemented with 20% FCS (Hyclone), 10 mM HEPES, 1 mM sodium pyruvate, 2 mM L-glutamine, 5 × 10-5 M 2-ME, non-essential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin. In some experiments, 20 ng/ml TNFα (R&D Systems) was added for 3 to 6 h.
Cd73 and adenosine receptor gene expression
Cd73 and Ado receptor expression were analyzed by PCR in a full length cDNA library derived from MACS® (Miltenyi Biotec)-isolated PNAd+ endothelial cells from lymph nodes (37) using previously described primers (38). In other experiments, RNA was prepared from KOP2.16 and RT-PCR was performed as described using β-actin as an internal control (38).
Digestion of lymph nodes for characterization of HEV or enumeration of CD11c+ dendritic cells by flow cytometry
Lymph nodes were dissected from mice, minced with scissors and digested in RPMI 1640 containing 10% FCS, 1 mg/ml collagenase B (Roche) and 2 μg/ml DNase I (Roche) for 30 min at 37°C with shaking at 50 rpm. The cell suspension was passed through a Pasteur pipette 40 times followed by digestion with 0.2% trypsin (Cellgro) and 0.5 mM EDTA at 37°C for 10 min. Cells were then passed through a 70-μm filter, washed, and stained.
Immunofluorescence
Lymphoid cells or PNAd+ cells were stained with the following monoclonal antibodies: FITC anti-CD4, FITC anti-CD8, FITC anti-MHC Class II, PE anti-CD11c, PE Cy5.5 anti-CD19 and allophycocyanin anti-CD45R (Caltag); PE anti-TCRβ (BD Pharmingen); allophycocyanin anti-CD45 (Southern Biotech), and biotinylated anti-CD73 (TY/23) (39), according to standard methods. PE-streptavidin was from BD Pharmingen. Data were collected with a FACSCalibur (Becton-Dickinson) and analyzed with CellQuest software. For lymphocyte migration experiments and experiments to enumerate DCs, data were collected on 750,000 and 350,000 cells, respectively.
Immunohistochemistry
Frozen sections (7 μm) of lymph nodes were fixed with cold acetone and blocked with 3% BSA in PBS. Sections were stained with TY/23 (anti-CD73, IgG2a) followed by Alexa Fluor 488 conjugated donkey anti-rat IgG (Molecular Probes) and then blocked with purified mouse IgG at 500 μg/ml. After washing, they were then stained with either Alexa Fluor 594 conjugated anti-PNAd mAb MECA-79 (IgM, BD Pharmingen). Other slides were stained with a combination of TY/23 and rabbit anti-collagen IV (Chemicon) followed by a combination of Alexa Fluor 488 conjugated donkey anti-rat IgG plus Alexa Fluor 594 conjugated donkey anti-rabbit IgG (Molecular Probes).
Inflammatory stimuli
Anesthetized mice were injected with 1 μg E. coli LPS (055:B5, Sigma-Aldrich) or 5 μg poly(I:C) (Sigma-Aldrich) in 30 μl PBS in the left front footpad using an insulin syringe. The right footpad was injected with same volume of PBS. Twenty-four hours later, brachial lymph nodes were examined as draining lymph nodes. In other experiments, mice were injected in either the rear footpad or thigh and popliteal or inguinal lymph nodes, respectively, were studied as draining lymph nodes.
Lymphocyte homing assay
Total splenocytes were labeled with 0.25 μM 5-chloromethylfluorescein diacetate (CMFDA, Molecular Probes) for 30 min at 37°C. Ten million labeled cells were injected i.v. into mice and one h later, spleen and lymph nodes were harvested. In some experiments, cd73-deficient and wild type splenocytes were labeled with 0.25 μM CMFDA and 2 μM 5-(and-6)-(((4-chloromethyl)benzoyl)amino)tetramethylrhodamine) (CMTMR, Molecular Probes), respectively, for 30 min at 37°C. Equal numbers of labeled cells were co-injected i.v. into both strains of mice. Harvested lymph nodes were pushed through 70-μm filters to make single cell suspensions. Cells were then counted and the percentages of labeled cells were determined by flow cytometry. In selected experiments, anti-L-selectin antibody (MEL-14, Southern Biotech) was given to mice (50 μg/mouse i.v.) simultaneously with LPS. In other experiments, mice were pre-treated with the A2BAR agonist BAY 60-6583 (40) at 0.32 mg/kg (Bayer HealthCare, Wuppertal, Germany, dissolved in polyethylene glycol (PEG) 400 and diluted to 80 μg/ml in PBS for i.v. injection) 30 min prior to the injection of labeled splenocytes.
RESULTS
CD73 and Ado receptor expression in lymphoid tissues and HEV
We previously reported the expression pattern of CD73 in lymphoid tissues of BALB/c mice (39); however, experiments indicating that CD73 expression was strain-dependent prompted us to investigate CD73 expression in lymphoid cells of C57BL/6 mice prior to using this strain for the experiments described in this report. Staining with monoclonal antibody TY/23 revealed that approximately 50% of CD4+, 85% of CD8+ and 2% of CD19+ lymphocytes derived from lymph nodes of wild type mice expressed CD73 (Fig. 1A). We confirmed the findings of Kobie et al. (41) that CD73 is expressed on CD4+CD25+Foxp3+ regulatory T cells. Nevertheless, cd73+/+ and cd73-/- mice had similar proportions of T cells with this phenotype (data not shown). Lymphocytes from cd73-/- mice expressed no detectable CD73, confirming the deletion. We also analyzed CD73 expression on HEV by flow cytometry, gating on the rare population of CD45- PNAd+ cells (0.05-0.15% of cells from enzyme-digested whole lymph nodes). Relatively high CD73 expression was observed on HEV compared to lymphocytes (Fig. 1B). In order to substantiate the results, immunohistochemistry was performed (Fig. 1C). Sections stained with anti-PNAd antibody and TY/23 revealed that HEV expressed CD73 abundantly. In contrast, only a few lymphocytes expressed enough CD73 to be detectable by this method. In addition, double staining with anti-collagen IV antibody and TY/23 demonstrated that CD73 is also expressed homogenously on basal lamina (i.e., not polarized to the luminal or abluminal surface).
FIGURE 1.

CD73 expression on lymphocytes and HEV. (A) Single cell suspensions of lymph node cells from cd73+/+ and cd73-/- mice were stained with FITC anti-CD4, FITC anti-CD8, or PE Cy5.5 anti-CD19 plus biotinylated anti-CD73 (TY/23) and PE-streptavidin or the relevant isotype-matched control antibodies. CD73 expression in cd73+/+ mice is shown in the shaded histograms and that in cd73-/- mice is shown with solid lines. Staining with isotype control antibodies is shown with dotted lines. The dotted lines and solid lines are virtually overlapping. (B) Other lymph nodes from wild type mice were digested with collagenase, DNase I, and trypsin as described in Materials and Methods and cells were stained with APC anti-CD45, purified anti-PNAd + Alexa Fluor 488 anti-IgM, and biotinylated anti-CD73 + PE-streptavidin. HEV were identified as CD45- PNAd+ cells. (C) Frozen sections of lymph nodes were stained with anti-CD73 (TY/23) and either anti-PNAd or anti-collagen IV as described in Materials and Methods. (D) Cd73 and Ado receptor expression were assessed by RT-PCR in a cDNA library derived from PNAd+ endothelial cells. Representative results are shown from more than three experiments. (E) Ado receptor expression was assessed by RT-PCR in total wild type splenocytes. Representative results from more than three experiments are shown.
Steady-state mRNA levels of cd73 and the Ado receptors were measured in HEV by PCR, using a cDNA library derived from PNAd+ endothelial cells (Fig. 1D). Cd73 and A2BAR were detected, but A1AR, A2AAR and A3AR were not expressed at detectible levels. Steady-state levels of Ado receptor mRNA in murine splenocytes are shown in Fig. 1E. All Ado receptors except the A1AR were easily detected.
Cd73-deficient mice have large draining lymph nodes
CD73 has been proposed to modulate lymphocyte-endothelial cell interactions as an adhesion molecule (32,35). Therefore, we asked whether CD73 plays a role in lymphocyte homing to secondary lymphoid tissue in the steady state. The sizes of spleen, peripheral lymph nodes, Peyer’s patches and mesenteric lymph nodes in cd73-deficient mice were normal (27 and unpublished data). Furthermore, the migration of CMFDA-labeled cd73-/- splenocytes to lymphoid tissues (spleen and lymph node) of unmanipulated cd73-/- mice was also comparable to that of cd73+/+ splenocytes to lymphoid organs of unmanipulated wild type mice (unpublished data). These observations suggested that CD73 does not have an obvious function in lymphocyte homing under steady state conditions.
CD73 plays important roles in vivo in maintaining the integrity of the vascular endothelium during hypoxia (27-29) and in regulating endothelial adhesion molecule expression after wire-induced injury (30). Taking this information into account, and considering the well-known anti-inflammatory properties of Ado, we hypothesized that CD73 might also regulate lymphocyte-HEV interactions after an inflammatory stimulus. To address this issue, LPS was injected into front left footpads of cd73+/+ and cd73-/- mice and 24 h later, the brachial (draining) lymph node cellularity was examined (Fig. 2A). The same volume of PBS was administered to the contralateral side as a control. As expected from previous studies (14,15), the draining lymph nodes were dramatically enlarged compared to those on the control side in wild type mice. Consistent with our hypothesis, there was a further increase in the size of the draining lymph nodes from cd73-deficient mice, which were approximately 1.5-fold larger than those of wild type mice. To examine whether lymphocyte migration from the blood stream to lymph nodes is also accelerated in cd73-/- mice, CMFDA-labeled wild type splenocytes were injected i.v. 24 h after LPS injection and the accumulation of labeled cells in the lymph nodes was measured after 1 h by flow cytometry (Fig. 2B). Although no differences were observed between cd73+/+ and cd73-/- mice on the control side, the number of lymphocytes that migrated into the draining lymph nodes of cd73-deficient mice was 2.7-fold greater than in wild type mice. These results suggest that it is CD73 on HEV (rather than on lymphocytes) that is responsible for the larger sizes of draining lymph nodes in cd73-deficient mice. Similar results were seen when poly(I:C), a TLR3 ligand, was used instead of LPS as the inflammatory stimulus (Fig. 2C,D). Furthermore, staining with TCRβ (Fig. 2E) and B220 (Fig. 2F) antibodies revealed that migration of both T and B lymphocytes was increased in cd73-deficient draining lymph nodes. Similar results were observed when LPS or poly(I:C) were injected into the rear footpad or thigh as revealed by examination of popliteal or inguinal lymph nodes, respectively (unpublished data).
FIGURE 2.

Cd73-deficient mice have large draining lymph nodes and high rates of L-selectin-dependent lymphocyte migration after an inflammatory stimulus LPS (1 μg, A,B,E,F,G,H) or poly(I:C) (5 μg, C,D) was injected into the left front footpad (or thigh, C,D) of cd73+/+ and cd73-/- mice and an equivalent volume of PBS was injected into the right front footpad (or thigh, C,D). Twenty-four h later, the mice were injected with 107 CMFDA-labeled cd73+/+ splenocytes i.v. One h later, brachial (or inguinal, C,D) lymph nodes were harvested and the total numbers of cells in each lymph node were counted. (A,C) The lymph node cells were then stained with PE anti-TCRβ and allophycocyanin anti-B220 (CD45R). The percentages of fluorescent cells were determined by flow cytometry and the absolute numbers of total lymphoctyes (B,D), T cells (E), and B cells (F) that migrated to the lymph nodes in 1 h were calculated. In some experiments, the mice also received 50 μg anti-L-selectin antibody MEL-14 i.v. at the same time as LPS/PBS. Twenty-four h later, the mice were injected with 107 CMFDA-labeled cd73+/+ splenocytes i.v. One h later, lymph nodes were harvested and the total numbers of cells in each lymph node were counted (G). The percentages of CMFDA+ cells were determined by flow cytometry and the absolute numbers of total lymphocytes that migrated to the lymph nodes in 1 h were calculated (H). Data from mice not receiving anti-L-selectin antibody in panels G and H are the same as in panels A and B. All results are expressed as mean ± SD. and are representative of 3-10 experiments (n=4 to 5 mice of each genotype for each experiment, p<0.025 for all comparisons of WT vs. KO in draining lymph nodes in panels A-F, for control vs. MEL-14 in draining lymph nodes for panel G, and for control vs. MEL-14 in all groups in panel H).
Next, we asked whether the increased lymphocyte migration and enlarged lymph nodes seen in cd73-deficient mice were the result of increased migration across HEV. To address this question, mice were pre-treated with L-selectin antibody. This antibody was chosen because lymphocyte expressed L-selectin is known to initiate rolling on HEV (through its interaction with PNAd) and because administration of L-selectin antibody has been shown to diminish lymphocyte migration to peripheral lymph nodes under steady state conditions in vivo (3,42). We observed that treatment of mice with L-selectin antibody i.v. at the same time as LPS, abrogated CMFDA-labeled lymphocyte migration even after 24 h in both strains of mice (Fig. 2H) and abolished the hallmark increased size of cd73-deficient draining lymph nodes (Fig. 2G).
Contribution of lymphocyte CD73 expression to migration across HEV
Because lymphocyte CD73 has been reported to be a signaling molecule, an adhesion molecule, and a maturation and subpopulation marker (16,39), we next evaluated its role in lymphocyte migration into draining lymph nodes. We first examined the percentage of CD4+, CD8+, and CD19+ lymphocytes that co-expressed CD73 in wild type draining lymph nodes by flow cytometry 24 h after stimulation. LPS-induced lymph node hypertrophy did not affect the CD73 expression pattern compared with that in lymph nodes from the PBS-treated side (Fig. 3A). The CD73 expression pattern was also equivalent to that in naïve lymph nodes and spleen (unpublished data). We next evaluated migration of cd73-deficient lymphocytes compared to wild type lymphocytes in both wild type and cd73-deficient mice. This was done by injecting mice with a 1:1 mixture of splenocytes from cd73+/+ and cd73-/- mice labeled with either CMFDA or CMTMR. Virtually identical ratios of cd73-/-:cd73+/+ lymphocytes were observed in both wild type and cd73-deficient draining lymph nodes (Fig. 3B). These results suggest no bias between CD73 positive and negative lymphocytes in their ability to migrate after an inflammatory stimulus. They further suggest that it is a lack of CD73 expression on HEV that is responsible for the increased migration of lymphocytes into draining lymph nodes of cd73-deficient mice.
FIGURE 3.

CD73 positive and negative lymphocytes migrate equally well into inflamed lymph nodes. LPS (1 μg) was injected into the left rear footpad of cd73+/+ mice and an equivalent volume of PBS was injected into the right rear footpad. (A) Twenty-four h later, the popliteal lymph node cells were stained with FITC anti-CD4, FITC anti-CD8, or PE Cy5.5 anti-CD19 plus biotinylated anti-CD73 (TY/23) and PE-streptavidin or the relevant isotype-matched control antibodies and the percentages of CD4+, CD8+, and CD19+ lymphocytes that co-expressed CD73 were determined by flow cytometry (n=4 in each of two independent experiments). (B) LPS (1 μg) was injected into the left thigh of cd73+/+ and cd73-/- mice and an equivalent volume of PBS was injected into the right thigh. Twenty-four h later, the mice were injected i.v. with an equal mixture of 1 × 107 cd73+/+ and cd73-/- splenocytes labeled with CMFDA or CMTMR, respectively. One h later, inguinal lymph nodes were harvested and the percentages of CMFDA+ and CMTMR+ lymphocytes were determined. Representative data from 1 out of 2 experiments are shown.
Contribution of DC to increased draining lymph node size in cd73-/- mice
The accumulation of activated DC in draining lymph nodes is critical for the regulation of pro-inflammatory cytokine production and induction of vascular growth (15). Local injection of LPS is known to induce DC migration to draining lymph nodes through the lymphatics and also their maturation and cytokine production (13). As previous reports showed that Ado is one of the key regulators of DC function (43,44), we speculated that the hypertrophied draining lymph nodes in cd73-/- mice might be due to increased migration of DCs though the lymphatics. Therefore, we measured the absolute numbers of MHC Class IIhi CD11c+ DCs in the draining lymph nodes of wild type and cd73-/- mice 6 h after the injection of LPS (Fig.4A). As expected, the numbers of DCs were increased in the draining lymph nodes of both strains of mice compared to those on the contralateral side. There was a trend towards higher numbers of DCs in the draining lymph nodes of cd73-/- mice, as the average number was almost 50% higher than for wild type mice; however, this difference was not statistically significant (p=0.14). Nevertheless, these data suggest that increased cytokine production by DC could contribute to the larger size of draining lymph nodes in cd73-/- mice. It is interesting to note that the draining lymph nodes in the cd73-/- mice were significantly larger than those of wild type mice (p=0.034) even at this early time point (Fig. 4B), suggesting that the kinetics of the inflammatory response are accelerated when CD73 is absent.
FIGURE 4.
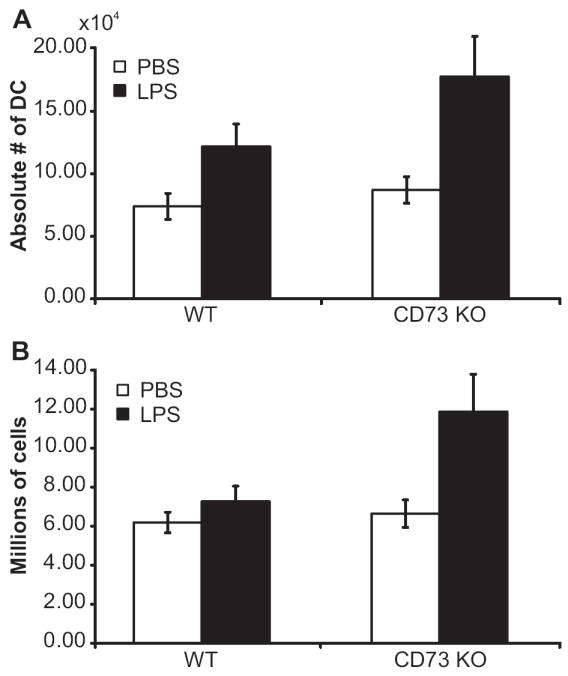
Migration of DC into draining lymph nodes after an inflammatory stimulus. LPS (1 μg) was injected into the left front footpad of cd73+/+ and cd73-/- mice and an equivalent volume of PBS was injected into the right front footpad. Six h later, lymph nodes were digested with collagenase, DNase I, and trypsin as described in Materials and Methods and cells were counted and stained with FITC anti-MHC Class II and PE anti-CD11c. Dead cells were excluded by propidium iodide staining. The average (± SEM) absolute numbers of MHC Class IIhi CD11c+ cells are shown (A) as well as the total number of leukocytes/lymph node (B). The results are combined from two experiments with a total of nine mice/group. P=0.14 for the number of MHC Class IIhi CD11c+ cells in draining lymph nodes of wild type vs. cd73-/- mice and p=0.034 for the comparison of total cell numbers/lymph node.
Up regulation of cd73 and A2BAR on HEV after an inflammatory stimulus
Previous studies showed CD73 expression can be regulated on HUVEC by mediators that are released during an inflammatory response, such as TNFα (45), IFNα (46), and Ado (47). Flow cytometry revealed a slight up regulation of cell surface CD73 on CD45-PNAd+ cells in draining lymph nodes relative to its level on HEV from control lymph nodes (Fig. 5A). Due to the lack of specific Ado receptor antibodies suitable for flow cytometry, we used KOP2.16, a cell line derived from lymph node endothelial cells, and semi-quantitative RT-PCR to examine the regulation of Ado receptor expression. Similar to what we observed in the HEV cDNA library (Fig. 1D), KOP2.16 expressed only the A2BAR. Expression increased 3- to 5-fold 3 h after TNFα stimulation in two independent experiments (Fig. 5B). These results suggest that elevated Ado, known to occur at sites of inflammation, could trigger the A2BAR on HEV in draining lymph nodes and that this could play a role in regulating lymphocyte migration into these nodes.
FIGURE 5.
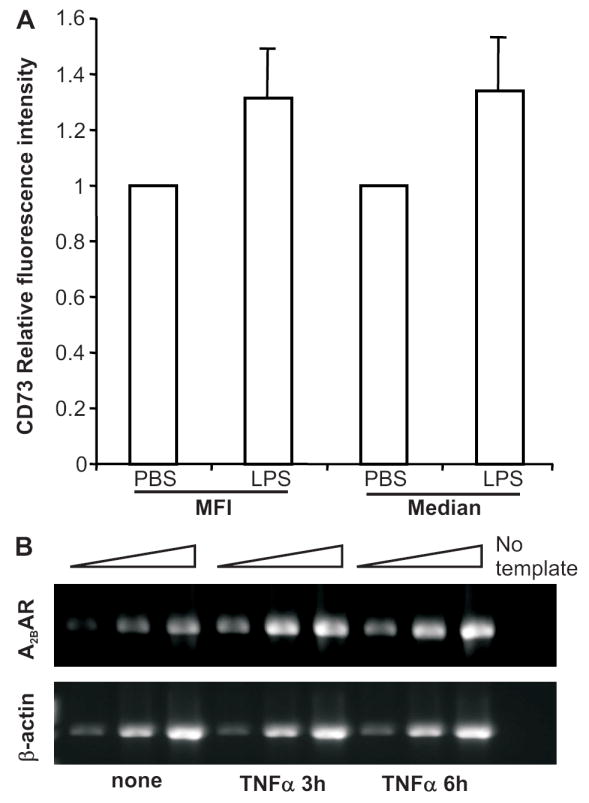
CD73 and the A2BAR are up regulated on HEV after an inflammatory stimulus. (A) LPS (1 μg) was injected into the left front footpad of cd73+/+ mice and an equivalent volume of PBS was injected into the right front footpad. Twenty-four h later, lymph nodes were digested with collagenase, DNase I, and trypsin as described in Materials and Methods and cells were stained with APC anti-CD45, purified anti-PNAd + Alexa Fluor 488 anti-IgM, and biotinylated anti-CD73 + PE-streptavidin. HEV were identified as CD45- PNAd+ cells. The relative mean (MFI) and median fluorescence intensities for CD73 staining are shown (mean ± S.D., total n= 8 from 3 independent experiments, p<0.01 for MFI and p<0.02 for median fluorescence intensities in paired t tests comparing PNAd+ cells from inflamed and control lymph nodes). (B) KOP2.16 cells were cultured ± TNFα for 3-6 h. RNA was isolated and A2BAR expression was determined by semi-quantitative RT-PCR on 5-fold serial dilutions of cDNA using β-actin expression as in internal standard. Data are representative of 1 out of 2 experiments. Quantitation of the band intensities revealed a 5-fold increase in A2BAR mRNA at 3 h and a 2.5-fold increase at 6 h relative to β-actin expression.
Adenosine receptor stimulation inhibits the increased lymphocyte migration into draining lymph nodes of cd73-deficient mice
Our previous findings and those of others suggest that Ado generated extracellularly by CD73 can modulate endothelial cell adhesion molecule expression and permeability via the A2AAR and A2BAR (27-30). Based on these findings, and our own observation that the A2BR was the only Ado receptor expressed on HEV, we asked whether the A2BAR-specific agonist BAY 60-6583 could influence lymphocyte homing after an inflammatory stimulus. As shown in Fig. 6A, treatment with BAY 60-6583 markedly decreased (p=0.0034) the number of lymphocytes that migrated into the draining lymph nodes of CD73-deficient mice. There was no impact on lymphocyte migration into lymph nodes on the contralateral side. The effect of BAY 60-6583 on lymphocyte migration into draining lymph nodes of wild type mice was much more modest and did not reach statistical significance (data not shown), perhaps because of higher concentrations of endogenous adenosine in these mice. All of these results support our hypothesis that CD73-generated Ado serves to regulate lymphocyte migration into draining lymph nodes after an inflammatory stimulus at least in part by triggering A2BAR signaling on HEV. Consistent with this hypothesis, A2BAR+/+ and A2BAR-/- lymphocytes showed equivalent rates of lymphocyte migration into draining lymph nodes of LPS-treated wild type and cd73-/- mice (Fig. 6B).
FIGURE 6.
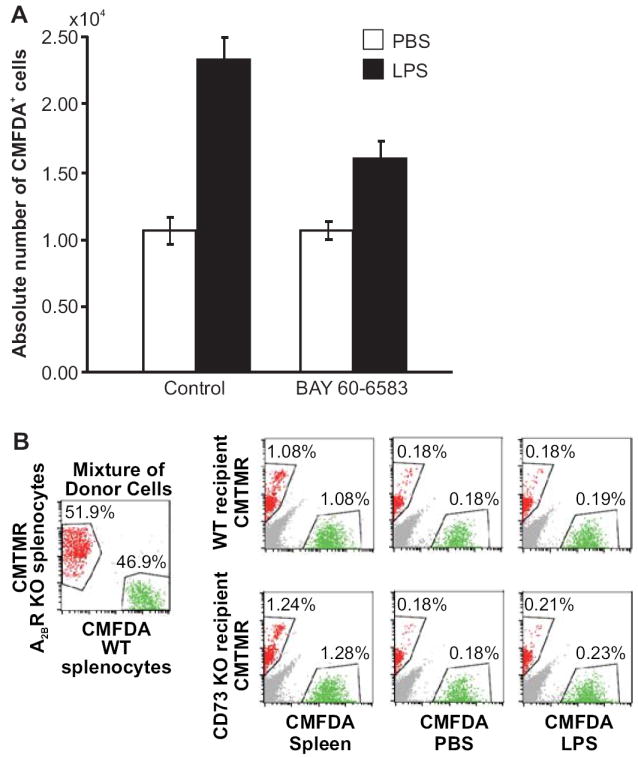
The A2B adenosine receptor agonist, BAY 60-6583, inhibits the increased lymphocyte migration into draining lymph nodes of CD73-deficient mice. A. LPS (1 μg) was injected into the left front footpad of cd73+/+ and cd73-/- mice and an equivalent volume of PBS was injected into the right front footpad. Twenty-two h and 30 min after the LPS injection, the mice were injected i.v. with BAY 60-6583 (320 μg/kg) or an equivalent volume of diluted PEG 400 carrier and 30 min later with 107 CMFDA-labeled cd73+/+ splenocytes. One h after the injection of labeled splenocytes, lymph nodes were harvested and the total numbers of cells in each lymph node were counted and the percentages of CMFDA+ cells were determined by flow cytometry (n=20-26, p=0.0034 for diluted PEG 400 vs. BAY 60-6583 in draining lymph nodes, data combined from 4 separate experiments). B. LPS (1 μg) was injected into the left front footpad of wild type and cd73-/- mice and an equivalent volume of PBS was injected into the right front footpad (5 mice/group). Twenty-three h later, the mice were injected i.v. with an equal mixture of 107 wild type and A2BR-/- splenocytes labeled with CMFDA or CMTMR, respectively. One h later, brachial lymph nodes were harvested and the percentages of CMFDA+ and CMTMR+ lymphocytes were determined by flow cytometry. Data are representative of 1 out of 2 experiments.
DISCUSSION
Lymphocyte homing to peripheral lymph nodes depends on interactions with HEV, specialized blood vessels that express chemokines and adhesion molecules required for lymphocyte transmigration. Although numerous reports have demonstrated the importance of L-selectin, PNAd, LFA-1, ICAM-1, and specific chemokines and chemokine receptors in the steady state, the way in which lymphocyte migration across HEV is regulated during an inflammatory response is not fully understood. The goal of this investigation was to determine if CD73, which is expressed on both PNAd+ endothelial cells and the basal lamina comprising HEV, plays a role in this process. The source of CD73 expressed on basal lamina is not known. However, CD73 is a GPI-anchored protein and previous studies by Mehul et al. (48) showed that CD73 can bind to laminin, one of the components of the basal lamina. Therefore, we hypothesize that CD73 may be synthesized in cells such as endothelial cells, cleaved from the cell surface by a phospholipase, and then bind to a component of the basal lamina such as laminin.
The contribution of CD73 to the formation of extracellular Ado, a well-known anti-inflammatory mediator, has been revealed in several experimental models. For example, the anti-inflammatory action of methotrexate in the carrageenan-treated air pouch model of inflammation is dependent upon CD73 (49). Similarly, CD73-generated Ado is necessary for ischemic preconditioning in both the heart (40) and kidney (50), and protects mice from bleomycin-induced lung injury (51). Furthermore, cd73-deficient mice exhibit a vascular leak syndrome characterized by neutrophil infiltration into tissues when exposed to normobaric hypoxia, suggesting a critical role for CD73-generated Ado in vascular barrier function (27-29). On the other hand, several in vitro studies suggested that CD73 functions as a costimulatory molecule on T lymphocytes (33,34) and an adhesion molecule that is important for lymphocyte binding to endothelium (35). The possibility that CD73 could impact lymphocyte interactions with HEV by multiple mechanisms prompted us to examine the role of this molecule in lymphocyte homing to lymph nodes. We showed here that Ado generated by CD73 on HEV negatively regulates lymphocyte migration from the blood stream into LPS-induced draining lymph nodes. As cd73+/+ and cd73-/- splenocytes showed equivalent rates of migration into draining lymph nodes, it is unlikely that any signaling or adhesive function of CD73 on lymphocytes plays a role in regulating migration of lymphocytes across HEV.
Although no abnormalities were observed in the cellularity of lymphoid organs of cd73-/- mice or in the migratory capacity of cd73-/- lymphocytes under steady state conditions, cd73-/- mice had larger draining lymph nodes when LPS, a TLR 4 agonist, was injected into a local site. Short term assays with CFMDA-labeled lymphocytes administered i.v., showed increased rates of migration into the draining lymph nodes of cd73-/- mice. This observation, coupled with the forward vs. side scatter profile of the lymphocytes (unpublished data), suggested that the lymph node hypertrophy induced by LPS was not due to the proliferation of lymphocytes, but rather to the accumulation of non-dividing lymphocytes. Furthermore, anti-L-selectin antibody treatment demonstrated that CD73 modulates lymphocyte migration into draining lymph nodes by an L-selectin-dependent pathway. Interestingly, both T and B lymphocyte entrance was promoted in cd73-/- mice, suggesting that a common pathway for both cell types is modulated by CD73. In addition, the percentages of CD73+ and CD73- lymphocytes did not change when splenocytes from cd73+/+ mice were used as donors in migration experiments in cd73+/+ mice, indicating that the ability to migrate across HEV was not influenced by the CD73 expression status of lymphocytes. Furthermore, the migration of splenocytes from cd73+/+ and cd73-/- mice into draining lymph nodes of cd73-/- mice was comparable, demonstrating that CD73 expression on lymphocytes cannot compensate for a lack of CD73 on HEV.
Information in the literature concerning the regulation of endothelial CD73 expression by pro-inflammatory cytokines is conflicting. For example, Kalsi et al. (45) showed a decrease in its expression and in its enzyme activity after TNFα treatment of HUVEC. On the other hand, Niemela and co-workers demonstrated that IFNα and IFNγ, but not other inflammatory cytokines such as IL-1β, IL-4, or TNFα could increase CD73 expression on HUVEC (46). Furthermore, Ado has been implicated in an increase in CD73 expression in microvascular endothelial cells that is mediated by a paracrine pathway (47). Our experiments revealed that the expression of CD73 on HEV in draining lymph nodes is up regulated compared to HEV in the contralateral side. Although our analysis has the advantage of evaluating changes in CD73 expression in vivo, the mechanism by which CD73 expression is modulated is still unknown.
To determine whether the enhanced lymphocyte migration in cd73-deficient mice was caused by a lack of Ado receptor signaling, we treated mice with the A2BR agonist BAY 60-6583. This approach was taken because of the four known subtypes of Ado receptors, only the A2BAR was found in a cDNA library derived from PNAd+ cells or in cDNA from the HEV-like cell line KOP2.16 (36). We also observed an up regulation of A2BAR expression in KOP2.16 cells after exposure to TNFα. Indeed, BAY 60-6583 treatment markedly reduced the rate of migration of labeled splenocytes into draining lymph nodes of cd73-/- mice. These data are consistent with the hypothesis that the A2BAR is at least partially responsible for the regulation of lymphocyte migration across HEV by CD73-generated Ado. Furthermore, the Ado is likely derived from CD73 on HEV, as lymphocytes from cd73+/+ and cd73-/- mice show similar increased rates of migration across HEV in draining lymph nodes of cd73-/- mice (i.e., Ado produced by cd73+/+ lymphocytes does not appear able to trigger adenosine receptors on HEV to regulate lymphocyte migration). Similarly, lymphocytes from A2BR+/+and A2BR-/- mice showed similar rates of migration into draining lymph nodes of wild type mice, suggesting that it is triggering of the A2BR on HEV that is relevant.
The expression of the adhesion molecules ICAM-1 and VCAM-1 is normal on cd73-/- HEV in the steady state (unpublished data). Our findings differ from those in a previous report (30) which concluded that CD73 deficiency resulted in increased VCAM-1 expression and decreased ICAM-1 expression on carotid arteries due to the lack of A2AAR signaling. This discrepancy could be explained by the fact that different cell types were being examined and that the A2BAR, rather than the A2AAR, seems to be the predominant AR on HEV. We did find that VCAM-1, but not ICAM-1, expression was up regulated on HEV in draining lymph nodes after LPS administration and this effect was more pronounced in cd73-/- mice. However, neither anti-VLA-4 nor anti-VCAM-1 antibody treatment reversed the increased rates of lymphocyte migration into draining lymph nodes of cd73-/- mice after LPS treatment (unpublished data), suggesting that the increase in VCAM-1 expression did not augment cell adhesion between lymphocytes and HEV. Furthermore, although the migration of CMFDA-labeled lymphocytes to draining lymph nodes was inhibited by treatment with an anti-LFA-1 antibody, the effect was the same in both cd73+/+ and cd73-/- mice (unpublished data). Taken together, these data support the conclusion that increased lymphocyte migration into draining lymph nodes of cd73-deficient mice is not mediated by increases in cell adhesion.
We propose instead that Ado generated by endothelial cell (and/or basal lamina) CD73 regulates lymphocyte migration across HEV through A2BAR signaling. The A2BAR is a seven transmembrane-spanning G protein-coupled receptor that is coupled to Gs and uses cAMP as a second messenger (52). It has been firmly established that cAMP can modulate endothelial cell-cell junctions through the protein kinase A and/or Epac-Rap1 pathways (53,54). Other reports suggest that the A2BAR can also be coupled to Gq (55). In the 1970s, several studies concluded that lymph node vasculature changed within 24 h after an inflammatory stimulus and that this was associated with changes in vascular integrity (56,57). Our studies do not address the mechanisms by which rates of lymphocyte migration are increased after an inflammatory stimulus, but do suggest that CD73-generated Ado may trigger a feedback mechanism to keep increases in permeability under control. Additional experiments with endothelial cell lines will be required to determine whether Ado receptor signaling modulates the ability of lymphocytes to migrate across HEV through changes in myosin light chain phosphorylation and decreased formation of stress fibers and/or through Rap1/Epac mediated increases in VE-cadherin based cell-cell contacts. Future work will also address the consequences of increased lymphocyte migration into cd73-/- draining lymph nodes during an immune response.
Acknowledgments
The authors acknowledge Ms. Mary Flynn for manuscript preparation, the excellent technical assistance of Scott Hooker and Patrick Marble, and the expertise of Ms. Julie Maier in the OMRF Imaging Facility. The authors also thank Dr. Paul Kincade for critical comments during manuscript preparation, Dr. Rob Welner for assistance with i.v. injections, and Drs. Almut Grenz and Tobias Eckle for advice regarding the administration of BAY 60-6583.
Footnotes
This work was supported by NIH Grants AI18220 (L.F.T.), P01 HL085607 (R.P.M.), and AI43472 (M.R.B.) and was part of the 21st Century COE entitled “Origination of Frontier BioDentistry” at Osaka University Graduate School of Dentistry supported by the Ministry of Education, Culture, Sports, Science and Technology. L.F.T. holds the Putnam City Schools Distinguished Chair in Cancer Research. R.P.M. holds the Eli Lilly Distinguished Chair in Biomedical Research.
Abbreviations used: Ado, adenosine; ADA, adenosine deaminase; AR, adenosine receptor; CFMDA, 5-chloromethylfluorescein diacetate; CMTMR, 5-(and-6)-(((4-chloromethyl)benzoyl)amino)tetramethylrhodamine); DC, dendritic cell; GPI, glycosyl phosphatidyl inositol; HEV, high endothelial venule; PEG, polyethylene glycol; PNAd, peripheral lymph node addressin.
DISCLOSURES Thomas Krahn is an employee of Bayer HealthCare AG, the manufacturer of BAY 60-6583. All other authors have no conflicting financial interests.
References
- 1.Kraal G, Mebius RE. High endothelial venules: Lymphocyte traffic control and controlled traffic. Adv Immunol. 1997;65:347–395. [PubMed] [Google Scholar]
- 2.Miyasaka M, Tanaka T. Lymphocyte trafficking across high endothelial venules: dogmas and enigmas. Nat Rev Immunol. 2004;4:360–370. doi: 10.1038/nri1354. [DOI] [PubMed] [Google Scholar]
- 3.Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129–156. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- 4.Hamann A, Jablonski-Westrich D, Duijvestijn A, Butcher EC, Baisch H, Harder R, Thiele HG. Evidence for an accessory role of LFA-1 in lymphocyte-high endothelium interaction during homing. J Immunol. 1988;140:693–699. [PubMed] [Google Scholar]
- 5.Campbell JJ, Hedrick J, Zlotnik A, Siani MA, Thompson DA, Butcher EC. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science. 1998;279:381–384. doi: 10.1126/science.279.5349.381. [DOI] [PubMed] [Google Scholar]
- 6.Arbones ML, Ord DC, Ley K, Ratech H, Maynard-Curry C, Otten G, Capon DJ, Tedder TF. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity. 1994;1:247–260. doi: 10.1016/1074-7613(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 7.Berlin-Rufenach C, Otto F, Mathies M, Westermann J, Owen MJ, Hamann A. Lymphocyte migration in lymphocyte function-associated antigen (LFA)-1-deficient mice. J Exp Med. 1999;189:1467–1478. doi: 10.1084/jem.189.9.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uchimura K, Gauguet J-M, Singer MS, Tsay D, Kannagi R, Muramatsu T, von Adrian UH, Rosen SD. A major class of L-selectin ligands is eliminated in mice deficient in two sulfotransferases expressed in high endothelial venules. Nat Immunol. 2005;6:1105–1113. doi: 10.1038/ni1258. [DOI] [PubMed] [Google Scholar]
- 9.Butcher EC, Williams M, Youngman K, Rott L, Briskin M. Lymphocyte trafficking and regional immunity. Adv Immunol. 1999;72:209–253. doi: 10.1016/s0065-2776(08)60022-x. [DOI] [PubMed] [Google Scholar]
- 10.Mitoma J, Bao X, Petryniak B, Schaerli P, Gauguet JM, Yu SY, Kawashima H, Saito H, Ohtsubo K, Marth JD, Khoo KH, von Adrian UH, Lowe JB, Fukuda M. Critical functions of N-glycans in L-selectin-mediated lymphocyte homing and recruitment. Nat Immunol. 2007;8:409–418. doi: 10.1038/ni1442. [DOI] [PubMed] [Google Scholar]
- 11.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 12.Roake JA, Rao AS, Morris PJ, Larsen CP, Hankins DF, Austyn JM. Dendritic cell loss from nonlymphoid tissues after systemic administration of lipopolysaccharide, tumor necrosis factor, and interleukin 1. J Exp Med. 1995;181:2237–2247. doi: 10.1084/jem.181.6.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsujimoto H, Uchida T, Efron PA, Scumpia PO, Verma A, Matsumoto T, Tschoeke SK, Ungaro RF, Ono S, Seki S, Clare-Salzler MJ, Baker HV, Mochizuki H, Ramphal R, Moldawer LL. Flagellin enhances NK cell proliferation and activation directly and through dendritic cell-NK cell interactions. J Leukoc Biol. 2005;78:888–897. doi: 10.1189/jlb.0105051. [DOI] [PubMed] [Google Scholar]
- 14.Soderberg KA, Payne GW, Sato A, Medzhitov R, Segal SS, Iwasaki A. Innate control of adaptive immunity via remodeling of lymph node feed arteriole. PNAS. 2005;102:16315–16320. doi: 10.1073/pnas.0506190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Webster B, Ekland EH, Agle LM, Chyou S, Ruggieri R, Lu TT. Regulation of lymph node vascular growth by dendritic cells. J Exp Med. 2005 doi: 10.1084/jem.20052272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson LF, Ruedi JM, Glass A, Moldenhauer G, Moller P, Low MG, Klemens MR, Massaia M, Lucas AH. Production and characterization of monoclonal antibodies to the glycosyl phosphatidylinositol-anchored lymphocyte differentiation antigen ecto-5’-nucleotidase (CD73) Tissue Antigens. 1990;35:9–19. doi: 10.1111/j.1399-0039.1990.tb01750.x. [DOI] [PubMed] [Google Scholar]
- 17.Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol. 2001;41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 18.Franco R, Valenzuela A, Lluis C, Blanco J. Enzymatic and extraenzymatic role of ecto-adenosine deaminase in lymphocytes. Immunol Rev. 1998;161:27–42. doi: 10.1111/j.1600-065x.1998.tb01569.x. [DOI] [PubMed] [Google Scholar]
- 19.Dong RP, Kameoka J, Hegen M, Tanaka T, Xu XH, Schlossman SF, Morimoto C. Characterization of adenosine deaminase binding to human CD26 on T cells and its biologic role in immune response. J Immunol. 1996;156:1349–1355. [PubMed] [Google Scholar]
- 20.Hashikawa T, Hooker SW, Maj JG, Knott-Craig CJ, Takedachi M, Murakami S, Thompson LF. Regulation of adenosine receptor engagement by ecto-adenosine deaminase. FASEB J. 2004;18:131–133. doi: 10.1096/fj.03-0011fje. [DOI] [PubMed] [Google Scholar]
- 21.Sun D, Samuelson LC, Yang T, Huang Y, Paliege A, Saunders T, Briggs J, Schnermann J. Mediation of tubuloglomerular feedback by adenosine: evidence from mice lacking adenosine 1 receptors. Proc Natl Acad Sci U S A. 2001;98:9983–9988. doi: 10.1073/pnas.171317998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ledent C, Vaugeois J-M, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen J-J, Costentin J, Heath JK, Vassart G, Parmentier M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature. 1997;388:674–678. doi: 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- 23.Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D, Moskowitz MA, Fink JS, Schwarzschild MA. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999;19:9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang D, Zhang Y, Nguyen HG, Koupenova M, Chauhan AK, Makitalo M, Jones MR, St Hilaire C, Seldin DC, Toselli P, Lamperti E, Schreiber BM, Gavras H, Wagner DD, Ravid K. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest. 2006;116:1913–1923. doi: 10.1172/JCI27933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salvatore CA, Tilley SL, Latour AM, Fletcher DS, Koller BH, Jacobson MA. Disruption of the A3 adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Biol Chem. 2000;275:4429–4434. doi: 10.1074/jbc.275.6.4429. [DOI] [PubMed] [Google Scholar]
- 26.Cronstein BN. Adenosine, an endogenous anti-inflammatory agent. J Appl Physiol. 1994;76:5–13. doi: 10.1152/jappl.1994.76.1.5. [DOI] [PubMed] [Google Scholar]
- 27.Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, Colgan SP. Crucial role for ecto-5’-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;200:1395–1405. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, Colgan SP. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 29.Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111:2024–2025. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zernecke A, Bidzhekov K, Ozuyaman B, Fraemohs L, Liehn EA, Luscher-Firzlaff JM, Luscher B, Schrader J, Weber C. CD73/ecto-5’-nucleotidase protects against vascular inflammation and neointima formation. Circulation. 2006;113:2120–2127. doi: 10.1161/CIRCULATIONAHA.105.595249. [DOI] [PubMed] [Google Scholar]
- 31.Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev. 1998;161:95–109. doi: 10.1111/j.1600-065x.1998.tb01574.x. [DOI] [PubMed] [Google Scholar]
- 32.Airas L, Niemela J, Jalkanen S. CD73 engagement promotes lymphocyte binding to endothelial cells via a lymphocyte function-associated antigen-1-dependent mechanism. J Immunol. 2000;165:5411–5417. doi: 10.4049/jimmunol.165.10.5411. In Process Citation. [DOI] [PubMed] [Google Scholar]
- 33.Massaia M, Perrin L, Bianchi A, Ruedi J, Attisano C, Altieri D, Rijkers GT, Thompson LF. Human T cell activation: synergy between CD73 (ecto-5’-nucleotidase) and signals delivered through CD3 and CD2 molecules. J Immunol. 1990;145:1664–1674. [PubMed] [Google Scholar]
- 34.Dianzani U, Redoglia V, Bragardo M, Attisano C, Bianchi A, DiFranco D, Ramenghi U, Wolff H, Thompson LF, Pileri A, Massaia M. Co-stimulatory signal delivered by CD73 molecule to human CD45RAhiCD45ROlo (naive) CD8+ T lymphocytes. J Immunol. 1993;151:3961–3970. [PubMed] [Google Scholar]
- 35.Airas L, Hellman J, Salmi M, Bono P, Puurunen T, Smith DJ, Jalkanen S. CD73 is involved in lymphocyte binding to the endothelium: Characterization of lymphocyte vascular adhesion protein 2 identifies it as CD73. J Exp Med. 1995;182:1603–1608. doi: 10.1084/jem.182.5.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toyama-Sorimachi N, Miyake K, Miyasaka M. Activation of CD44 induces ICAM-1/LFA-1-independent, Ca2+, Mg2+-independent adhesion pathway in lymphocyte-endothelial cell interaction. Eur J Immunol. 1993;23:439–446. doi: 10.1002/eji.1830230221. [DOI] [PubMed] [Google Scholar]
- 37.Umemoto E, Tanaka T, Kanda H, Jin S, Tohya K, Otani K, Matsutani K, Matsumoto M, Ebisuno Y, Jang MH, Fukuda M, Hirata T, Miyasaka M. Nepmucin, a novel HEV sialomucin, mediates L-selectin-dependent lymphocyte rolling and promotes lymphocyte adhesion under flow. J Exp Med. 2006;203:1603–1614. doi: 10.1084/jem.20052543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van De Wiele CJ, Vaughn JG, Blackburn MR, Ledent C, Jacobson M, Jiang H, Thompson LF. Adenosine kinase inhibition promotes survival of fetal adenosine deaminase-deficient thymocytes by blocking dATP accumulation. J Clin Invest. 2002;110:395–402. doi: 10.1172/JCI15683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamashita Y, Hooker SW, Jiang H, Laurent AB, Resta R, Khare K, Coe A, Kincade PW, Thompson LF. CD73 expression and Fyn-dependent signaling on murine lymphocytes. Eur J Immunol. 1998;28:2981–2990. doi: 10.1002/(SICI)1521-4141(199810)28:10<2981::AID-IMMU2981>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 40.Eckle T, Krahn T, Grenz A, Köhler D, Mittelbronn M, Ledent C, Jacobson M, Osswald H, Thompson LF, Unertl K, Eltzschig HK. Cardioprotection by ecto-5’-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- 41.Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol. 2006;177:6780–6786. doi: 10.4049/jimmunol.177.10.6780. [DOI] [PubMed] [Google Scholar]
- 42.Lepault F, Gagnerault MC, Faveeuw C, Boitard C. Recirculation, phenotype and functions of lymphocytes in mice treated with monoclonal antibody MEL-14. Eur J Immunol. 1994;24:3106–3112. doi: 10.1002/eji.1830241229. [DOI] [PubMed] [Google Scholar]
- 43.Panther E, Idzko M, Herouy Y, Rheinen H, Gebicke-haerter PJ, Mrowietz U, Dichmann S, Norgauer J. Expression and function of adenosine receptors in human dendritic cells. FASEB J. 2001;15:1963–1970. doi: 10.1096/fj.01-0169com. [DOI] [PubMed] [Google Scholar]
- 44.Panther E, Corinti S, Idzko M, Herouy Y, Napp M, La Sala A, Girolomoni G, Norgauer J. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood. 2003;101:3985–3990. doi: 10.1182/blood-2002-07-2113. [DOI] [PubMed] [Google Scholar]
- 45.Kalsi K, Lawson C, Dominguez M, Taylor P, Yacoub MH, Smolenski RT. Regulation of ecto-5’-nucleotidase by TNF-alpha in human endothelial cells. Mol Cell Biochem. 2002;232:113–119. doi: 10.1023/a:1014806916844. [DOI] [PubMed] [Google Scholar]
- 46.Niemela J, Henttinen T, Yegutkin GG, Airas L, Kujari AM, Rajala P, Jalkanen S. IFN-alpha induced adenosine production on the endothelium: a mechanism mediated by CD73 (ecto-5’-nucleotidase) up-regulation. J Immunol. 2004;172:1646–1653. doi: 10.4049/jimmunol.172.3.1646. [DOI] [PubMed] [Google Scholar]
- 47.Narravula S, Lennon PF, Mueller BU, Colgan SP. Regulation of endothelial CD73 by adenosine: paracrine pathway for enhanced endothelial barrier function. J Immunol. 2000;165:5262–5268. doi: 10.4049/jimmunol.165.9.5262. [DOI] [PubMed] [Google Scholar]
- 48.Mehul B, Doyennette-Moyne M, Aubery M, Mannherz H, Codogno P. 5’-Nucleotidase is involved in chick embryo myoblast spreading on laminin. Cell Bio Int Rep. 1990;2:155–164. doi: 10.1016/0309-1651(90)90032-t. [DOI] [PubMed] [Google Scholar]
- 49.Montesinos MC, Takedachi M, Thompson LF, Wilder TF, Fernández P, Cronstein BN. The anti-inflammatory mechanism of methotrexate depends on extracellular conversion of adenine nucleotides to adenosine by ecto-5’-nucleotidase (CD73) Arthritis Rheum. 56:1440–1445. doi: 10.1002/art.22643. [DOI] [PubMed] [Google Scholar]
- 50.Grenz A, Zhang H, Eckle T, Wehrmann M, Köhle C, Thompson L, Osswald H, Eltzschig HK. Protective role of ecto-5’-nucleotidase (CD73) in renal ischemia. J Am Soc Nephrol. 18:833–845. doi: 10.1681/ASN.2006101141. [DOI] [PubMed] [Google Scholar]
- 51.Volmer JB, Thompson LF, Blackburn MR. Ecto-5’-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J Immunol. 2006;176:4449–4458. doi: 10.4049/jimmunol.176.7.4449. [DOI] [PubMed] [Google Scholar]
- 52.Schulte G, Fredholm BB. The G(s)-coupled adenosine A(2B) receptor recruits divergent pathways to regulate ERK1/2 and p38. Exp Cell Res. 2003;290:168–176. doi: 10.1016/s0014-4827(03)00324-0. [DOI] [PubMed] [Google Scholar]
- 53.Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, Saito Y, Kangawa K, Mochizuki N. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol. 2005;25:136–146. doi: 10.1128/MCB.25.1.136-146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patterson CE, Lum H, Schaphorst KL, Verin AD, Garcia JG. Regulation of endothelial barrier function by the cAMP-dependent protein kinase. Endothelium. 2000;7:287–308. doi: 10.3109/10623320009072215. [DOI] [PubMed] [Google Scholar]
- 55.Linden J, Thai T, Figler H, Jin X, Robeva AS. Characterization of human A(2B) adenosine receptors: radioligand binding, western blotting, and coupling to G(q) in human embryonic kidney 293 cells and HMC-1 mast cells. Mol Pharmacol. 1999;56:705–713. [PubMed] [Google Scholar]
- 56.Herman PG, Yamamoto I, Mellins HZ. Blood microcirculation in the lymph node during the primary immune response. J Exp Med. 1972;136:697–714. doi: 10.1084/jem.136.4.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anderson ND, Anderson AO, Wyllie RG. Microvascular changes in lymph nodes draining skin allografts. Am J Pathol. 1975;81:131–160. [PMC free article] [PubMed] [Google Scholar]