

Abstract
Treatment of degenerating basal forebrain cholinergic neurons with nerve growth factor (NGF) in Alzheimer’s disease has long been contemplated, but an effective and safe delivery method has been lacking. Towards achieving this goal, we are currently developing CERE-110, an adeno-associated virus-based gene delivery vector that encodes for human NGF, for stereotactic surgical delivery to the human nucleus basalis of Meynert. Results indicate that NGF transgene delivery to the targeted brain region via CERE-110 is reliable and accurate, that NGF transgene distribution can be controlled by altering CERE-110 dose, and that it is possible to achieve restricted NGF expression limited to but covering the target brain region. Results from animals examined at longer time periods of 3, 6, 9 and 12 months after CERE-110 delivery indicate that NGF transgene expression is stable and sustained at all time points, with no loss or build-up of protein over the long-term. In addition, results from a series of experiments indicate that CERE-110 is neuroprotective and neurorestorative to basal forebrain cholinergic neurons in the rat fimbria-fornix lesion and aged rat models, and has bioactive effects on young rat basal forebrain cholinergic neurons. These findings, as well as those from several additional non-clinical experiments conducted in both rats and monkeys, led to the initiation of a Phase I clinical study to evaluate the safety and efficacy of CERE-110 in Alzheimer’s disease subjects, which is currently ongoing.
Keywords: adeno-associated virus, Alzheimer’s disease, basal forebrain cholinergic neurons, CERE-110, dose-response, gene delivery, nerve growth factor, neurotrophin, nucleus basalis of Meynert, trophic activity
INTRODUCTION
Protecting and restoring the basal forebrain cholinergic neurons (BFCNs) of the nucleus basalis of Meynert (NBM) is a logical approach to treating mild to moderate Alzheimer’s disease. Although the etiology of Alzheimer’s disease is largely unknown, three primary neuroanatomical features characterize the Alzheimer’s diseased brain: amyloid plaques, neurofibrillary tangles, and the loss of neurons. Severe cholinergic neuronal death occurs in the NBM of the basal forebrain in Alzheimer’s disease (Whitehouse et al., 1981; Whitehouse et al., 1982; Coyle et al., 1983; Lyness et al., 2003), and cholinergic loss in Alzheimer’s disease correlates significantly with severity of dementia and synapse loss (Perry et al., 1978a; Perry et al., 1978b; Bierer et al., 1995). Cholinergic blockade in humans and monkeys impairs cognition in ways that are qualitatively similar to cognitive dysfunction associated with mild to moderate Alzheimer’s disease (Bartus et al., 1976; Robbins et al., 1997; Taffe et al., 1999; Taffe et al., 2002), and modest pharmacological augmentation of cholinergic function improves Alzheimer’s disease symptoms (Birks, 2006; Burns and O’Brian, 2006; Hansen et al., 2007). In addition, cholinergic axonal projections of BFCNs regulate neuronal activity in the cortex and hippocampus, which places BFCNs in a unique position to influence a diverse array of executive functions (Bucci et al., 1998; Kilgard and Merzenich, 1998; Baxter and Chiba, 1999; Furey et al., 2000).
Treatment of basal forebrain cholinergic degeneration in Alzheimer’s disease with NGF was proposed many years ago (e.g. Chen, et al., 1989; Phelps et al., 1989; Hefti and Schneider, 1991; Hefti et al., 1996). In animal models, NGF prevents the death of BFCNs after axonal injury (Williams et al., 1986; Hefti, 1986; Rosenberg et al., 1988; Koliatsos et al., 1990; Tuszynski et al., 1990; Koliatsos et al., 1991a and 1991b; Tuszynski and Gage, 1995), reverses their spontaneous age-related atrophy (Chen and Gage, 1995; Lindner et al., 1996; Smith et al., 1999; Conner et al., 2001), and improves learning and memory in lesioned and aged rats (Fischer et al., 1987; Fischer et al., 1991; Williams et al., 1991; Markowska et al., 1994; Tuszynski and Gage, 1995; Chen and Gage 1995; Martinez-Serrano et al., 1996). In Alzheimer’s disease, NGF levels are reduced in the BFCNs of the NBM (Mufson et al., 1995; Scott et al., 1995). At the same time, NGF levels in the natural target of BFCN axons, the cortex, have generally found to be elevated in Alzheimer’s disease brains (Crutcher et al., 1993; Mufson et al., 1995; Scott et al., 1995; Hellweg et al., 1998; Fahnestock et al., 2001; Peng et al., 2004) This suggests retrograde axonal transport of NGF from the cortex to the BFCN somata is defective in Alzheimer’s disease (Mufson et al., 1995; Scott et al., 1995; Mufson et al., 1999). Supporting this concept, defective retrograde transport of NGF by BFCNs has been demonstrated in a mouse model of Alzheimer’s pathology (Cooper et al., 2001; Salehi et al., 2003; Salehi et al., 2006). Therefore, it is possible that circumventing this putative retrograde transport defect by administering NGF directly to BFCNs could both prevent loss of BFCNs and augment function of remaining BFCNs in Alzheimer’s disease (i.e. Hellweg et al., 1990).
While administration of NGF to BFCNs could be an effective treatment for Alzheimer’s disease, a safe and effective means of accurately delivering NGF to BFCNs has been lacking. NGF needs to be continuously administered since non-clinical studies have found that when NGF is withdrawn its effects are not maintained (Montero and Hefti, 1988; Niewiadomska, et al., 2002). NGF protein does not readily cross the blood-brain barrier when administered systemically (Lapchak et al., 1993) and therefore must be administered directly to the brain in order to be most effective. Because NGF administration needs to bypass the blood-brain barrier, and be continuous, an initial clinical trial of NGF for Alzheimer’s disease tested continuous infusion of NGF into the cerebral ventricles. Unfortunately, adverse side effects arose in subjects in that trial, and the trial was halted without observation of substantial benefits (Olson et al., 1992; Eriksdotter Jonhagen et al., 1998). Subsequent non-clinical research has revealed that neuroanatomical changes occur in response to the broad distribution of NGF in the cerebrospinal fluid (CSF) following intracerebroventricular administration, and that these changes are related to the physiological side effects induced by the action of NGF on non-target cells. Importantly, it has also been established that both the neuroanatomical changes and the physiological side effects can be avoided by direct administration of NGF into the brain parenchyma (Olson et al., 1991; Day-Lollini et al., 1997; Winkler et al., 1997; Pizzo, et al., 2002). Thus, effective delivery of NGF requires both consistent exposure of the protein to the targeted NBM, while avoiding non-targeted exposure to other brain regions.
To this end, gene transfer may be the most effective and practical delivery method available, in that it can provide controlled and sustained delivery of a therapeutic protein such as NGF to a targeted brain region following a single surgical procedure, without the complications of indwelling hardware. In support of this concept, a Phase I clinical study of ex vivo gene transfer (autologous fibroblasts transfected with a retroviral vector to express NGF) has shown none of the adverse effects associated with off-target NGF delivery, and suggests potential effects on brain metabolism and, possibly, cognition (Tuszynski et al., 2005). However, the ex vivo gene transfer approach is limited by a decline in NGF protein expression from the cells over 18 months post-implantation, while manufacturing complexities and costs make this approach impractical for application to larger numbers of patients. Therefore, CERE-110, a genetically engineered, replication defective adeno-associated virus serotype 2 (AAV2) vector that contains the full-length human β-nerve growth factor (NGF) cDNA, is currently being developed for the delivery of NGF for Alzheimer’s disease. The AAV2 vector was chosen because it preferentially transduces neurons, delivers the transgene predominantly as non-integrated DNA (thereby reducing the possibility of insertional mutagenesis), and results in long-lasting gene expression following a single administration to the brain parenchyma. A series of non-clinical experiments were performed to: (1) examine NGF transgene kinetics and pattern of expression, (2) determine the dose-response relationship between CERE-110 and resulting NGF expression, and (3) test the hypothesis that NGF delivered via CERE-110 provides the expected trophic activity to NBM basal forebrain cholinergic neurons. Findings from these studies, described herein, support accurate, reliable targeting and control of NGF transgene expression, sustained and stable transgene expression over long time periods, bioactivity of NGF expressed from the viral vector, and efficacy in the rodent models of BFCN degeneration relevant to AD. Results from these studies provide support for a Phase I clinical study of CERE-110 in subjects with mild to moderate AD, which is currently ongoing (Arvanitakis et al., 2007).
MATERIALS AND METHODS
Vector plasmid constructs
A prototype vector, AAV2-NGF-wPRE, was used for the fimbria-fornix lesion experiment. The wPRE element was subsequently removed from the vector due to potential safety concerns (for example, see Kingsman et al., 2005) and this resulting vector, called CERE-110, was used in all other experiments. The AAV2-NGF-wPRE vector genome contains the AAV2 inverted terminal repeats (ITRs) flanking a transgene expression cassette containing the CAG promoter (Niwa et al., 1991), the human NGF cDNA, the woodchuck hepatitis post-transcriptional regulatory element (wPRE) (Donello et al., 1998) and the human growth hormone gene (hGH) polyadenylation signal (polyA) (Stratagene). To generate the AAV2-NGF-wPRE plasmid, the CAG-NGF-wPRE cassette (Blesch, et al., 2005) was isolated by restriction digest and cloned into the pAAV-MCS (Stratagene) plasmid. The CERE-110 expression cassette was created by removing the wPRE from AAV2-NGF-wPRE. In addition, the ampicillin selection cassette was replaced by the kanamycin selection cassette. Restriction digestions and nucleotide sequence determination confirmed plasmid integrity.
Cell culture and vector production
Unless otherwise specified, cells were cultured in phenol red and antibiotic-free Iscove’s Modified Dulbecco’s Medium (IMDM)-5% FBS supplemented with 4mM L-Glutamine (Irvine Scientific) at 37°C in 5% CO2. Both vectors were produced by overnight triple plasmid calcium phosphate transfection of subconfluent 293 cells using an equimolar cocktail of the following plasmids: a vector genome plasmid, an AAV2 rep/cap plasmid, and an adenovirus helper plasmid, which encodes adenoviral genes necessary for AAV2 particle production. Medium was replaced the following morning, and 2 to 3 days post-transfection cells were harvested and lysed by mechanical disruption in a deoxycholate containing buffer to release vector particles. Cellular DNA, RNA and residual plasmid DNA were then digested with 100U/mL of Benzonase (Merck) for 3 hours at 37°C. AAV2-NGF-wPRE vector was purified by filtration and affinity (heparin) chromatography followed by dialysis in an isotonic saline formulation buffer (FB; 2mM MgCl2 in PBS) using a Slide-A-Lyzer 10,000 MWCO (Pierce). CERE-110 vector was purified by filtration, affinity (heparin) and ion exchange chromatography. CERE-110 vector particles were then subjected to centrifugal filtration on 50 KDa filter units (Millipore) and concentrated into FB. Upon concentration the bulk product was sterilized-filtered on a 0.2μm filter unit and then filled into 0.5 mL polypropylene cryovials. Vector titers were determined by dot blot or quantitative PCR using specific primers and are expressed in vector genomes (vg).
Experimental subjects
One hundred and thirty young male Sprague Dawley rats and twenty-one 21-month old male Fischer 344 rats (Harlan) were used in these studies. Rats were provided food and water ad libitum and maintained on a 12-hour light/dark cycle. All experiments were conducted in accordance with the guidelines of the Office of Laboratory Animal Welfare and the Ceregene, Inc. Institutional Animal Care and Use Committee.
Experimental details
For all surgeries, rats were anesthetized with a mixture consisting of xylazine (3.25mg/kg), acepromazine (0.62mg/kg) and ketamine (62.5mg/kg), and placed in a stereotaxic frame (Stoelting). The skull was exposed and burr holes made with a Dremel drill above the injection sites. Vector or formulation buffer (FB) control was delivered via a 10 μL Hamilton syringe attached to a 26 gauge beveled stainless steel needle. Injections were made at a rate of 0.5 μL/minute, via an injection pump (Stoelting). Following each injection, the needle was left at the injection site for 1 minute, retracted 1–3 mm, and then held in place an additional minute, followed by removal from the brain. Details of the injection parameters for each experiment (i.e. target brain structure, stereotaxic injection coordinates, injection volume, injection dose) are summarized in Table 1.
Table 1.
Summary of Injection Parameters used in Different Experiments
| Experiment | Target BFCN Structure | Injection Site Coordinates | Injection Volume (μL) | AAV2-NGF Dose per Hemisphere (vg) | ||
|---|---|---|---|---|---|---|
| AP1 (mm) | ML1 (mm) | DV (mm) | ||||
| Fimbria-Fornix Lesion Model | MS | 0.3 | −0.7 | −8.01 | 2 | 5.2×109 |
| Dose Range Testing | NBM | −1.5 | ±2.5 | −8.01 | 0.5, 1 or 2 | 8.8×107to 1.1×1010 |
| Aged Rat Model | NBM | −1.5 | −2.5 | −7.02 | 1 | 1×108 |
| −2.5 | −3.8 | −6.82 | 1 | |||
| Long-term Young Rat Model | NBM | −1.5 | ±2.5 | −8.01 | 1 | 1×108; 2×109 |
| −2.5 | ±3.8 | −7.21 | 1 | |||
Measured from bregma;
Relative to dura
BFCN = Basal Forebrain Cholinergic neuron; MS = Medial Septum; NBM = Nucleus Basalis of Meynert; AP = anterior-posterior; ML = medial-lateral; DV = dorsal-ventral; vg = vector genomes
Fimbria-fornix lesion model experiment
In the fimbria-fornix lesion model experiment, 4 male Sprague Dawley rats were injected with 2 μL of AAV-NGF-wPRE (5.2×109 vg) in the right hemisphere medial septum. Nine days later, an ipsilateral aspirative lesion of the fimbria-fornix was performed on vector injected animals as well as 4 control (lesion only) animals. At 2 weeks post-lesion, all animals were sacrificed for histological analysis.
CERE-110 dose-range testing experiment
In the dose-range testing experiment, 38 male Sprague Dawley rats were injected with a range of doses of CERE-110 or FB control in the NBM. Animals for NGF immunohistochemical analysis were injected in the right hemisphere NBM with one of the following CERE-110 doses: 1.8×108 vg, 2.7×108 vg, 5.3×108 vg, 1.1×109 vg, 1.8×109 vg, or 5.3×109 vg (n = 3 animals/dose). Animals for NGF ELISA analysis were injected in the NBM bilaterally with one of the following CERE-110 doses: 8.8×107 vg, 1.8×108 vg, 2.7×108 vg, 3.5×108 vg, 5.3×108 vg, 8.8×108 vg, 1.1×109 vg, 1.8×109 vg, 2.7×109 vg, 3.5×109 vg, 5.3×109 vg, or 1.1×1010 vg (n = 3 NBM/dose). Two additional animals were injected bilaterally with 1 μL of FB control. All animals were sacrificed at 2 weeks post injection for NGF analysis by immunohistochemistry or ELISA.
Aged rat model experiment
Twenty-one 21-month old male Fischer 344 rats were injected with 1×108vg CERE-110 (11 animals) or FB control (10 animals) in the right hemisphere NBM. During the course of the study, 4 animals were found dead and 1 animal was euthanized 2 days prematurely due to his moribund condition. Four of the 5 animals had evidence of neoplasia and 1 animal had evidence of chronic renal failure. Both of these are common causes of spontaneous death in aged rats. Of the 5 animals that died or were sacrificed prematurely, 2 were in the formulation buffer control group and 3 were in the CERE-110 treated group. Mortality rates were not significantly different between treatment and control groups (2/10 vs. 3/11, X2 test, P>0.05). After a 3 month post-injection period, all remaining animals were sacrificed for histological analysis.
Long-term experiment
A total of 84 young male Sprague Dawley rats were injected with FB control or one of 2 different doses of CERE-110 (1×108or 2×109vg per NBM) bilaterally in the NBM. Six animals per group were sacrificed at 3 and 6 months post-injection and 8 animals per group were sacrificed at 9 and 12 months post injection for histological analysis. One animal in the 12-month FB control group died prematurely, due to spontaneously occurring pulmonary carcinoma.
Histology
Perfusion and tissue processing for immunohistochemistry
Animals were overdosed with an anesthetic cocktail and transcardially perfused with ice-cold 0.9% saline followed by 2% paraformaldehyde (PFA) with 0.2% parabenzoquinone (PBQ). Brains were removed, post-fixed for 2 hours in 2% PFA with 0.2% PBQ, and cryoprotected in 30% sucrose at 4°C. Brains were coronally sectioned on a sliding microtome at 40 μm and sections stored in cryoprotectant at −20°C.
Immunohistochemistry
Immunohistochemistry was performed on separate 1-in-6 series of sections using antibodies raised against NGF (rabbit anti-NGF, used at 1:1000, a gift from Dr. J. Conner, UCSD, La Jolla, CA) or ChAT (goat polyclonal anti-ChAT) used at 1:500, Chemicon). Free-floating sections through the forebrain were blocked with 5% horse serum in TBS/0.25% Triton X-100 for 1–2 hours and then incubated overnight at 4°C with the primary antibody. This was followed by incubation with biotinylated secondary antibody (donkey anti-rabbit (used at 1:500, Jackson ImmunoResearch) for NGF and horse anti-goat (used at 1:333, Vector Laboratories) for ChAT for 3 hours at room temperature. Sections were visualized with avidin-biotinylated peroxidase complex procedure (Vector Laboratories) using 3,3-diaminobenzidine (DAB) as the chromogen. After sections were mounted onto glass slides they were dehydrated and coverslipped with DPX mounting media.
Nissl staining and AChE histochemistry
Successful lesion of the fimbria-fornix pathway was confirmed by examination of cresyl violet stained sections through the lesioned area and by loss of acetylcholinesterase (AChE) staining in the medial septum cholinergic neuron target region, the hippocampus. Cresyl violet stain was performed by immersing sections in 0.2% cresyl violet solution followed by dehydration with ethanol and xylene and coverslipping with DPX. For AChE histochemistry, sections were incubated in a solution containing 24% Sodium Sulfate (Na2SO4), 0.15% Glycine, 0.002% Copper Sulfate (CuSO4), 0.12% Acethylthiocholine iodide and 0.000037% tetraisopropylpyrophosphosphoramide (Iso-OMPA; Sigma) in a 0.05M maleate buffer overnight at 37°C. After rinsing with 20% Na2SO4 followed by 10% Na2SO4, staining was visualized using 4% (NH4)2S for 1 minute. Sections were then rinsed with distilled water and incubated in 10% formalin for 20 minutes. Slides were dehydrated and coverslipped with DPX.
Quantitation of NGF by ELISA
At scheduled sacrifice, brains were collected fresh and flash-frozen on dry ice. Three 1-mm thick hemi-coronal slices centered on the injection site were collected from each hemisphere. Each slice was homogenized in a buffer (Phosphate Buffer, pH 7.0, 400mM NaCl, 0.1% Triton X-100, 5mM EDTA and 0.5% BSA) at a tissue concentration of 10μL of buffer per 1mg tissue. A protease inhibitor cocktail (Sigma) was added at 1μL per 20mg tissue. Quantification of NGF was performed using the NGF Emax Immunoassay System kit (Promega), which is specific for NGF, exhibiting typically less than 3% cross-reactivity with other neurotrophic factors (Promega, 2007). Each sample was analyzed in duplicate at two dilutions (1/20 and 1/50) and optical density was measured at 450nm using an optical plate reader (Versamax, Molecular Devices). All groups were compared in the same assay to reduce the influence of interassay variances; measured NGF-levels were uncorrected for recovery of NGF-spiked samples. Data were analyzed using SOFTmax PRO 4.0 and the highest value of the three hemi-coronal sections from each hemisphere was reported.
Morphometric analysis
Using unbiased stereological techniques (optical fractionator, StereoInvestigator software v5.0, MicroBrightField Inc.) an estimation of the total number and size of ChAT positive cells was determined on a 1-in-6 series of sections through the basal forebrain medial septum (fimbria-fornix lesion model experiment) or NBM (aged rat model and long-term experiments). Quantitative analyses were performed by an individual blind with respect to treatment group.
Statistical analysis
Statistical analyses were performed using SigmaStat v2.03 (SPSS). Preliminary analyses were performed to verify that the data were normally distributed. Differences between groups were assessed using Student’s t-test or analysis of variance (ANOVA) followed by Tukey multiple comparison post-hoc testing, when appropriate. A significant difference between groups was established if p < 0.05.
RESULTS
AAV2-NGF prevents degeneration of BFCNs in the rat fimbria-fornix lesion model
Complete lesioning of the fimbria-fornix pathway was confirmed in all animals by examination of cresyl violet stained sections covering the lesion area and by loss of acetylcholinesterase staining in the axonal target region of the medial septal cholinergic neurons, the hippocampus (data not shown). Robust expression of NGF protein was observed in the medial septum of all AAV2-NGF injected animals, as determined by NGF immunohistochemistry (Figure 1A). As shown in Figure 1B and C, in uninjected rats the lesion reduced the number of ChAT-positive cells in the medial septum to 26.1 ± 0.5 % of the control (unlesioned) side, and diminished the size of remaining ChAT-positive cells to 81.9 ± 1.6 % of control side (Figure 1D). AAV2-NGF significantly prevented the loss of ChAT-positive BFCNs (83.7 ± 1.0% of control side; p < 0.05 compared with lesion-only animals; Figure 1B and C) and completely prevented the reduction in cell size (99.2 ± 2.4% of control side; p < 0.05 compared with lesion-only animals; Figure 1D). These results demonstrate that AAV2-mediated delivery is effective in providing biologically active NGF protein to BFCNs and yielded the characteristic trophic response, in a standard well-characterized model of cholinergic neuronal degeneration.
Figure 1. AAV2-NGF is neuroprotective in the rat fimbria-fornix lesion model of basal forebrain cholinergic neuronal degeneration.
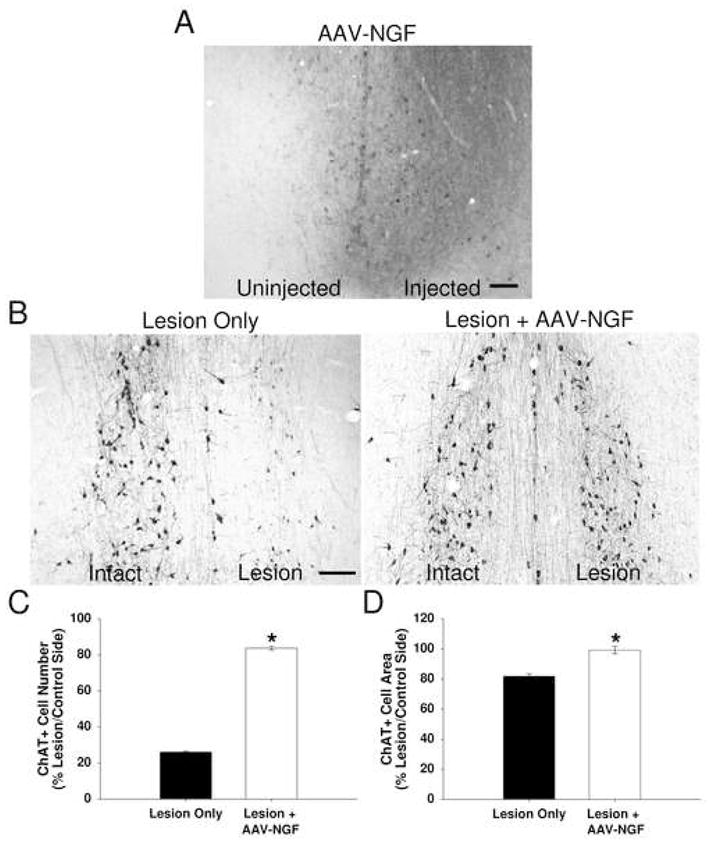
(A) Robust NGF protein expression, as detected by NGF immunohistochemistry, is evident in the AAV2-NGF injected side of the medial septum. (B) Following unilateral lesion of the fimbria-fornix pathway of the rat brain, ChAT immunostained cells in the medial septum (MS) are shown in uninjected control and AAV2-NGF injected brains. Lesion and vector injection were performed on the right side. (C) The number of ChAT-positive medial septum neurons is significantly increased in AAV2-NGF injected brains compared to controls. (D) The cell area of ChAT-positive medial septum neurons is also significantly increased in AAV2-NGF injected brains compared to controls. Error bars represent standard error of the mean (SEM). *p < 0.05. Scale bars = 100 um.
CERE-110 results in targeted and controlled NGF delivery to the NBM
Figure 2A shows representative images of NGF immunohistochemistry, illustrating the resulting NGF protein expression in the NBM from a range of CERE-110 doses. A noticeable increase in resulting NGF protein distribution was evident when the dose of CERE-110 was increased. In addition, a qualitative difference in the intensity of NGF immunohistochemical staining between CERE-110 dose groups was evident such that the intensity of staining increased, both intracellularly (closed arrows) and extracellularly (open arrows), as the dose of CERE-110 delivered to the NBM increased. Analysis of NGF immunohistochemistry over the range of doses tested also indicated that it was possible to achieve targeted and localized delivery of NGF to the rat NBM. Review of the NGF immunohistochemical data and the anatomy of the rat NBM indicated that a dose of 1×108 vg/NBM was optimal for providing as much exposure of the rat NBM to vector-derived NGF as possible without having significant diffusion of protein to brain regions distal to the NBM. This dose was operationally defined as the ‘optimal’ dose of CERE-110 for the rat NBM. Quantification of NGF protein levels by ELISA confirmed that the amount of NGF produced following CERE-110 administration could be controlled by altering delivered vector dose (Figure 2B). Regression analysis indicated that parenchymal brain NGF concentration was significantly positively correlated with the dose of CERE-110 injected (R2 = 0.60, p < 0.001). The defined optimal dose of CERE-110 (1×108 vg/NBM) corresponds to an NGF dose of approximately 6 ng/mg tissue, as measured by ELISA. Taken together, these results demonstrate that the amount and distribution of NGF protein delivered via CERE-110 is dependent upon the vector dose injected and that it is possible to achieve targeted NGF expression restricted to but covering the target NBM.
Figure 2. Dose-related expression of NGF protein following CERE-110 delivery to the NBM of the young rat.
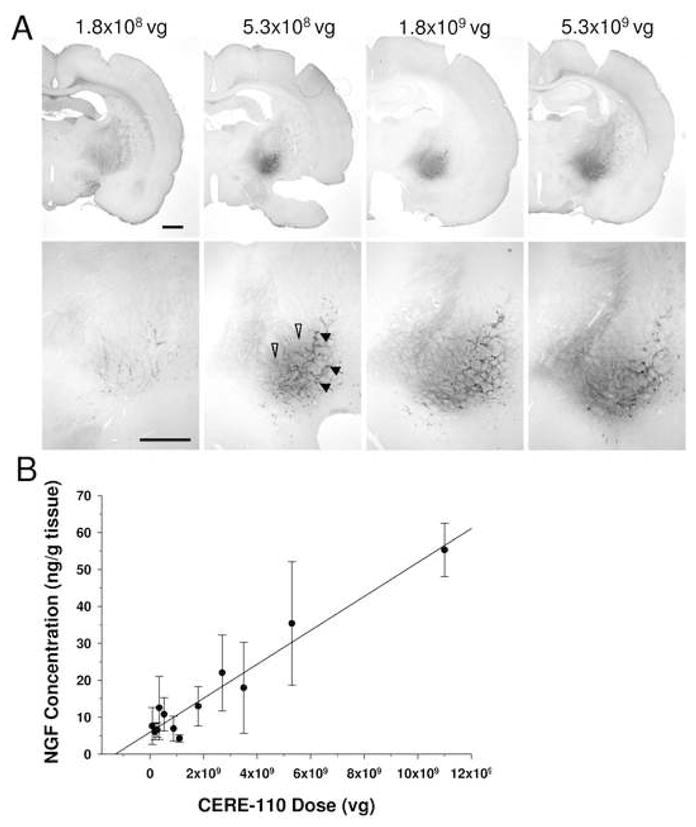
(A) Immunohistochemical staining for NGF shows increased protein expression both intracellularly (closed arrows) and extracellularly (open arrows) with escalating doses of CERE-110 delivered to the NBM of the rat brain. Injection volume was held constant at 1 μL for all injections and CERE-110 vector dose was manipulated by varying vector concentration. Scale bar = 1 mm. As shown by ELISA (B), brain tissue NGF concentration is a function of the number of vector genomes of CERE-110 delivered to the NBM (R2 = 0.60, P<0.001). Data are expressed as ng NGF protein/gram wet brain weight; Error bars represent standard error of the mean (SEM). vg = vector genomes.
CERE-110 restores the cholinergic phenotype of BFCNs in the aged rat
The efficacy of CERE-110 was also tested in the aged (21-month old Fischer 344) rat. As determined from the rat dose-range testing study described above, the ‘optimal’ dose of CERE-110 (1×108 vg/NBM) was used. Injections of 1×108 vg CERE-110 into the NBM resulted in robust expression of NGF protein over the NBM target brain region, as assessed by NGF immunohistochemistry (Figure 3B). As intended with this dose, NGF immunohistochemistry was generally confined to the target NBM and did not spread to the ventricular boundary, where NGF could potentially leak into the CSF. As shown in Figure 4, in control rats, FB injection did not have a significant effect on the number or size of ChAT-positive cells in the NBM. In FB injected animals, ChAT-positive cell number was 102.7 ± 7.7 % of the number on the uninjected (contralateral) side NBM, and cell size was 99.9 ± 2.2 % of the uninjected side (Figure 4B and C). In contrast, injection of CERE-110 resulted in trophic effects on NBM neurons, as measured by ChAT-positive cell number (135.9 ± 3.7 % of uninjected side; p < 0.001 compared with FB injected; Figure 4A and B) and ChAT-positive cell size (167.9 ± 5.1 % of uninjected side; p < 0.005 compared with FB injected; Figure 4A and C). These results demonstrate that CERE-110 is efficacious in providing sustained production of biologically active NGF and imparting significant trophic support to cholinergic neurons in the aged brain.
Figure 3. NGF expression at three months following CERE-110 delivery to the NBM of the aged rat brain.

NGF immunohistochemical signal in the FB control injected NBM (A) is similar in pattern and intensity to endogenous NGF expression normally seen in uninjected brains. In the CERE-110 injected aged rat brain (B), robust expression of NGF protein above endogenous levels is observed, providing NGF coverage of the target NBM and diffuse expression in the cortex, in the region of terminal fields of NBM neurons. Scale bar = 1 mm. vg = vector genomes.
Figure 4. Bioactivity of CERE-110 on NBM basal forebrain cholinergic neurons in the aged rat.

(A) ChAT-positive immunostained cells in the NBM in control FB injected and CERE-110 injected aged rat brains. Injection of FB or CERE-110 was performed on the right side, while the left side remained uninjected for comparison purposes. (B) The number of aged rat NBM neurons positively stained for ChAT is significantly increased in CERE-110 injected brains compared to FB injected controls. (C) The cell area of aged rat NBM ChAT-immunopositive neurons is significantly increased in CERE-110 injected brains compared to FB injected controls. Error bars represent standard error of the mean (SEM). *p < 0.05. Scale bar = 100 um. vg = vector genomes.
CERE-110 produces the characteristic hypertrophic response of BFCNs in the young rat
It has previously been shown that NGF protein delivery to the NBM results in hypertrophy of cholinergic neurons in young rodents (Pizzo, et al., 2002). In order to test whether CERE-110 has similar effects and whether these effects persist over long time periods, an experiment was conducted in which CERE-110 was injected to the NBM in young, healthy rats and resulting transgene expression and potential cholinergic neuron hypertrophy assessed at 3, 6, 9 and 12 months post-administration. In FB control animals at 3, 6, 9 and 12 months, endogenous NGF expression was detected in the NBM and was not greater than NGF levels normally seen in uninjected rat NBM using this assay (data not shown). Substantial NGF expression above endogenous levels was detected in the NBMs of all CERE-110 injected animals at 3, 6, 9 and 12 months following CERE-110 administration (Figure 5). The distribution and degree of immunohistochemical signal in high dose (2×109 vg/NBM) animals (Figure 5, bottom panels) far exceeded that in ‘optimal’ dose (1×108 vg/NBM) animals (Figure 5, top panels), and was evident in many of the natural targets of NBM cells (i.e., cortex, amygdala, thalamus). The extent of NGF immunohistochemical signal in optimal dose animals was largely confined to the NBM, with a small amount observed in the cortex, a natural target of NBM neurons (Figure 5, top panels). NGF immunohistochemical signals were similar across all time points at both dose levels (Figure 5), indicating that transgene expression was sustained and stable up to 1 year after CERE-110 delivery.
Figure 5. NGF expression at 3, 6, 9 and 12 months following CERE-110 delivery to the NBM of the young rat.
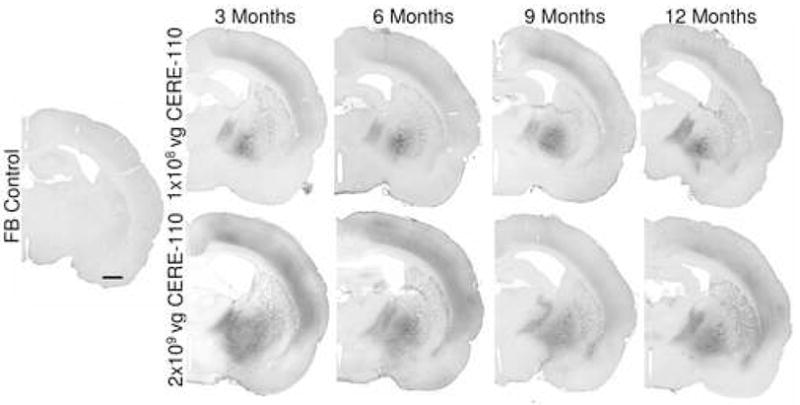
NGF immunostained cells and secreted protein is shown in (A) control formulation buffer (FB) injected rats at 3 months, (B) optimal dose (1 × 108 vg/NBM) CERE-110 injected rats at 3, 6, 9 and 12 months, and (C) high dose (2×109 vg/NBM) CERE-110 injected rats at 3, 6, 9 and 12 months (C). Scale bar = 1 mm. vg = vector genomes.
A consistent significant difference in cholinergic NBM cell size between FB control injected and CERE-110 injected groups was observed at all time points examined (Figure 6B). A significant difference in cell size was observed at 3 months (means ± SEM: FB control = 174 ± 8 μm2; optimal dose CERE-110 = 298 ± 9 μm2; high dose CERE-110 = 321 ± 6 μm2; one-way ANOVA, p < 0.001), 6-months (FB control = 178 ± 8 μm2; optimal dose CERE-110 = 307 ± 8 μm2; high dose CERE-110 = 320 ± 13 μm2; one-way ANOVA, p < 0.001), 9-months (FB control = 168 ± 6 μm2; optimal dose CERE-110 = 279 ± 10 μm2; high dose CERE-110 = 307 ± 10 μm2; one-way ANOVA, p < 0.001), and 12-months (FB control = 159 ± 3 μm2; optimal dose CERE-110 = 288 ± 9 μm2; high dose CERE-110 = 287 ± 11 μm2; one-way ANOVA, p < 0.001). Post-hoc analyses indicated a significant difference in cholinergic NBM cell size between FB control and CERE-110 high dose groups and FB control and CERE-110 optimal dose groups (Tukey least significant difference, p’s < 0.001) and no difference between CERE-110 optimal and high dose groups at all time points. At each of 3, 6, 9 or 12 months, no significant differences in cholinergic NBM cell number were observed between groups (p > 0.05; Figure 6A). NBM cholinergic cell number was equivalent between groups at 3 months (means ± SEM: FB control = 2721 ± 177; optimal dose CERE-110 = 2515 ± 152; high dose CERE-110 = 2624 ± 301; one-way ANOVA, p > 0.05), 6 months, (FB control = 3455 ± 237; optimal dose CERE-110 = 4081 ± 189; high dose CERE-110 = 3696 ± 390; one-way ANOVA, p > 0.05), 9 months (FB control = 4037 ± 292; optimal dose CERE-110 = 4704 ± 201; high dose CERE-110 = 4148 ± 218; one-way ANOVA, p > 0.05), and 12 months (FB control = 3596 ± 168; optimal dose CERE-110 = 3663 ± 215; high dose CERE-110 = 3863 ± 176; one-way ANOVA, p > 0.05). Thus, CERE-110 delivery resulted in long-term biologically active NGF transgene product that had a consistent, stable trophic effect on basal forebrain cholinergic neurons of the NBM for up to 1 year, with no cell loss.
Figure 6. Increased ChAT positive cell area but no change in cell number following CERE-110 delivery to the NBM of the young rat.

(A) NBM ChAT-positive cell number at 3 months, 6 months, 9 months, and 12 months following delivery of formulation buffer control and optimal and high dose CERE-110 to the NBM. (B) NBM ChAT-positive cross-sectional cell area at 3 months, 6 months, 9 months and 12 months following FB control and optimal and high dose CERE-110 delivery to the. Error bars represent standard error of the mean (SEM). *Significantly different compared to FB control (p < 0.05).
DISCUSSION
Gene delivery of the neurotrophic factor NGF to degenerating and dying basal forebrain cholinergic neurons is being developed as a potential treatment for mild to moderate Alzheimer’s disease. In support of this concept, research over the past three decades has supported the importance of basal forebrain cholinergic neurons in the cognitive symptoms that characterize the earlier stages of this disease (Bartus, 2000). Similarly, research published over the past two decades has shown that BFCNs respond positively when they are supplied with exogenous NGF, often returning function to aged or injured neurons and enabling these neurons to withstand significant neurodegenerative perturbations. Studies reported herein clearly indicate that AAV2 vector-mediated delivery is effective in providing NGF protein to BFCNs, with in vivo biological consequences, in the fimbria-fornix lesion model, the aged rat model of cholinergic neuronal degeneration, and bioactive neuronal responses in young animals.
In the clinic, the goal is to expose as much of the human NBM as possible to vector-derived NGF, without significant diffusion of NGF to distal brain regions, especially CSF-containing ventricles and meninges. Studies were performed in which various doses of CERE-110 were injected into the rat NBM, and results indicate that NGF transgene delivery to the targeted brain region is reliable and accurate, and that NGF transgene distribution can be controlled by altering CERE-110 dose. We demonstrate that it is possible to achieve restricted NGF expression limited to but covering the target brain region, and that a dose of 1×108 vg/NBM is optimal for providing as much exposure of the rat NBM to vector-derived NGF as possible without having significant diffusion of protein to brain regions distal to the NBM or the CSF. Results from animals examined at longer time periods of 3, 6, 9 and 12 months after CERE-110 delivery indicate that NGF transgene expression is stable and sustained at all time points, with no loss or build-up of protein over the long-term.
In addition, we provide evidence that CERE-110 mediated delivery of NGF protein provides neuroprotective or neurorestorative effects in several rodent models of BFCN degeneration directly relevant to Alzheimer’s disease. Multiple studies have shown that fimbria-fornix lesion-induced degeneration of BFCNs can be prevented by treatment of the neuronal cell somata with NGF (Hefti, 1986; Williams et al., 1986; Kromer, 1987; Rosenberg et al., 1988; Tuszynski et al., 1990; Koliatsos, et al., 1991a and 1991b). Results in the fimbria-fornix lesion model of cholinergic neuronal degeneration demonstrate that the use of an AAV2-based vector is an effective means of delivering NGF protein to prevent degeneration of BFCNs. Results from this experiment are consistent with similar published reports of in vivo neuroprotection of BFCNs after AAV vector-mediated delivery of NGF in this model (Mandel et al., 1999; Wu et al., 2003; Wu et al., 2005). In aged rats, BFCNs are not lost, but they both shrink in size and lose expression of phenotypic markers such as p75, TrkA, and ChAT (Smith et al., 1993; Greferath et al., 2000; Stemmelin et al., 2000; Saragovi, 2005). These pathologic changes, also observed in the Alzheimer’s disease brain, can be reversed in aged animals by administration of NGF. This response has been linked to improved cognitive function in aged rats (Fischer et al., 1987; Fischer et al., 1991; Chen and Gage, 1995; Martinez-Serrano et al., 1996), as well as enhanced cortical reinnervation in aged monkeys (Conner et al., 2001). Results in the aged rat demonstrate that CERE-110, delivered at the operationally defined optimal dose of 1×108 vg/NBM, is an effective means of delivering NGF to provide significant trophic support to aging cholinergic neurons, as assessed by enhanced cholinergic cell size and cell number in the NBM of 24-month old rats. This dose was also effective in producing a biological response (cell hypertrophy) in NBM neurons in young rats without adversely affecting NBM cholinergic cell numbers at each of 3, 6, 9 and 12 months following vector injection. The biological response provided by the optimal dose of CERE-110 (1×108 vg/NBM) was not significantly less than the response produced by a 20x greater dose of CERE-110 (2×109 vg/NBM). This result is consistent with the concept that a maximal response occurs at the dose that exposes a preponderance of the cholinergic neurons of the NBM to NGF protein, as assessed by immunohistochemistry, and that increasing the CERE-110 dose above the operationally defined optimal dose results in no greater biological effect. Notably, in this study, NBM cellular hypertrophy was not associated with adverse behavioral changes in young rats as assayed by daily cage side observations or the functional observation battery (Moser, 2000; Bishop et al., manuscript in preparation).
Based on these proof-of-concept data in animal models of cholinergic neuronal degeneration, one can test the hypothesis that CERE-110 delivered to the basal forebrain region containing the NBM in subjects with Alzheimer’s disease may have a positive effect on BFCN function and cognitive functions regulated by the cholinergic system. BFCN loss has been linked to cognitive dysfunction in Alzheimer’s disease, and further, is presumed to contribute to the progression of Alzheimer’s disease symptoms. This hypothesis has been validated by the many controlled clinical trials that have demonstrated the efficacy of drugs that marginally augment CNS cholinergic function in Alzheimer’s disease (Hake, 2001). The first four drugs approved by the Food and Drug Administration for Alzheimer’s disease, the cholinesterase inhibitors (ChEIs), act through the common mechanism of prolonging cholinergic synaptic signaling. CERE-110-mediated NGF delivery might show cognitive benefit similar to the ChEIs, since NGF is predicted to effectively increase availability of acetylcholine (ACh) in the cerebral cortex. However, CERE-110 may provide significant additional cognitive benefit by: 1) preventing the death of BFCNs, 2) increasing the vitality of remaining BFCNs, and 3) generating more ACh in the cortex than do ChEIs (especially since the systemically administered ChEIs have significant dose-limiting toxicity associated with peripheral cholinesterase inhibition). Neither of the first two of these benefits of NGF has been clearly associated with the use of ChEIs. Moreover, preserving and revitalizing the BFCNs themselves would be expected to preserve and restore the natural spatial and temporal patterns of ACh transmission in the cortex, which ChEIs do not.
In addition to the loss of BFCNs, other neuroanatomical changes occur in Alzheimer’s disease. These include the accumulation of amyloid plaques and neurofibrillary tangles, as well as loss of other neuronal populations. Currently it is unclear how the various factors related to those anatomical changes interact, and to what extent those changes might also contribute to the cognitive dysfunction in Alzheimer’s disease (Mesulam, 1999; Bartus, 2000; Blusztajn and Berse, 2000; Hardy and Selkoe, 2002; Isaacson et al., 2002). Nor is it known how bolstering cholinergic function might affect those pathogenic processes, although use of ChEIs does not appear to significantly alter the course of the underlying disease. It is anticipated that preventing the loss of, and/or augmenting the function of, remaining BFCNs will improve symptoms of Alzheimer’s disease and reduce cognitive decline. This could occur directly if the loss of BFCNs significantly contributes to the progression of Alzheimer’s disease, and/or indirectly if the loss of BFCNs contributes significantly to other aspects of Alzheimer’s disease, such as accumulation of β-amyloid, that may be relevant to disease progression. Towards testing these hypotheses, clinical testing of CERE-110 is currently underway. A small Phase 1 clinical study to examine the safety of CERE-110 delivered to the NBM in Alzheimer’s disease patients is ongoing (Arvanitakis et al., 2007), and a double-blind, sham surgery-controlled, Phase II study to further examine the efficacy and safety of CERE-110 is planned.
Acknowledgments
This work was supported by NINDS grants 1R43NS44652-1 and 1R43NS44652-2 (K.B.). We thank Lamar Brown, Chris Herzog, Brian Kruegel, Shokry Lawandry, Denise Rius, and Alistair Wilson for expert technical assistance. We are extremely grateful to Jim Conner (UCSD) for assistance with the fimbria-fornix lesion technique and for providing NGF antibodies for immunohistochemistry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arvanitakis Z, Tuszynski MH, Potkin S, Bartus RT, Bennett D. A Phase 1 Clinical Trial of CERE-110 (AAV-NGF) Gene Delivery in Alzheimer’s Disease; American Academy of Neurology Annual Meeting; Boston, MA. 2007. [Google Scholar]
- Bartus RT. On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp Neurol. 2000;163(2):495–529. doi: 10.1006/exnr.2000.7397. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Johnson HR. Short-term memory in the rhesus monkey: disruption from the anti-cholinergic scopolamine. Pharmacol Biochem Behav. 1976;5(1):39–46. doi: 10.1016/0091-3057(76)90286-0. [DOI] [PubMed] [Google Scholar]
- Baxter MG, Chiba AA. Cognitive functions of the basal forebrain. Curr Opin Neurobiol. 1999;9(2):178–83. doi: 10.1016/s0959-4388(99)80024-5. [DOI] [PubMed] [Google Scholar]
- Bierer LM, Haroutunian V, Gabriel S, Knott PJ, Carlin LS, Purohit DP, Perl DP, Schmeidler J, Kanof P, Davis KL. Neurochemical correlates of dementia severity in Alzheimer’s disease: relative importance of the cholinergic deficits. J Neurochem. 1995;64(2):749–60. doi: 10.1046/j.1471-4159.1995.64020749.x. [DOI] [PubMed] [Google Scholar]
- Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev. 2006;25(1):CD005593. doi: 10.1002/14651858.CD005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesch A, Conner J, Pfeifer A, Gasmi M, Ramirez A, Britton W, Alfa R, Verma I, Tuszynski M. Regulated lentiviral NGF gene transfer controls rescue of medial septal cholinergic neurons. Mol Ther. 2005;11(6):916–25. doi: 10.1016/j.ymthe.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Blusztajn JK, Berse B. The cholinergic neuronal phenotype in Alzheimer’s disease. Metab Brain Dis. 2000;15(1):45–64. doi: 10.1007/BF02680013. [DOI] [PubMed] [Google Scholar]
- Bucci DJ, Holland PC, Gallagher M. Removal of cholinergic input to rat posterior parietal cortex disrupts incremental processing of conditioned stimuli. J Neurosci. 1998;18(19):8038–46. doi: 10.1523/JNEUROSCI.18-19-08038.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns A, O’Brien J. Clinical practice with anti-dementia drugs: a consensus statement from British Association for Psychopharmakology. J Psychopharmacol. 2006;20:732–55. doi: 10.1177/0269881106068299. [DOI] [PubMed] [Google Scholar]
- Chen KS, Gage FH. Somatic gene transfer of NGF to the aged brain: behavioral and morphological amelioration. J Neurosci. 1995;15(4):2819–25. doi: 10.1523/JNEUROSCI.15-04-02819.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KS, Tuszynski MH, Gage FH. Role of neurotrophic factors in Alzheimer’s disease. Neurobiol Aging. 1989;10(5):545–6. doi: 10.1016/0197-4580(89)90124-3. discussion 552–3. [DOI] [PubMed] [Google Scholar]
- Conner JM, Darracq MA, Roberts J, Tuszynski MH. Nontropic actions of neurotrophins: subcortical nerve growth factor gene delivery reverses age-related degeneration of primate cortical cholinergic innervation. Proc Natl Acad Sci U S A. 2001;98(4):1941–6. doi: 10.1073/pnas.98.4.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JD, Salehi A, Delcroix JD, Howe CL, Belichenko PV, Chua-Couzens J, Kilbridge JF, Carlson EJ, Epstein CJ, Mobley WC. Failed retrograde transport of NGF in a mouse model of Down’s syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc Natl Acad Sci U S A. 2001;98(18):10439–44. doi: 10.1073/pnas.181219298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Price DL, DeLong MR. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science. 1983;219(4589):1184–90. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- Crutcher KA, Scott SA, Liang S, Everson WV, Weingartner J. Detection of NGF-like activity in human brain tissue: increased levels in Alzheimer’s disease. J Neuroscience. 1993;13(6):2540–550. doi: 10.1523/JNEUROSCI.13-06-02540.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day-Lollini PA, Stewart GR, Taylor MJ, Johnson RM, Chellman GJ. Hyperplastic changes within the leptomeninges of the rat and monkey in response to chronic intracerebroventricular infusion of nerve growth factor. Exp Neurol. 1997;145(1):24–37. doi: 10.1006/exnr.1997.6448. [DOI] [PubMed] [Google Scholar]
- Donello JE, Loeb JE, Hope TJ. Woodchuck hepatitis virus contains a tripartite posttranscriptional regulatory element. J Virol. 1998;72(6):5085–92. doi: 10.1128/jvi.72.6.5085-5092.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksdotter Jonhagen M, Nordberg A, Amberla K, Backman L, Ebendal T, Meyerson B, Olson L, Seiger Shigeta M, Theodorsson E, et al. Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 1998;9(5):246–57. doi: 10.1159/000017069. [DOI] [PubMed] [Google Scholar]
- Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol Cell Neuroscience. 2001;18:210–220. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- Fischer W, Bjorklund A, Chen K, Gage FH. NGF improves spatial memory in aged rodents as a function of age. J Neurosci. 1991;11(7):1889–906. doi: 10.1523/JNEUROSCI.11-07-01889.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W, Wictorin K, Bjorklund A, Williams LR, Varon S, Gage FH. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature. 1987;329(6134):65–8. doi: 10.1038/329065a0. [DOI] [PubMed] [Google Scholar]
- Furey ML, Pietrini P, Haxby JV. Cholinergic enhancement and increased selectivity of perceptual processing during working memory. Science. 2000;290(5500):2315–9. doi: 10.1126/science.290.5500.2315. [DOI] [PubMed] [Google Scholar]
- Greferath U, Bennie A, Kourakis A, Barrett GL. Impaired spatial learning in aged rats is associated with loss of p75-positive neurons in the basal forebrain. Neurosci. 2000;100(2):363–73. doi: 10.1016/s0306-4522(00)00260-8. [DOI] [PubMed] [Google Scholar]
- Hake AM. Use of cholinesterase inhibitors for treatment of Alzheimer disease. Cleve Clin J Med. 2001;68(7):608–9. 613–4, 616. doi: 10.3949/ccjm.68.7.608. [DOI] [PubMed] [Google Scholar]
- Hansen RA, Gartlehner G, Lohr KN, Kaufer DI. Functional outcomes of drug treatment in Alzheimer’s disease. A systematic review and meta-analysis. Drugs Aging. 2007;24(2):155–67. doi: 10.2165/00002512-200724020-00007. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hefti F, Armanini MP, Beck KD, Caras IW, Chen KS, Godowski PJ, Goodman LJ, Hammonds RG, Mark MR, Moran P, et al. Development of neurotrophic factor therapy for Alzheimer’s disease. Ciba Found Symp. 1996;196:54–63. doi: 10.1002/9780470514863.ch5. discussion 63–9. [DOI] [PubMed] [Google Scholar]
- Hefti F, Schneider LS. Nerve growth factor and Alzheimer’s disease. Clin Neuropharmacol. 1991;14(Suppl 1):S62–76. doi: 10.1097/00002826-199114001-00008. [DOI] [PubMed] [Google Scholar]
- Hefti F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J Neurosci. 1986;6(8):2155–62. doi: 10.1523/JNEUROSCI.06-08-02155.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellweg R, Gericke CA, Jendroska K, Hartung H-D, Cervos-Navarro J. NGF content in the cerebral cortex of non-demented patients with amyloid-plaques and in symptomatic Alzheimer’s disease. Int J Devl Neuroscience. 1998;16(78):787–94. doi: 10.1016/s0736-5748(98)00088-4. [DOI] [PubMed] [Google Scholar]
- Hellweg R, Fischer W, Hock C, Gage FH, Bjorklund A, Thoenen H. Nerve growth factor levels and choline acetyltransferase activity in the brain of aged rats with spatial memory impairments. Brain Research. 1990;537:123–30. doi: 10.1016/0006-8993(90)90348-f. [DOI] [PubMed] [Google Scholar]
- Isacson O, Seo H, Lin L, Albeck D, Granholm AC. Alzheimer’s disease and Down’s syndrome: roles of APP, trophic factors and ACh. Trends Neurosci. 2002;25(2):79–84. doi: 10.1016/s0166-2236(02)02037-4. [DOI] [PubMed] [Google Scholar]
- Kilgard MP, Merzenich MM. Cortical map reorganization enabled by nucleus basalis activity. Science. 1998;279(5357):1714–18. doi: 10.1126/science.279.5357.1714. [DOI] [PubMed] [Google Scholar]
- Kingsman SM, Mitrophanous K, Olsen JC. Potential oncogene activity of the woodchuck hepatitis post-transcriptional regulatory element (WPRE) Gene Ther. 2005;12:3–4. doi: 10.1038/sj.gt.3302417. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Applegate MD, Knusel B, Junard EO, Burton LE, Mobley WC, Hefti FF, Price DL. Recombinant human nerve growth factor prevents retrograde degeneration of axotomized basal forebrain cholinergic neurons in the rat. Exp Neurol. 1991a;112(2):161–73. doi: 10.1016/0014-4886(91)90066-l. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Clatterbuck RE, Nauta HJ, Knusel B, Burton LE, Hefti FF, Mobley WC, Price DL. Human nerve growth factor prevents degeneration of basal forebrain cholinergic neurons in primates. Ann Neurol. 1991b;30(6):831–40. doi: 10.1002/ana.410300613. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Nauta HJ, Clatterbuck RE, Holtzman DM, Mobley WC, Price DL. Mouse nerve growth factor prevents degeneration of axotomized basal forebrain cholinergic neurons in the monkey. J Neurosci. 1990;10(12):3801–13. doi: 10.1523/JNEUROSCI.10-12-03801.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kromer LF. Nerve growth factor treatment after brain injury prevents neuronal death. Science. 1987;235(4785):214–6. doi: 10.1126/science.3798108. [DOI] [PubMed] [Google Scholar]
- Lapchak PA, Araujo DM, Carswell S, Hefti F. Distribution of [125I]nerve growth factor in the rat brain following a single intraventricular injection: correlation with the topographical distribution of trkA messenger RNA-expressing cells. Neuroscience. 1993;54(2):445–60. doi: 10.1016/0306-4522(93)90265-h. [DOI] [PubMed] [Google Scholar]
- Lindner MD, Kearns CE, Winn SR, Frydel B, Emerich DF. Effects of intraventricular encapsulated hNGF-secreting fibroblasts in aged rats. Cell Transplant. 1996;5(2):205–23. doi: 10.1177/096368979600500210. [DOI] [PubMed] [Google Scholar]
- Lyness SA, Zarow C, Chui HC. Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: a meta-analysis. Neurobiol Aging. 2003;24(1):1–23. doi: 10.1016/s0197-4580(02)00057-x. [DOI] [PubMed] [Google Scholar]
- Mandel RJ, Gage FH, Clevenger DG, Spratt SK, Snyder RO, Leff SE. Nerve growth factor expressed in the medial septum following in vivo gene delivery using a recombinant adeno-associated viral vector protects cholinergic neurons from fimbria-fornix lesion-induced degeneration. Exp Neurol. 1999;155(1):59–64. doi: 10.1006/exnr.1998.6961. [DOI] [PubMed] [Google Scholar]
- Markowska AL, Koliatsos VE, Breckler SJ, Price DL, Olton DS. Human nerve growth factor improves spatial memory in aged but not in young rats. J Neurosci. 1994;14(8):4815–24. doi: 10.1523/JNEUROSCI.14-08-04815.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Serrano A, Fischer W, Soderstrom S, Ebendal T, Bjorklund A. Long-term functional recovery from age-induced spatial memory impairments by nerve growth factor gene transfer to the rat basal forebrain. Proc Natl Acad Sci U S A. 1996;93(13):6355–60. doi: 10.1073/pnas.93.13.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM. Neuroplasticity failure in Alzheimer’s disease: bridging the gap between plaques and tangles. Neuron. 1999;24(3):521–9. doi: 10.1016/s0896-6273(00)81109-5. [DOI] [PubMed] [Google Scholar]
- Montero CN, Hefti F. Rescue of lesioned septal cholinergic neurons by nerve growth factor: specificity and requirement for chronic treatment. J Neurosci. 1988;8(8):2986–99. doi: 10.1523/JNEUROSCI.08-08-02986.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser VC. The functional observational battery in adult and developing rats. Neurotoxicology. 2000;21(6):989–96. [PubMed] [Google Scholar]
- Mufson EJ, Conner JM, Kordower JH. Nerve growth factor in Alzheimer’s disease: defective retrograde transport to nucleus basalis. Neuroreport. 1995;6(7):1063–6. doi: 10.1097/00001756-199505090-00028. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Kroin JS, Sendera TJ, Sobreviela T. Distribution and retrograde transport of trophic factors in the central nervous system: functional implications for the treatment of neurodegenerative diseases. Prog Neurobiol. 1999;57(4):451–84. doi: 10.1016/s0301-0082(98)00059-8. [DOI] [PubMed] [Google Scholar]
- Niewiadomska G, Komorowski S, Baksalerska-Pazera M. Amelioration of cholinergic neurons dysfunction in aged rats depends on the continuous supply of NGF. Neurobiol Aging. 2002;23(4):601–13. doi: 10.1016/s0197-4580(01)00345-1. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108(2):193–9. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Olson L, Backlund EO, Ebendal T, Freedman R, Hamberger B, Hansson P, Hoffer B, Lindblom U, Meyerson B, Stromberg I, et al. Intraputaminal infusion of nerve growth factor to support adrenal medullary autografts in Parkinson’s disease. One-year follow-up of first clinical trial. Arch Neurol. 1991;48(4):373–81. doi: 10.1001/archneur.1991.00530160037011. [DOI] [PubMed] [Google Scholar]
- Olson L, Nordberg A, von Holst H, Backman L, Ebendal T, Alafuzoff I, Amberla K, Hartvig P, Herlitz A, Lilja A, Lundqvist H, Langstrom B, Meyerson B, Persson A, Viitanen M, Winblad B, Seiger A. Nerve growth factor affects 11C-nicotine binding, blood flow, EEG, and verbal episodic memory in an Alzheimer patient (case report) J Neural Transm. 1992;4:79–95. doi: 10.1007/BF02257624. [DOI] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J Neuropath Exp Neurology. 2004;63(6):641–9. doi: 10.1093/jnen/63.6.641. [DOI] [PubMed] [Google Scholar]
- Perry EK, Tomlinson BE, Blessed G, Bergmann K, Gibson PH, Perry RH. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br Med J. 1978a;2(6150):1457–9. doi: 10.1136/bmj.2.6150.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry EK, Perry RH, Blessed G, Tomlinson BE. Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathol Appl Neurobiol. 1978b;4(4):273–7. doi: 10.1111/j.1365-2990.1978.tb00545.x. [DOI] [PubMed] [Google Scholar]
- Phelps CH, Gage FH, Growdon JH, Hefti F, Harbaugh R, Johnston MV, Khachaturian ZS, Mobley WC, Price DL, Raskind M, et al. Potential use of nerve growth factor to treat Alzheimer’s disease. Neurobiol Aging. 1989;10(2):205–7. doi: 10.1016/0197-4580(89)90032-8. [DOI] [PubMed] [Google Scholar]
- Pizzo DP, Winkler J, Sidiqi I, Waite JJ, Thal LJ. Modulation of sensory inputs and ectopic presence of Schwann cells depend upon the route and duration of nerve growth factor administration. Exp Neurol. 2002;178(1):91–103. doi: 10.1006/exnr.2002.8010. [DOI] [PubMed] [Google Scholar]
- Promega. Technical Bulletin: NGF Emax® ImmunoAssay System. 2007. [Google Scholar]
- Robbins TW, Semple J, Kumar R, Truman MI, Shorter J, Ferraro A, Fox B, McKay G, Matthews K. Effects of scopolamine on delayed-matching-to-sample and paired associates tests of visual memory and learning in human subjects: comparison with diazepam and implications for dementia. Psychopharmacology (Berl) 1997;134(1):95–106. doi: 10.1007/s002130050430. [DOI] [PubMed] [Google Scholar]
- Rosenberg MB, Friedmann T, Robertson RC, Tuszynski M, Wolff JA, Breakefield XO, Gage FH. Grafting genetically modified cells to the damaged brain: restorative effects of NGF expression. Science. 1988;242(4885):1575–8. doi: 10.1126/science.3201248. [DOI] [PubMed] [Google Scholar]
- Salehi A, Delcroix JD, Mobley WC. Traffic at the intersection of neurotrophic factor signaling and neurodegeneration. Trends Neurosci. 2003;26(2):73–80. doi: 10.1016/S0166-2236(02)00038-3. [DOI] [PubMed] [Google Scholar]
- Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, Xia W, Villar A, Campbell WA, Kulnane LS, Nixon RA, Lamb BT, Epstein CJ, Stokin GB, Goldstein LS, Mobley WC. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51(1):29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Saragovi HU. Progression of age-associated cognitive impairment correlates with quantitative and qualitative loss of TrkA receptor protein in nucleus basalis and cortex. J Neurochem. 2005;95:1472–80. doi: 10.1111/j.1471-4159.2005.03479.x. [DOI] [PubMed] [Google Scholar]
- Scott SA, Mufson EJ, Weingartner JA, Skau KA, Crutcher KA. Nerve growth factor in Alzheimer’s disease: increased levels throughout the brain coupled with declines in nucleus basalis. J Neurosci. 1995;15(9):6213–21. doi: 10.1523/JNEUROSCI.15-09-06213.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ML, Deadwyler SA, Booze RM. 3-D reconstruction of the cholinergic basal forebrain system in young and aged rats. Neurobiol Aging. 1993;14:389–92. doi: 10.1016/0197-4580(93)90126-v. [DOI] [PubMed] [Google Scholar]
- Smith DE, Roberts J, Gage FH, Tuszynski MH. Age-associated neuronal atrophy occurs in the primate brain and is reversible by growth factor gene therapy. Proc Natl Acad Sci U S A. 1999;96(19):10893–8. doi: 10.1073/pnas.96.19.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmelin J, Lazarus C, Cassel S, Kelche C, Cassel J-C. Immunohistochemical and neurochemical correlates of learning deficits in aged rats. 2000;96(2):275–89. doi: 10.1016/s0306-4522(99)00561-8. [DOI] [PubMed] [Google Scholar]
- Taffe MA, Weed MR, Gutierrez T, Davis SA, Gold LH. Differential muscarinic and NMDA contributions to visuo-spatial paired-associate learning in rhesus monkeys. Psychopharmacology (Berl) 2002;160(3):253–62. doi: 10.1007/s00213-001-0954-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taffe MA, Weed MR, Gold LH. Scopolamine alters rhesus monkey performance on a novel neuropsychological test battery. Brain Res Cogn Brain Res. 1999;8(3):203–12. doi: 10.1016/s0926-6410(99)00021-x. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R, Patel P, Blesch A, Vahlsing HL, Ho G, Tong G, Potkin SG, Fallon J, Hansen L, Mufson EJ, Kordower JH, Gall C, Conner J. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nature Med. 2005;11:551–555. doi: 10.1038/nm1239. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, Gage FH. Bridging grafts and transient nerve growth factor infusions promote long-term central nervous system neuronal rescue and partial functional recovery. Proc Natl Acad Sci U S A. 1995;92(10):4621–5. doi: 10.1073/pnas.92.10.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuszynski MH, U HS, Amaral DG, Gage FH. Nerve growth factor infusion in the primate brain reduces lesion-induced cholinergic neuronal degeneration. J Neurosci. 1990;10(11):3604–14. doi: 10.1523/JNEUROSCI.10-11-03604.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215(4537):1237–9. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol. 1981;10(2):122–6. doi: 10.1002/ana.410100203. [DOI] [PubMed] [Google Scholar]
- Williams LR, Rylett RJ, Moises HC, Tang AH. Exogenous NGF affects cholinergic transmitter function and Y-maze behavior in aged Fischer 344 male rats. Can J Neurol Sci. 1991;18(3 Suppl):403–7. doi: 10.1017/s0317167100032546. [DOI] [PubMed] [Google Scholar]
- Williams LR, Varon S, Peterson GM, Wictorin K, Fischer W, Bjorklund A, Gage FH. Continuous infusion of nerve growth factor prevents basal forebrain neuronal death after fimbria fornix transection. Proc Natl Acad Sci U S A. 1986;83(23):9231–5. doi: 10.1073/pnas.83.23.9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J, Ramirez GA, Kuhn HG, Peterson DA, Day-Lollini PA, Stewart GR, Tuszynski MH, Gage FH, Thal LJ. Reversible Schwann cell hyperplasia and sprouting of sensory and sympathetic neurites after intraventricular administration of nerve growth factor. Ann Neurol. 1997;41(1):82–93. doi: 10.1002/ana.410410114. [DOI] [PubMed] [Google Scholar]
- Wu K, Klein RL, Meyers CA, King MA, Hughes JA, Millard WJ, Meyer EM. Long-term neuronal effects and disposition of ectopic preproNGF gene transfer into the rat septum. Hum Gene Ther. 2003;14(15):1463–72. doi: 10.1089/104303403769211673. [DOI] [PubMed] [Google Scholar]
- Wu K, Meyer EM, Bennett JA, Meyers CA, Hughes JA, King MA. AAV2/5-mediated NGF gene delivery protects septal cholinergic neurons following axotomy. Brain Research. 2005;1061:107–113. doi: 10.1016/j.brainres.2005.08.056. [DOI] [PubMed] [Google Scholar]