

Abstract
Cyclic guanosine 3′,5′-monophosphate (cGMP) mediates a wide spectrum of physiologic processes in multiple cell types within the cardiovascular system. Dysfunctional signaling at any step of the cascade--- cGMP synthesis, effector activation, or catabolism--- have been implicated in numerous cardiovascular diseases, ranging from hypertension to atherosclerosis to cardiac hypertrophy and heart failure. In this review, we outline each step of the cGMP signaling cascade and discuss its regulation and physiologic effects within the cardiovascular system. In addition, we illustrate how cGMP signaling becomes dysregulated in specific cardiovascular disease states. The ubiquitous role cGMP plays in cardiac physiology and pathophysiology presents great opportunities for pharmacologic modulation of the cGMP signal in the treatment of cardiovascular diseases. We detail the various therapeutic interventional strategies that have been developed or are in development, summarizing relevant preclinical and clinical studies.
Keywords: cGMP, cardiovascular disease, soluble guanylyl cyclase, natriuretic peptide receptor, PKG, Phosphodiesterases
1. Introduction
Cyclic guanosine 3′,5′-monophosphate (cGMP) is a ubiquitous intracellular second-messenger that mediates a vast array of physiologic processes, from ion channel conductance to cell growth and apoptosis to cellular mobility and contractility. In the cardiovascular system, cGMP signaling is vital to endothelial, vascular smooth muscle, and cardiac myocyte function. Generated by guanylyl cyclase isoforms in response to natriuretic peptides (NPs) and nitric oxide (NO), cGMP exerts its actions through cGMP-gated cation channels, cGMP-dependent protein kinases (PKGs), and cGMP-regulated phosphodiesterases (PDEs) that in turn hydrolyze cyclic nucleotides. Since the discovery of cGMP in rat urine nearly 50 years ago (Ashman et. al, 1963), the field of cGMP signaling research has grown exponentially. Abnormalities at each step of the cGMP signaling cascade, from cGMP synthesis to its degradation, have been implicated in cardiovascular disease and thus represent potential targets for pharmacologic therapies.
cGMP has two distinct pathways that regulate its synthesis, one coupled to natriuretic peptide hormone, and the other a simple gas (nitric oxide) (Fig. 1). No other second messenger, not even cyclic adenosine monophosphate (cAMP), is activated by a gas. The significance of NO-cGMP signaling was recognized by the 1998 Nobel Prize in Physiology and Medicine that was awarded for the major discoveries surrounding nitric oxide (Arnold et al., 1977; Ignarro et al., 1987a; Ignarro et al., 1987b; Katsuki et al., 1977; Schultz et al., 1977). Natriuretic peptide-mediated cGMP signaling was discovered in the early 1980s, when a polypeptide hormone was isolated from heart atrial muscle tissue and found to have potent diuretic (natriuretic) and hypotensive properties (Ackermann et al., 1984; Atarashi et al., 1984; Atlas et al., 1984; Bloch et al., 1985; de Bold, 1982; de Bold, 1985). The discovery of atrial natriuretic peptide was momentous in its implication of the heart as more than a circulatory pump or electrically conductive tissue but also an endocrine organ; a finding which ultimately helped shift the conceptual paradigm of heart failure to the current neurohormonal model.
Figure 1.

cGMP signaling cascade. cGMP is produced by particulate (pGC) and soluble (sGC) guanylyl cyclases, upon natriuretic peptide and nitric oxide activation, respectively. cGMP can then activate cGMP-dependent protein kinase (PKG) and either activate (green arrow) or inhibit (red arrow bar) various phophodiesterase isoforms. PKG-I phosphorylates several protein targets, including phospholamban (PLB), vasodilatory-stimulated phosphoprotein (VASP), regulator of G protein signaling 2 (RGS2), and the L-type calcium channel. PDE2 and PDE3 catabolize both cAMP and cGMP, whereas PDE5 specifically catabolizes cGMP. Upon cGMP binding to its regulatory GAF domain, PDE2 undergoes a conformational change and increases its enzymatic activity for cAMP. PDE5 similarly increases its catalytic activity for cGMP by an order of magnitude upon cGMP binding to its regulatory GAF domain.
Each of these pathways couples to a distinct guanylyl cyclase isoform--- soluble (sGC) and particulate (pGC) guanylyl cyclase respectively. These cyclases differ in their intracellular distribution, with sGC historically described as a cytosolic protein and pGC being a membrane bound protein. However, their intracellular localization is more nuanced, and recent studies support distinct pools of cGMP generation with different downstream effects (Castro et al., 2006; Nausch et al., 2008; Takimoto et al., 2007). Cyclic GMP activates three types of effector molecules, with cGMP-dependent protein kinases (PKGs) and phosphodiesterases (PDEs) predominating in the cardiovascular system. A third type of effector molecule, cGMP-gated cation channels, exists in retinal and olfactory neuroepithelium and nephrons, but neither protein expression nor physiological function of these channels have been established in the cardiovascular system. Cyclic GMP-PKG signaling within the vascular endothelium stimulates cell proliferation and increases permeability (Draijer et al., 1995a; Draijer et al., 1995b; Holschermann et al., 1997; Hood et al., 1998; Kook et al., 2003; Smolenski et al., 2000; Vaandrager et al., 1996a); it inhibits cell proliferation and mediates vasorelaxation in vascular smooth muscle (Archer et al., 1994; Bolotina et al., 1994; Cornwell et al., 1994; Murad et al., 1985); while in cardiac myocardium, it inhibits hypertrophy and modulates contractility (Kinugawa et al., 1997; Lohmann et al., 1991; Shah et al., 1994; Takimoto et al., 2005b; Tatsumi et al., 2000; Vila-Petroff et al., 1999). In all three tissues, cGMP-PKG signaling also mediates cellular apoptosis (Arstall et al., 1999; DeMeester et al., 1998; Fukuo et al., 1996; Suenobu et al., 1999; Taimor et al., 2000; Wu et al., 1997).
Lastly, cGMP catabolism is regulated by a subgroup of the 11 member phosphodiesterase superfamily. PDEs play a role in not only spatiotemporal regulation of cGMP signal but also cross-regulation of the cAMP signal. The strategy of inhibiting PDEs to enhance cGMP and related signaling has already been harnessed with the PDE5A inhibitor sildenafil, a common treatment for erectile dysfunction (Boolell et al., 1996). PDE inhibition has been further examined for the treatment of a variety of cardiovascular diseases, including pulmonary hypertension and now chronic heart failure, and this continues to be a highly active and promising field of research (Attina et al., 2008; Baliga et al., 2008; Bethke et al., 1992a; Bethke et al., 1992b; Eddahibi et al., 1998; Gillies et al., 2002; Guazzi et al., 2007a; Lewis et al., 2007a; Park, 2008; Reffelmann et al., 2003).
This review summarizes our current understanding of cGMP signaling within the cardiovascular system, specifically in vascular endothelial and smooth muscle cells and cardiac myocytes. Several others have already reviewed in greater detail specific aspects of cGMP signaling, from the upstream ‘first messengers’ nitric oxide and natriuretic peptides to the downstream effectors PKG and PDEs (Birschmann et al., 2004; D'Souza et al., 2004; Rastaldo et al., 2007; Saraiva et al., 2006; Schulz et al., 2008; Vaandrager et al., 1996a; Woodard et al., 2008). After discussing the major elements of the cGMP signaling pathway, we focus on the role of dysfunctional cGMP signaling in cardiovascular disease and the potential targets that this poses for the pharmacological treatment of cardiovascular diseases.
2. cGMP Signaling
2.1 Generation by guanylyl cyclases
The biosynthesis of cyclic GMP from guanosine triphosphate (GTP) is catalyzed by two different isoforms of guanylyl cyclase, one which functions as the biosensor for nitric oxide and the other, as the plasma membrane receptor for natriuretic peptides.
2.1.1 Nitric oxide-mediated biosynthesis of cGMP
Nitric oxide was long considered to be merely a toxic air pollutant until its identification as a labile factor released from endothelial cells, initially termed “endothelial derived relaxant factor” (EDRF) (Cherry et al., 1982; Furchgott et al., 1980). After seminal work demonstrating that EDRF, as induced by stimulating cells with acetylcholine, increased cGMP levels, activated PKG, and phosphorylated the same vascular smooth muscle proteins as did nitrovasodilators (Rapoport et al., 1983a; Rapoport et al., 1983b), EDRF and nitric oxide were proposed to be one and the same (Furchgott et al., 1987; Ignarro et al., 1987a; Ignarro et al., 1987b).
Nitric oxide is produced by nitric oxide synthase (NOS) which exists as three isoforms---neuronal nitric oxide synthase (NOS-1 or nNOS), inducible nitric oxide synthase (NOS-2 or iNOS), and endothelial nitric oxide synthase (NOS-3 or eNOS)(Alderton et al., 2001). The NOS isoforms were named by order of their discovery and initially reported expression pattern. However, all three isoforms have been detected in cardiac myocytes, vascular smooth muscle cells, and vascular endothelial cells (Balligand et al., 1995; Gyurko et al., 2000; Koide et al., 1993; Kurihara et al., 1998; MacNaul et al., 1993). Inducible NOS expression is, as its name implies, inducible, whereas eNOS and nNOS expression are constitutive and also inducible. Capable of associating with soluble, membrane, or cytoskeletal proteins, the NOS isoforms can be mobile within the cell and vary in their subcellular localization. They are active as homodimers with a central heme prosthetic group and require a complex array of cofactors and co-substrates to effectively generate NO. These factors include tetrahydrobiopterin (BH4), oxygen, calmodulin, NADPH, flavin mononucleotide, and flavin adenine dinucleotide. NOS catalyzes the oxidation of a guanidino nitrogen of L-arginine, whereby NO is produced as a byproduct. Inducible NOS synthesizes NO in much larger amounts than eNOS and nNOS (Shah et al., 2000).
The biosensor of NO, soluble guanylyl cyclase (sGC) is a heterodimer with an α subunit and β subunit (Kamisaki et al., 1986) and exists in various isoforms with two different α subunits (α1 and α2) and two different β subunits (β1 and β2). The α1β1 heterodimer is the most prevalent sGC isoform. The α2β1 isoform is a less active cyclase, and neither the α1β2 nor α2β2 isoforms have any reported guanylyl cyclase activity (Behrends et al., 1995; Harteneck et al., 1991; Yuen et al., 1990). The amino-terminal of the β1 subunit of sGC contains an evolutionarily conserved protoporphyrin-IX heme domain to which nitric oxide binds with distinct specificity. Nitric oxide can activate sGC at low nanomolar concentrations. Oxygen does not bind to the heme moiety, and carbon monoxide (CO) binding is at least 106-fold weaker than NO binding. When nitric oxide binds to the ferrous heme iron, the catalytic domain of the α and β subunits is activated, resulting in a 200-400 fold increase in Vmax and decrease in the GTP substrate concentration at half-maximal enzyme velocity (Km). Even at high micromolar concentrations, carbon monoxide binding to the iron of sGC results in only a two- to four-fold activation of the enzyme.
Nitric oxide activates soluble guanylyl cyclase (sGC) via a complex interplay between binding of NO to both heme and non-heme sites of sGC. Moreover, the two-step activation process results in two distinct NO-bound forms of sGC that are characterized by low and high enzymatic activity. Initially NO binds to the ferrous, five-coordinate heme moiety of sGC, forming an inactive but NO-responsive six-coordinate nitrosyl intermediate. In the presence of magnesium, cGMP, and pyrophosphate, this nitrosyl intermediate sGC species immediately converts to a five-coordinate nitroxyl complex upon further NO-binding (Russwurm et al., 2004; Tsoukias et al., 2004; Zhao et al., 1999). This second NO-binding step breaks the bond between the heme iron and the protein histidine axial ligand, triggering a conformational change in the catalytic domain of the enzyme and accelerating the basal rate of conversion of GTP to cGMP by several hundred fold. However, in the absence of magnesium, cGMP, or pyrophosphate, NO binding to the six-coordinate nitrosyl intermediate sGC species does not activate the enzyme. Instead this NO-bound form of sGC has only low or basal enzymatic activity. Thus, at low levels of NO, sGC remains in a low-activity state, whereas at high levels of NO and substrates/products, even the low-activity state sGC can be converted to the highly active state.
Alternatively, NO can also activate sGC by binding to a non-heme site, as initially suggested by the observation that the rate of NO dissociation from sGC is much slower than the rate of sGC deactivation (Cary et al., 2005). Again the initial step of sGC activation involves NO binding to the ferrous heme moiety of sGC. However, the second NO-binding event involves binding of NO at a non-heme site, which ruptures the histidine-iron bond and fully activates sGC. At low NO levels, NO dissociates from the non-heme site to give a low-activity state of sGC. This non-heme NO-binding site is thought to be the mechanism by which an acute increase in NO leads to a rapid rise in cGMP production. At continual low levels of NO, sGC produces cGMP at long-lasting, low levels. With acute bursts of NO, sGC produces a rapid rise in cGMP level.
Soluble GC activity is adversely affected by oxidant stress, the proposed mechanism of which is multifactorial and debatable. In the presence of excess ROS, sGC enzymatic function may be compromised by: a) a peroxynitrite (ONOO-) –mediated decrease in sGC specific activity (Weber et al., 2001); b) oxidation of the β1-associated prosthetic heme group and conversion of sGC to its NO-insensitive state (Stasch et al., 2006); c) oxidation-induced disulfide formation of β1-thiol groups (Mingone et al., 2006); or, d) NO-dependent post-translational modification (e.g., S-nitrosylation) of a β1 -thiol (Sayed et al., 2008). Regulation of sGC expression and activity by oxygen is controversial as various studies using models of chronic hypoxia have given conflicting results. Whereby some studies demonstrate decrease in sGC expression and activity in the pulmonary vasculature with chronic hypoxia (He et al., 2007; Williams et al., 2006), others have shown increase in protein expression and enzyme activity (He et al., 2007; Li et al., 1999; Vermeersch et al., 2007).
2.1.2 Natriuretic peptide-mediated biosynthesis of cGMP
Natriuretic peptides comprise a family of polypeptide mediators secreted by the heart and vasculature with fundamental roles in the regulation of blood volume, systemic vascular resistance, central venous pressure, and cardiac contractility (D'Souza et al., 2004). The principal natriuretic peptides are atrial natriuretic peptide (ANP, also A-type), brain natriuretic peptide (BNP, also B-type), and C-type natriuretic peptide (CNP). ANP and BNP are secreted primarily by the atria and ventricles of the heart, respectively, while CNP is secreted by the vascular endothelium. Cardiac production and release of ANP and BNP is triggered mainly by increases in myocardial wall stretch and/or pressure (de Bold et al., 2001) but may also be influenced by neurohumoral factors such as cathecholamines, arginine vasopression, angiotensin II, endothelin, and glucocorticoids (Silberbach et al., 2001). Natriuretic peptides exert their biological effects by binding to membrane-associated guanylyl cyclase receptors (alternatively known as NPRs or particulate guanylyl cyclases, pGCs). There are at least seven mammalian membrane-associated guanylyl cyclases, but two subtypes, NPR-A and NPR-B (also known as GC-A and GC-B), are responsible for the majority of the physiological effects of natriuretic peptides. NPR-A is activated by ANP and BNP, thereby mediating their endocrine actions in regulating body volume homeostasis and blood pressure and their local antihypertrophic effects in the myocardium. Both ANP and BNP bind to NPR-A with relatively high affinity, but ANP is many times more potent than BNP in receptor activation (Kambayashi et al., 1990; Mukoyama et al., 1990; Nakao et al., 1990). NPR-B mediates the paracrine action of CNP in vascular regeneration and endochondral ossification. NPR-A is widely expressed throughout the cardiovascular system, in vascular smooth muscle, vascular endothelium, and heart, as well as the kidney. While NPR-B is also highly abundant in vascular endothelium and smooth muscle (Hutchinson et al., 1997), its presence in the heart is thought to predominantly localize to the non-myocyte population and mostly in fibroblasts (Doyle et al., 2002). However, studies have also reported CNP/NPR-B signaling in myocytes, and have shown antihypertrophic and pro-apoptotic effects in cardiac myocytes (Han et al., 2003; Rosenkranz et al., 2003; Tokudome et al., 2004).
NPR-A consists of an extracellular ligand-binding domain, transmembrane domain, and intracellular domain. The intracellular domain has a kinase homology domain which is central to regulating guanylyl cyclase activity and modulating receptor sensitivity. In the basal state, NPR-A is phosphorylated and the guanylyl cyclase catalytic domain of the receptor is repressed. Upon hormone ligand binding, the kinase homology domain of NPR-A becomes ephosphorlyated, the catalytic domain is activated, and cGMP generated.
2.2 Activation of effector molecules
Cyclic GMP exerts its physiologic actions in the cardiovascular system by activating cGMP-dependent protein kinases and phosphodiesterases.
2.2.1 cGMP-dependent protein kinases
Three isotypes of cGMP-dependent protein kinases (PKGs) have been identified, two of which are splice variants of a single gene. PKG type I (PKG-I), which consists of an α and a β isoform (76kDa), is the prominent isotype in the cardiovascular system. It is expressed at very high levels (>0.1μM) in vascular smooth muscle cells (both PKG-Iα and PKG-Iβ) and endothelial cells (PKG-Iβ) and at lower levels in cardiac myocytes (PKG-Iα). PKG type II (PKG-II, 86kDa) is mainly expressed in the kidney, brain, and intestine. Both PKG-I and PKG-II exist as homodimers with identical structures. Each subunit consists of three functional domains--- an N-terminal domain, a regulatory domain, and a kinase domain. The N-terminal domain mediates PKG homodimerization, suppresses kinase domain activity in the absence of cGMP, and interacts with target substrate proteins. Amino-terminal modifications, acetylation in the case of PKG-I and myristoylation in the case of PKG-II, appear to control intracellular localization and hence function of PKG. Myristoylation is thought to be a major determinant in the membrane-association of PKG-II, whereas acetylation of PKG-I renders it soluble and thus cytosolic in distribution (Vaandrager et al., 1996b). Upon cGMP binding to specific sites in the regulatory domain, PKG undergoes a conformational change, resulting in the release of the N-terminus inhibition of the kinase domain. The kinase domain then catalyzes the phosphorylation of a serine/threonine side chain of the target substrate protein. PKG-Iα and PKG-Iβ isoforms differ in their N-terminus domain, which also regulates cooperativity between the cGMP-binding sites of PKG-Iα (Hofmann et al., 2006). Consequently, PKG-Iβ requires ten-fold higher concentrations of cGMP for kinase activation than does PKG-Iα.
2.2.2 Phosphodiesterases
The cyclic nucleotide phosphodiesterases (PDE) comprise a 21-gene super family categorized into 11 isoenzymes (PDE1-PDE11) with a total of 48 isoforms. Each break the phosphodiester bond in cGMP and/or cAMP resulting in the linear GMP or AMP. The various isoenzymes are differentially expressed in tissues, are selective or non-selective for the two cyclic nucleotides, and can be activated or inhibited by them as well. cGMP regulates the activity of PDE-2, -3, -5, and -9. Of these, PDE-2, -3, and -5 are known to be expressed in cardiac myocytes; PDE-3 and -5 are expressed in vascular smooth muscle cells; and PDE-2, -3, and -5 are expressed in vascular endothelial cells (Lugnier et al., 1999; Maurice, 2005; Netherton et al., 2005; Pauvert et al., 2002; Phillips et al., 2005; Sadhu et al., 1999; Thompson et al., 2002). While expressed in the heart, the cardiovascular role of PDE9 remains unknown at present. The molecular biology of the PDE isoforms and their role in physiologic regulation have been detailed in excellent reviews elsewhere (Bender et al., 2006; Conti et al., 2007; Osadchii, 2007; Rybalkin et al., 2003).
2.2.2.1 cGMP-activated PDEs
cGMP selectively activates PDE2 and PDE5 by binding to regulatory GAF domains in the N-terminus. PDE2 hydrolyzes both cAMP and cGMP at a high Vmax and low Km. Upon cGMP-GAF binding, PDE2 undergoes a conformational change and increases its enzymatic activity for cAMP (Martins et al., 1982). Its dual substrate specificity allows it to mediate negative cross-talk between the cGMP and cAMP signaling pathways. PDE2 exists as three different N-terminal splice variants (PDE2A1-3), of which PDE2A3 is the human variant and thought to be membrane associated. The co-localization of PDE2 in plasma membrane lipid rafts (cholesterol-rich microdomains) suggests coupling of PDE2 with other lipid raft-localized cyclic nucleotide signaling molecules, such as β-adrenoceptor, adenylyl cyclase, and nitric oxide synthase (Mongillo et al., 2006). In fact, PDE2 has been shown to regulate the L-type calcium channel in cardiac myocytes. L-type calcium channels are activated by β-adrenergic receptor-stimulated cAMP and cAMP-dependent protein kinase (PKA), exerting chronotropic and inotropic effects on the heart. Through PDE2 activation, cGMP is able to reduce the cAMP signal and affect cardiac function (Fischmeister et al., 2005; Fischmeister et al., 2006; Leroy et al., 2008; Mery et al., 1995; Vandecasteele et al., 2001). This effect appears coupled to co-activation by β-adrenergic stimulants of cGMP synthesis via β3-adrenergic receptor agonism coupled to NOS3 activation (Mongillo et al., 2006).
Whereas PDE2 can hydrolyze both cAMP and cGMP, PDE5 selectively hydrolyzes cGMP. PDE5 has high affinity for cGMP, and its catalytic activity increases by an order of magnitude when cGMP binds to its regulatory GAF domain. Catalytic activity is further enhanced by phosphorylation, mostly by PKG, at a S92, which stabilizes cGMP binding. PKA can also phosphorylate PDE5, particularly when cGMP levels are high and cGMP is already bound to PDE5. This provides a positive feedback mechanism that can be initially triggered by cGMP synthesis; hence PDE5 activity can be prolonged. PDE5 also has 3 N-terminal variants, PDE5A1-3, all of which have similar Km values and have been identified in humans. PDE5A1 and PDE5A2 are widely distributed; PDE5A3 has been suggested to be specifically expressed in smooth muscle cells and cardiac myocytes. There is no known difference in activity among these isoforms. PDE5 is not localized at the sarcolemmal membrane but is more cytosolic, with particular localization in the cardiac myocyte Z bands, suggesting an association with other Z band-localized proteins such as PKG (Takimoto et al., 2007; Zhang et al., 2008a).
PDE5A is well established as a regulator of vascular smooth muscle contraction through regulation of cGMP, with high levels of expression in the lung and corpus cavernosum. PDE5 inhibitors are now widely used to treat pulmonary hypertension and erectile dysfunction (Croom et al., 2008; Driscoll et al., 2008; Rosen et al., 2003). More recently, PDE5 was implicated as an important regulator of cGMP in cardiac myocytes, including the hypertrophic response to pressure-overload stress (Borlaug et al., 2005; Nagayama et al., 2008; Takimoto et al., 2007; Takimoto et al., 2005a; Takimoto et al., 2005b; Zhang et al., 2008a), cell survival signaling and apoptosis associated with ischemia/reperfusion (Das et al., 2006), post-infarction remodeling (Salloum et al., 2008), and doxorubicin toxicity (Fisher et al., 2005). While controversy has long existed over whether PDE5A expression in the heart was relevant to the cardiac myocyte itself, a gene silencing model recently confirmed PDE5A protein expression as well as its role in myocyte hypertrophy (Zhang et al., 2008a). Other studies have shown that the sub-cellular localization of PDE5A to myocyte z-bands depends upon NOS-NO-cGMP signaling (Kass et al., 2007; Nagayama et al., 2008; Takimoto et al., 2005). In mice genetically lacking eNOS, or those with NOS chronically inhibited by L-NAME, PDE5A distribution within the myocyte becomes diffuse. This modification impacts the capacity of PDE5A inhibition to counter acute and chronic cardiac stress (Nagayama et al., 2008; Senzaki et al., 2001; Takimoto et al., 2005a).
2.2.2.2 cGMP-inhibited PDE
Like PDE2, PDE3 is also a dual substrate enzyme and can hydrolyze both cAMP and cGMP with relatively high affinities. However, the Vmax of PDE3 for cAMP is 10-fold greater than for cGMP, so the enzyme largely targets cAMP and can be competitively inhibited by cGMP. At low cGMP levels, PDE3 may have a larger role in controlling cGMP levels. Two isoforms, PDE3A and PDE3B, have been identified in humans, with three N-terminal variants of PDE3A (PDE3A-136, PDE3A-118 and PDE3A-94, named according to their molecular size) expressed in cardiac myocytes and vascular smooth muscle and the single isoform of PDE3B expressed in cardiac myocytes and adipocytes. PDE3A-136 is exclusively membrane associated, whereas PDE3A-118 and PDE3A-94 are distributed in both the cytosol and membrane fractions of cardiac myocytes and vascular smooth muscle cells. Both PDE3A and PDE3B are activated by PKA-mediated phosphorylation in response to a variety of hormone stimulants, including prostaglandin and epinephrine(Shakur et al., 2001). PDE3B and possibly PDE3A can also be activated by PI3K/PKB signaling triggered by insulin, insulin growth factor (IGF-1), and leptin (Patrucco et al., 2004; Shakur et al., 2001).
In the cardiovascular system, PDE3 is involved in regulating cardiac myocyte and vascular smooth muscle contractility as well as vascular smooth muscle phenotype switch and stress response. PDE3 regulates cardiac myocyte contractility and relaxation respectively via L-type calcium channels (Jurevicius et al., 2003; Malecot et al., 1986; Rochais et al., 2006; Vandecasteele et al., 2001; Verde et al., 1999) and the cardiac sarcoplasmic reticulum calcium pump (SERCA2) (Gaide et al., 1983; Gwathmey et al., 1985; Malecot et al., 1986; Yano et al., 2000).
In vitro studies in adult rat ventricular myocytes examined the spatiotemporal dynamics of cAMP signals under conditions of β-adrenergic activation. Using engineered cyclic nucleotide-gated channels as well as the fluorescence resonance energy transfer-based sensor, Epac2-camps, cAMP changes beneath the membrane and within the cytosol were respectively monitored in response to pulse administration of isoprenaline. Comparison of the cAMP kinetics in these subcellular compartments with the time course of the L-type calcium channel current revealed that cAMP changes are not rate-limiting in the phosphorylation/dephosphorylation of the channel. Furthermore, selective and non-selective inhibition of PDE3 and the cAMP-specific phosphodiesterase PDE4 demonstrated that, while PDE3 may regulate the constitutive cAMP pool coupled to contractility, it has a minor role in regulating the cAMP and L-type calcium current response to brief β-adrenergic stimulation. Instead, the PDE4 regulates the cAMP microdomains generated by β-adrenergic stimulation (Leroy et al., 2008).
PDE3A has also been shown to induce expression of ICER (inducible cAMP early repressor) in a positive feedback loop and thereby regulate myocyte apoptosis (Yan et al., 2007). Regulation of PDE3B activity by PI3Kγ in cardiac myocytes plays a role in pressure overload cardiac remodeling and negative modulation of cardiac contractility (Marcantoni et al., 2006; Patrucco et al., 2004). PDE3A and PDE3B likely also regulate vascular smooth muscle contractility. Their expression also varies with conditions of elevated cAMP levels, the switch from contractile to secretory/synthetic phenotype, and hypoxia (Dunkerley et al., 2002; Maurice et al., 2003; Murray et al., 2002).
2.3 Catabolism by PDEs
In addition to the above mentioned PDE isoforms (PDE-2, -3, and -5), two other PDE isoforms also hydrolyze cGMP and are relevant to the cardiovascular system--- PDE1 and PDE9. PDE1 has dual specificity for cAMP and cGMP catabolism, but the three PDE1 isoforms vary in their affinity for the nucleotides and their expression among mammalian species. PDE1A and PDE1B target cGMP over cAMP (but with less specificity than PDE5 or PDE9); while PDE1C hydrolyzes both cyclic nucleotides with equal Km (Bender et al., 2006; Bender et al., 2005; Hansen et al., 1988; Sharma et al., 1984; Sharma et al., 1986; Snyder et al., 1999; Sonnenburg et al., 1995). PDE1 isoforms are cytosolic, calcium- and calmodulin-dependent phosphodiesterases. PDE1A likely regulates vascular smooth muscle contraction (Wu et al., 2004); PDE1C is involved in VSMC proliferation and apoptosis (Nagel et al., 2006). PDE1C is also expressed in cardiac myocytes, and recent studies have found it plays an important role in both cAMP and cGMP hydrolysis in vitro in human myocardium, though its physiologic role remains unknown (Vandeput et al., 2007). PDE9 is expressed in brain and heart, exists in both cytosol and cell nucleus, and has the highest affinity for cGMP of all the cGMP-hydrolyzing PDEs. Its function remains unknown and is an area of active investigation.
2.4 Compartmentalization of cGMP signaling
Compartmentalization of cGMP signaling is accounted for by the differential spatial distribution of soluble guanylyl cyclase and particulate guanylyl cyclase, PKG isoforms, and PDE isoforms, as well as the distribution of other proteins that associate with cGMP signaling (Fig. 2A). In addition to spatial regulation by different components of the cGMP synthetic pathways, mechanisms for NO-cGMP signal compartmentalization also exist. Soluble GC has long been described as the cytosolic isoform of guanylyl cyclase, with the natriuretic peptide receptor as its membrane-associated or particulate counterpart. However, over the past several years, NO-sensitive sGC has also been noted to associate with the plasma membrane of various cell types (Linder et al., 2005; Russwurm et al., 2001; Schoser et al., 2001; Zabel et al., 2002). The co-localization of sGC with other proteins of the NO-cGMP signaling pathway at the plasma membrane suggests a plasmalemmal functional microdomain with enhanced NOS-NO-cGMP signaling. Phosphodiesterases also play a very large role in the compartmentation of the cGMP signal as described in the previous section on PDE isoforms. For example, in cardiac myocytes stimulated with either nitric oxide donors or natriuretic peptides, a membrane localized cGMP pool was detected predominantly with natriuretic peptide stimulation (Fig. 2B). Moreover, this pool was differentially enhanced by PDE2 (NP stimulation) versus PDE5 (NO stimulation) inhibition (Castro et al., 2006; Fischmeister et al., 2006; Maurice et al., 2003; Rybalkin et al., 2003).
Figure 2.
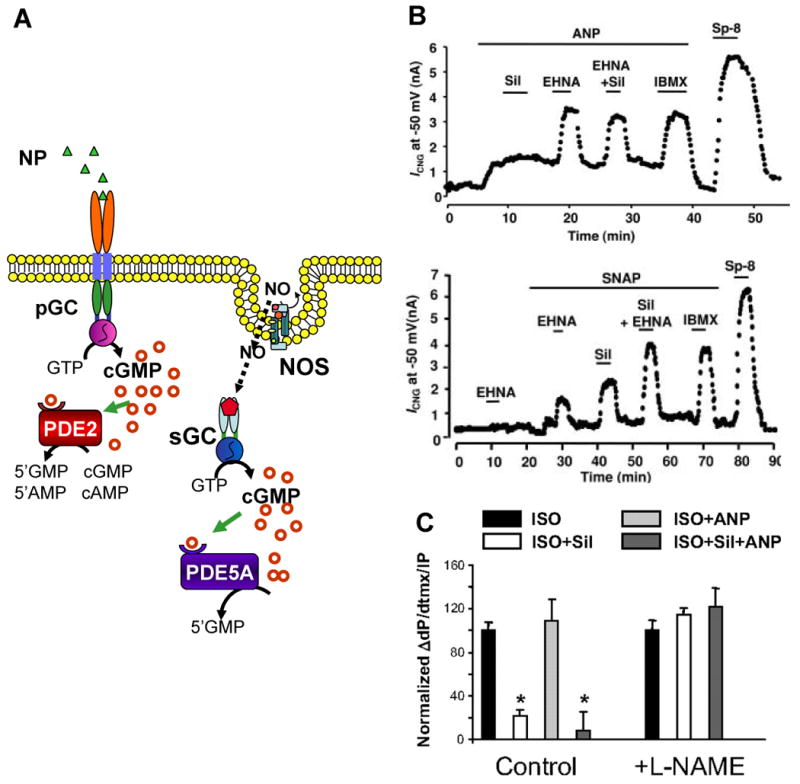
Compartmentalization of cGMP signal. (A) NP-induced cGMP pool is subplasmalemmal and specifically hydrolyzed by the PDE2 isoform. In contrast, NO-induced cGMP pool appears to be more cytosolic, not localized to the subplasmalemmal region, and is specifically hydrolyzed by PDE5 isoform. (B) In studies of adeno-CNGA2 infected adult rat cardiac myocytes, Castro et al.(2006) measured the CNG channel current in response to ANP and the NO-donor SNAP in the presence of selective (EHNA for PDE2; sildenafil for PDE5) and non-selective (IBMX) PDE inhibitors. A hydrolysis-resistent cGMP analog (Sp-8) was used as the positive control. These findings established the specificity of PDE2 and PDE5 for the distinct NP-cGMP and NO-cGMP pools, respectively. (C) Summary data of contractility studies of intact mouse hearts by Takimoto et al. (2007) demonstrated that the β-adrenergic response is modulated specifically by the NO-cGMP signal pool. These studies established that the distinct cGMP pools have distinct functions. (ISO, isoproterenol; Sil, sildenafil; ANP, atrial natriuretic peptide)
This model of spatially distinct cGMP pools was tested by examining the cyclic nucleotide-gated channel (CNG) activation response of cultured vascular smooth muscle cells to stimulation with an NO donor or atrial natriuretic peptide (Piggott et al., 2006). Vascular smooth muscle cells were infected with an adenovirus encoding the CNGA2 subunit. CNG channels were activated far more readily with ANP stimulation than NO stimulation, even in the presence of nonspecific PDE inhibitors. Furthermore, total cGMP levels as measured by enzyme immunoassays were not significantly different within the cells regardless of stimulation by the NO donor or ANP. Such findings suggest that cGMP signals are functionally localized to different subcellular compartments, and that the compartmentalization of the cGMP signal is regulated not only be PDE activity but also guanylyl cyclase activity.
The functional differences between the NP-cGMP and NO-cGMP pools was highlighted by studies of β-adrenergic response modulation in intact mouse hearts and isolated myocytes (Takimoto et al., 2007). Isoproterenol-stimulated cardiac contractility was compared under conditions of ANP stimulation (NP-cGMP) versus PDE5A inhibition (NO-cGMP). To confirm the specificity of NO-cGMP effects, the comparison was also perfomed with and without NOS inhibition (Fig. 2C). Whereas PDE5A inhibition and selective elevation of the NO-cGMP pool blunted the β-adrenergic cardiac response, ANP-triggered increase in the NP-cGMP pool did not affect the β-adrenergic cardiac response. Furthermore, ANP stimulation could not counter the effects of PDE5A inhibition. NOS inhibition completely abrograted the differential modulation of PDE5A inhibition on the β-adrenergic cardiac response. Compartmental regulation of cGMP signaling is an active area of investigation aided by the development of new fluorescent biosensors that can detect cGMP in living cells (Herget et al., 2008; Honda et al., 2001; Nausch et al., 2008). Improvement in signal sensitivity and specificity remain needed as levels of cGMP are low in myocytes.
3 cGMP regulation of cardiovascular system
3.1 Vascular system
3.1.1 Vascular smooth muscle cells
cGMP signaling directs vascular tone and smooth muscle cell (SMC) proliferation and differentiation (Fig. 3) (Munzel et al., 2003; Murad et al., 1985). Vascular tone is regulated by changes in intracellular free calcium concentrations within SMCs. In general, SMC contraction is triggered by the receptor-mediated generation of the second-messenger inositol 1,4,5-trisphosphate (IP3). IP3 induces release of free calcium from intracellular stores, further provoking influx of extracellular calcium via voltage- and non-voltage-gated calcium channels. The rise in intracellular calcium activates calcium/calmodulin-dependent myosin light chain kinase (MLCK) which phosphorylates myosin light chain (MLC) to activate myosin ATPase and trigger SMC contraction. Reduction in intracellular calcium concentration thus results in vasorelaxation.
Figure 3.
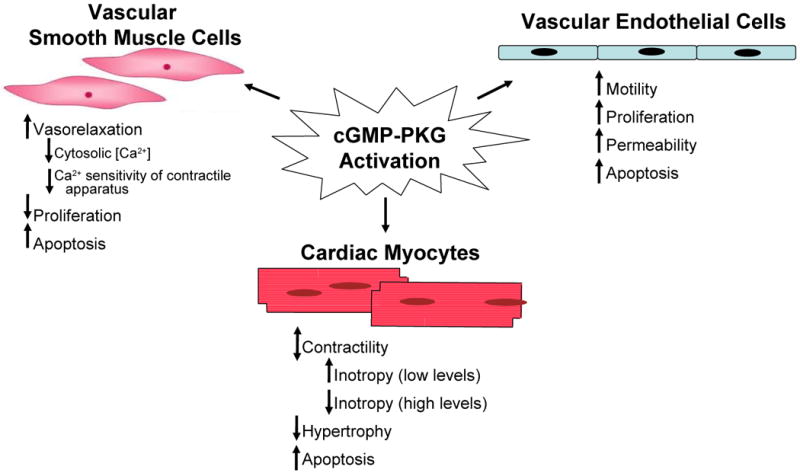
Physiological effects of cGMP-PKG activation in various cell types of the cardiovascular system.
cGMP regulates the cytosolic free calcium level within vascular smooth muscle cells through several mechanisms: (a) inhibition of IP3-mediated calcium release from intracellular stores; (b) removal and sequestration of intracellular calcium through calcium pump mechanisms; and (c) both direct and indirect inhibition of the influx of extracellular calcium through voltage-gated calcium channels. PKG-Iβ phosphorylates both the IP3 receptor (IP3R) and the IP3R-associated PKG-I substrate (IRAG), resulting in decreased calcium release from the sarcoplasmic and endoplasmic reticulum (Ammendola et al., 2001; Schlossmann et al., 2000). PKG-Iα phosphorylates phospholamban, an important modulator of calcium transport of sarcoplasmic reticulum, in vascular smooth muscle cells and cardiac myocytes (Raeymaekers et al., 1988). Phosphorylated phospholamban is unable to inhibit SERCA2 and calcium is taken up into the endoplasmic reticulum, reducing intracellular free calcium levels. cGMP-PKG mediated phosphorylation of the plasmalemmal calcium ATPase pump similarly stimulates the extrusion of calcium with resultant decrease of cytoplasmic calcium concentration (Eggermont et al., 1988). PKG also phosphorylates the large conductance, calcium-activated potassium channel (BKCa), thereby opening the channel, hyperpolarizing the cell membrane, and closing a number of other channels, including the L-type calcium channel. This ultimately reduces extracellular calcium influx. BKCa activation by PKG has been shown to be involved in the relaxation of several different types of smooth muscle, including human pulmonary artery smooth muscle cells and rat mesenteric smooth muscle cells (Carrier et al., 1997; Peng et al., 1996). BKCa channels are also indirectly regulated by PKG via PKG-activation of an associated protein phosphatase 2A (White et al., 1993; Zhou et al., 1996).
Aside from regulating intracellular calcium concentrations, cGMP-PKG signaling modulates vascular tone by altering calcium sensitization and thin filament interaction. PKG-Iα phosphorylates the myosin-binding subunit (MBS) of MLC phosphatase, thus activating the catalytic subunit of the phosphatase (Nakamura et al., 1999; Surks et al., 2003; White et al., 1993; Zhou et al., 1996). Dephosphorylation of MLC reduces the calcium sensitivity of the smooth muscle contractile machinery to inhibit smooth muscle contraction. Two thin filament-actin binding proteins, vasodilatory-stimulated phosphoprotein (VASP) and the 20-kDa heat shock-related protein (HSP20), are also target substrates of PKG. VASP is integral to cell adhesion and motility, and binds to actin filaments and stress fibers, suggesting a potential though unproven role in smooth muscle contraction. PKG-mediated phosphorylation of VASP decreases VASP binding to actin filaments. Though the exact mechanism of this remains unclear, modulation of the interaction between actin and the actin-binding regulatory protein profilin has been proposed (Reinhard et al., 1995). Phosphorylation of HSP20, which can be mediated by either cGMP-PKG activation or cAMP-protein kinase C (PKC) activation, is associated with vasorelaxation (Beall et al., 1997; McLemore et al., 2005; Rembold et al., 2000; Rembold et al., 2003). The importance of PKG in modulating vascular tone is emphasized by PKG-I deficient mice, which die at a young age and are afflicted by impaired smooth muscle relaxation, amongst other defects (Koeppen et al., 2004; Pfeifer et al., 1998; Sausbier et al., 2000; Wegener et al., 2002).
Another important role of cGMP-PKG signaling in vascular smooth muscle cells (VSMC) is its regulation of proliferation and differentiation. Several reviews have already detailed cGMP-PKG regulation of the multiple genes involved in VSMC proliferation and differentiation (Lincoln et al., 2006; Pilz et al., 2003). Briefly, VSMC proliferation involves mitogen-activated protein kinases (MAPK), cyclins, cyclin-dependent protein kinases (Cdk), Cdk inhibitors, the retinoblastoma protein (Rb), and E2F, the transcription factor that induces S-phase gene expression. cGMP-PKG activation directs an anti-proliferative signal in VSMCs, downregulating mRNA levels of cyclins-A, -D1, and E, and vascular endothelial growth factor (VEGF, under hypoxic conditions) and upregulating mRNA levels of MAPK phosphatase-1 (MKP-1) and p16 (gene encoding Cdk inhibitor 2A). The phenotypic switch of VSMC between a contractile, “differentiated” state and a synthetic or secretory, “dedifferentiated” state involves the differential expression of contractile proteins (i.e. smooth muscle myosin heavy chain -2, SM-α-actin, SM-calponin), extracellular matrix proteins (i.e. thrombospondin, osteopontin), signal transduction proteins, and growth factors as well as their receptors (i.e. fibroblast growth factor receptors-1/2). VSMCs dedifferentiate from the “contractile” phenotype to the “synthetic” or “secretory” phenotype in response to vascular injury or during in vitro culture. During the dedifferentiation process, VSMCs can proliferate, migrate, and produce extracellular matrix proteins. This process is associated with loss of PKG expression and transcriptional down-regulation of contractile proteins. The details of VSMC-specific gene expression are not completely understood but are thought to involve muscle cell-specific serum-response factor (SRF), which binds to DNA sequences known as CArG boxes, in cis elements of VSMC-specific genes such as smooth muscle α-actin. SRF then recruits the co-transcriptional regulator myocardin. cGMP stimulation and PKG activation in VSMCs thus favor the contractile phenotype.
3.1.2 Vascular endothelium
While cGMP-PKG signaling has been extensively studied in vascular smooth muscle cells, the details and physiological consequences of cGMP-PKG signaling in vascular endothelium are only beginning to be understood. In vascular endothelial cells, cGMP-PKG signaling regulates cell motility, migration, and proliferation, all of which are vital to angiogenesis, and vascular permeability (Fig. 3).
Angiogenesis involves vascular endothelial cell motility, migration, and proliferation, physiological processes which are mediated by cGMP signaling. The central role of cGMP signaling in angiogenesis has been demonstrated by both in vitro and in vivo studies examining NO-induced and ANP-induced cGMP pathways. In eNOS-/- knockout mice, VEGF fails to induce neovascularization (Fukumura et al., 2001; Murohara et al., 1998). Furthermore, a model of hind-limb ischemia in eNOS-/- knockout mice demonstrated defective neovascularization due to reduced mobilization of endothelial progenitor cells (Aicher et al., 2003). Similarly, in vitro studies showed that VEGF-stimulated endothelial cell capillary-like tube network formation requires eNOS activation and NO-cGMP signaling (Bussolati et al., 2001; Papapetropoulos et al., 1997b; Papapetropoulos et al., 1997a). siRNA-mediated knockdown of NPR-A, PKG, or VASP all prevent ANP-induced endothelial tube formation, demonstrating the importance of ANP-mediated cGMP-signaling in endothelial function (Chen et al., 2008). By mediating cross-talk between cAMP and cGMP signaling, PDEs have also been shown to play a significant role in regulating endothelial cell motility, migration, proliferation, and hence angiogenesis (Netherton et al., 2005).
Vascular permeability and endothelial barrier function are predominantly determined by endothelial cell contraction. Both NO- and ANP-triggered cGMP signaling have been shown to improve endothelial barrier function and protect against vascular injury in the systemic (Furst et al., 2008; Sabrane et al., 2005; Surapisitchat et al., 2007) and pulmonary vasculature (Brovkovych et al., 2008; Irwin et al., 2001; Irwin et al., 2005; Klinger et al., 2006; Klinger et al., 1998; Mitaka et al., 1998; Tanabe et al., 1996; Yin et al., 2008).
As in vascular smooth muscle cells, the physiological actions of cGMP in vascular endothelial cells are determined by the downstream phosphorylation target substrates of PKG. In vascular endothelial cells, PKG has been shown to phosphorylate eNOS (Butt et al., 2000), 6-pyruvovyl-tetrahydropterin synthase which produces the essential eNOS cofactor BH4(Scherer-Oppliger et al., 1999), and VASP (Chen et al., 2008; Schafer et al., 2003). The proliferative effect of cGMP in vascular endothelial cells also correlates with increased activity of extracellular signal-related kinases Erk-1/2, and may be related to increased VEGF production (Hood et al., 1998; Parenti et al., 1998; Zaragoza et al., 2002; Zhang et al., 2003). Many studies have proposed that cGMP regulation of endothelial barrier function is mediated by PKG phosphorylation of VASP (Draijer et al., 1995b; Mehta et al., 2006; Price et al., 2000). However, a recent study of cGMP-PKG signaling in hydrogen peroxide-induced endothelial barrier dysfunction of pulmonary artery endothelial cells could not confirm that VASP phosphorylation was central (Rentsendorj et al., 2008). Others have proposed a mechanism by which cGMP cross-regulation of cAMP signaling may account for ANP-mediated protective effects against agonist-induced pulmonary endothelial cell barrier dysfunction; ANP protective effects in thrombin-induced endothelial cell hyper-permeability were linked with activation of cAMP and PKA signaling cascades (Birukova et al., 2008; Lorenowicz et al., 2008).
3.2 Cardiac myocytes
In cardiac myocytes, cGMP negatively modulates contractility and hypertrophy and mediates apoptosis (Fig. 3). The role of cGMP-PKG signaling in cardiac contractility was determined by studies of isolated myocytes from conventional and cardiac-specific PKG-I knockout mice (Wegener et al., 2002). Whereas cGMP analogues reduced the force of contraction in electrically stimulated myocardium from wild-type control mice, they had no effect on the force of contraction in myocardium from conventional or cardiac-specific PKG-I knockout mice. Furthermore, the difference between the wild-type and knockout mice myocardium persisted even in experiments done in the presence of forskolin, an activator of the β-adrenergic-cAMP pathway, thereby verifying the specificity of cGMP signaling for reducing contractile force.
cGMP modulation of myocyte contractility can be initiated by either nitric oxide or natriuretic peptide (Kojda et al., 1999; Kojda et al., 1996; Mohan et al., 1996; Zhang et al., 2005). Interestingly, NO has bimodal actions on myocardial contractility; low concentrations of NO increase myocardial contractility, while high concentrations exert a negative inotropic effect. Low concentrations of NO can activate adenylyl cyclase without activating guanylyl cyclase (Vila-Petroff et al., 1999), inducing production of cAMP and not cGMP. At low concentrations, cGMP also inhibits the activity of PDE3, preventing the hydrolysis of cAMP. Accumulation of cAMP then activates PKA, leading to the opening of sarcolemmal voltage-gated and sarcoplasmic ryanodine receptors calcium channels, and ultimately improved myocyte contractility (Marx et al., 2000).
The negative inotropic effect of NO has in part been attributed to a cGMP-PKG mediated reduction of myofilament calcium responsiveness (Layland et al., 2002; Shah, 1996; Vila-Petroff et al., 1999), though the exact mechanism was initially unclear. Phosphorylation of troponin I by PKG had been suggested by in vitro studies (Blumenthal et al., 1978), but others demonstrated inconsistent effects of PKG on calcium sensitivity of skinned cardiac muscle (Mope et al., 1980; Pfitzer et al., 1982). The role of PKG in the contractile response to nitric oxide was elegantly established by isolated myocyte contractility studies in which the intracellular calcium transient was simultaneously assessed as the cell was exposed to an NO donor (diethylamine NONOate; DEA/NO), with and without inhibitors of soluble guanylyl cyclase (1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one; ODQ) and PKG (Rp-8-Br-cGMPs)(Layland et al., 2002). The NO donor significantly increased resting myocyte cell length and accelerated the relaxation time, without changing either the amplitude or kinetics of the intracellular calcium transient. Inhibiting either soluble guanylyl cyclase or PKG abrogated the effect of DEA/NO on the myofilaments. Additionally, direct activation of PKG with 8-pCTP-cGMP mimicked the myocyte relaxation induced by DEA/NO. DEA/NO treated hearts also demonstrated increased phophorylation of troponin I.
The L-type voltage-gated calcium channel has also been identified as a phosphorylation target of PKG (Yang et al., 2007). Phosphorylation sites within the α1c and β2a subunits of the L-type calcium channel were identified by a glutathione S-transferase (GST) fusion protein screen, and the sites were confirmed by phospho-epitope specific antibodies which detected immunoreactive bands in cGMP-stimulated myocyte extracts. Heterologous expression of rabbit α1c and β2a subunits in HEK cells showed that PKG activation does indeed inhibit the L-type calcium current. Furthermore, mutation of a β2a PKG-phosphorylation site reduced PKG-mediated inhibition of the L-type calcium current.
Differential activation of PDEs is also believed to modulate the contractile effect of NO-cGMP. At high NO levels, not only is cGMP production triggered, but cGMP-activation of PDE2 leads to hydrolysis of cAMP and the cGMP signal predominantes. Some have suggested that the variable effects of NO are not simply due to its concentration but relate also to its source, the specific NOS isoform generating the NO. For example, cardiac nNOS localizes to the sarcoplasmic reticulum (Khan et al., 2003; Xu et al., 1999) where it would be in close proximity of activating sources of calcium and calcium cycling ion channels as well as phospholamban and ryanodine receptors (Martin et al., 2006; Sears et al., 2003; Zhang et al., 2008b).
That cGMP has an antihypertrophic cardiac effect was initially suggested by the exacerbation of cardiac hypertrophy in various knockout mice lacking elements of the cGMP signaling pathway. The global NPR-A-/- knockout mice demonstrated salt-resistent arterial hypertension with a disproportionate degree of cardiac hypertrophy and fibrosis (Holtwick et al., 2003; Kuhn et al., 2002; Lopez et al., 1995; Oliver et al., 1997). In the face of pressure overload induced by transverse aortic constriction, cardiac-selective NPR-A-/- knockout mice had an exaggerated hypertrophic response to stress, along with slightly enhanced cardiac fibrosis and marked cardiac dysfunction (Holtwick et al., 2003). The Nppa-/- knockout mice, deficient in preproANP, developed salt-sensitive arterial hypertension and cardiac hypertrophy (John et al., 1995). Mice lacking eNOS and/or nNOS also develop cardiac hypertrophy and dysfunction (Flaherty et al., 2007; Li et al., 2004; Massion et al., 2003; Ruetten et al., 2005; Wenzel et al., 2007). Consistent with the cardiac hypertrophy phenotype of these mouse models of NOS, NPR-A, and NPPA deficiency, in vitro cardiac myocyte studies have also shown that natriuretic peptides and cGMP analogues suppress phenylephrine-induced hypertrophy and its associated re-induction of the fetal gene program (Horio et al., 2000). Thus enhancement of the cGMP signaling cascade indeed exerts antihypertrophic effects.
The exact role of cGMP-PKG activation in protecting against hypertrophic signaling remains to be confirmed. Conventional PKG-I knockout mice die young and perhaps too early to develop any cardiac hypertrophy (Pfeifer et al., 1998). A cardiac-specific PKG-I knockout mice designed upon the MLC2a-Cre transgenic mouse line had been studied with respect to cardiac contractility but not hypertrophic response (Wegener et al., 2002). A conditional, cardiac-specific PKG-I knockout mouse remains to be generated in order to fully examine the role of PKG in the adult myocyte hypertrophic signaling. However, in vitro adenoviral overexpression of PKG-Iβ in neonatal rat cardiac myocytes enhanced the antihypertrophic effect of NO in phenylephrine-stimulated cardiac myocytes (Wollert et al., 2002).
The cardiac hypertrophic response involves a complex web of signaling pathways that have been reviewed elsewhere (Clerk et al., 2007; Heineke et al., 2006; Mudd et al., 2008). The best studied downstream mechanism by which cGMP-PKG signaling exerts its antihypertrophic action is the inhibition, at multiple levels, of the calcium-calcineurin-NFAT pathway (Fiedler et al., 2002; Kato et al., 2000; Molkentin et al., 1998; Taigen et al., 2000; Zhu et al., 2000). Calcium activation of calcineurin results in the dephosphorylation and nuclear translocation of cytoplasmic NFAT (nuclear factor of activated T-cells), resulting in expression of a hypertrophic gene program. cGMP-PKG signaling may reduce intracellular calcium concentrations to inhibit calcineurin activation, though other mechanisms appear to apply as well. This was recently shown in a study of CnAβ-deficient mice (the Aβ-subunit providing the majority of Cn activity). These mice develop less hypertrophy in response to pressure-overload than their wildtype counterparts, but importantly, the hypertrophy that does develop is still inhibited by enhancing PKG activity via PDE5A inhibitors (Hsu et al., 2008).
Most recently PKG activation has been demonstrated to stimulate regulator of G-protein coupled signaling 2 (RGS2), a GTPase that suppresses Gαq/11 coupled stimulation (Takimoto et al., in press). Mice lacking RGS2 display a profound exacerbated response to pressure-overload coupled with enhanced Cn and other Gq-coupled signaling cascades, and demonstrated early mortality, marked hypertrophy, fibrosis, and chamber dysfunction. Even though PKG activation was similarly enhanced, inhibition of PDE5A with chronic sildenafil treatment did not suppress pressure-overload induced hypertrophy in RGS2-/- animals as it does in mice expressing RGS2 (Takimoto et al., 2005b).
In addition to RGS2, cGMP-PKG signaling has been shown to inhibit a muscle LIM protein (MLP)-dependent pro-hypertrophic pathway in cultured cardiac myocytes (Heineke et al., 2003). The relevance of this pathway to in vivo cardiac structural remodeling remains unknown.
Recently, ANP-cGMP-PKG activation was shown to inhibit transforming growth factor (TGF)-β1–induced extracellular matrix expression in cardiac fibroblasts via phosphorylation of Smad3 (Li et al., 2008). Phosphorylation of Smad3 by PKG occurred at serine/threonine residues distinct from the serine residues typically phosphorylated by the TGF-β receptor kinase. Consequently, nuclear translocation of pSmad3 was disrupted and TGF-β1 induced downstream signaling of pSmad3 was inhibited. This mechanism is now thought to contribute to the antifibrogenic effects of natriuretic peptides in the heart.
Proapoptotic effects of cGMP have been reported in VSMCs, endothelial cells, and cardiac myocytes and appear to be mediated by PKG (Chiche et al., 1998; Kloss et al., 2000; Pollman et al., 1996; Wu et al., 1997). The downstream mechanisms have not been fully determined, but in vascular cells they may involve activation of c-Jun N-terminal kinase (JNK) and/or the phosphorylation and inactivation of β-catenin. In cardiac myocytes, cGMP decreases the mRNA levels of the anti-apoptotic Bcl-2 homologue Mcl-1 (Wu et al., 1997).
4 Pathophysiological role of cGMP signaling in cardiovascular disease
With cGMP signaling being as vital as it is to the physiologic functions of the heart and vasculature, it comes as no surprise that dysfunction at any level of the cGMP signaling pathway is a factor in many cardiovascular diseases. Endothelial cell dysfunction contributes to hypertensive disease, both systemic and pulmonary, and atherosclerosis; vascular smooth muscle dysfunction, systemic and pulmonary hypertensive and ischemic heart disease; and cardiac myocyte dysfunction, hypertrophic and ischemic heart disease as well as cardiomyopathy and heart failure (Fig. 4). Dysfunctional cGMP signaling has also been implicated in dysfunctional mitochondrial metabolism, an area that is now beginning to be explored for its role in heart disease.
Figure 4.

Dysfunctional cGMP signaling in cardiovascular diseases.
4.1 Hypertension
That NO release contributes to basal vascular tone is supported by data from eNOS knockout mice which display increased systemic resistance and blood pressure (Stauss et al., 1999; Yang et al., 1999), and results from humans and other experimental animals given NOS inhibitors such as L-NMMA. While the blood pressure rise itself is often modest (<5%), systemic resistance rises more (20-30%). The disparity is due to a decline in cardiac output (stroke volume and in some instances heart rate) due to the afterload effect on the heart (Clarkson et al., 1995; Sakuma et al., 1992; Stamler et al., 1994). Indeed, endothelial dysfunction occurs in many forms of vascular disease, independent of the etiology and vascular structure (Rizzoni et al., 1998) and is thought to be due to altered oxidative stress processes. However, the presence of endothelial dysfunction per se – as commonly observed in atherosclerotic models – does not necessarily translate to increased arterial pressures. Reduced NO function has been attributed to deficiencies in the NOS signaling enzymes and/or their function, as well as scavenging effects from the enhanced production of superoxide anion (O2-) (Grunfeld et al., 1995; Rajagopalan et al., 1996) or angiotensin-II (Rajagopalan et al., 1996)
NOS function and oxidant stress are linked by the phenomenon of NOS uncoupling. This involves a change in the enzyme configuration resulting in less tight molecular packing of the homodimer and production of superoxide rather than nitric oxide (Kuzkaya et al., 2003; Rosen et al., 2002; Xia et al., 1996). eNOS uncoupling, demonstrated by elevated eNOS expression but reduced NO production, was noted in an in vivo model of angiotensin-II induced hypertension in Wistar rats (Mollnau et al., 2002). eNOS uncoupling can occur due to cofactor or co-substrate deficiency, or to direct biochemical disruption of the enzyme. Depletion of cofactor BH4 can occur by its oxidatation to BH3- radical and BH2, neither of which can support NO-generation by NOS. Superoxide generated by several other sources including NADPH oxidase can result in eNOS uncoupling and thus contributes to hypertension induced by angiotensin II (Mollnau et al., 2002) and in DOCA-salt models (Landmesser et al., 2003). Other ROS sources including mitochondria or xanthine oxidase may also result in NOS uncoupling. The bioavailability of BH4 is further influenced by declines in its de novo synthesis by GTP cyclohydrolase or by inhibition of enzymes such as dihydrofolate reductase that are required to restore BH4 from BH2 (salvage pathway)(Nichol et al., 1983). BH2 can also competitively bind to eNOS; thus the ratio of BH2/BH4 appears to be an important factor for normal or abnormal NOS enzyme function (Mollnau et al., 2002; Vasquez-Vivar et al., 2002).
In addition to NO synthesis, expression of its downstream target sGC in endothelial cells also declines in hypertension (Bauersachs et al., 1998; Kloss et al., 2000; Mollnau et al., 2002). Furthermore, both superoxide (Brune et al., 1990) and peroxynitrite (Weber et al., 2001) may inhibit sGC directly. Downstream PKG activity measured by immunodetection of phosphorylated VASP is also markedly reduced in models of angiotensin II-induced hypertension (Mollnau et al., 2002). However, recent studies have found that oxidant stress modification of PKG can actually stimulate the kinase (Burgoyne et al., 2007), so whether this activity corresponds to the decline in upstream triggers or other changes at the enzyme itself is less clear.
4.2 Atherosclerosis
Atherosclerosis is a dynamic pathologic process involving vascular injury, endothelial dysfunction, cellular proliferation, inflammation, and accumulation of oxidized low density lipoproteins (LDL). Both vascular endothelial and smooth muscle cells become impaired secondary to increased oxidative stress. As in hypertension, superoxide production by xanthine oxidase (Ohara et al., 1993), NADPH oxidase(Hathaway et al., 2002), and eNOS (Laursen et al., 2001; Oelze et al., 2000) are increased in hypercholesterolemia and atherosclerosis. Inconsistent changes in the expression of sGC have been reported in different animal models of hypercholesterolemia and atherosclerosis (Laber et al., 2002; Mollnau et al., 2003). However, sGC activity appears dysfunctional as endothelium-dependent and –independent vasodilation are markedly inhibited (Laber et al., 2002). While PKG expression remains unchanged, its activity as assessed by phosphorylated-VASP is strongly reduced (Oelze et al., 2000). Increased local oxidative stress and endothelial dysfunction in the coronary circulation was demonstrated in humans with early atherosclerosis but without obstructive coronary artery disease (Lavi et al., 2007; Lavi et al., 2008). Assessment during angiography revealed enhanced generation of local superoxide and conserved NO production but reduced bioavailability of NO.
4.3 Pulmonary hypertension
As with the systemic vasculature, NO-cGMP signaling also modulates vascular tone in the pulmonary circulatory system. Unlike the systemic vasculature, the pulmonary vasculature is a low pressure system with a total resistance about an eighth of the systemic vasculature. Pulmonary vascular resistance occurs predominantly at the peripheral precapillary beds, where NO-cGMP signaling plays an even larger role in modulating changes in the pulmonary microcirculation. This is highlighted by the developmental regulation of pulmonary eNOS expression seen across multiple species, in which pulmonary eNOS and iNOS are markedly upregulated in fetal life, peak shortly after birth, and then decline (Arrigoni et al., 2002; Kawai et al., 1995; North et al., 1994; Parker et al., 2000; Shaul et al., 2002; Shaul et al., 1997). Congenital diseases such as persistent pulmonary hypertension of the newborn and congenital diaphragmatic hernia, in which the pulmonary circulation fails to switch to a low pressure system at birth, have been associated with reduced NOS expression, bioavailability of NOS cofactors, and NO production (Fineman et al., 1994; Vosatka et al., 1994). In fact, inhaled nitric oxide has a profound vasodilatory effect on the pulmonary hypertensive fetal circulation and is used for treatment of the disease in newborns.
The role of NO-cGMP signaling in the adult pulmonary circulatory system, however, is less clear. Under normoxic conditions, inhaled NO has little vasodilatory effect on the adult pulmonary vasculature, suggesting perhaps a maximally vasodilated state at baseline or a non-responsiveness to NO (Jiang et al., 2002; Koizumi et al., 1994; Pison et al., 1993). In models of hypoxia-, thromboxane-, or endotoxin-induced pulmonary hypertension or acute lung injury associated pulmonary hypertension, NO blunts vasoconstriction and lowers pulmonary arterial pressure (Bottiger et al., 1996; Frostell et al., 1993; Rich et al., 1993; Rossaint et al., 1993; Weitzberg et al., 1993). How dysregulation of the NO-cGMP pathway contributes to the development of pulmonary hypertension is not well understood; contradictory evidence has arisen from models of experimentally induced pulmonary hypertension and patients with pulmonary arterial hypertension. In experimentally induced pulmonary hypertension, endogenous NO production appears increased, and chronic hypoxia upregulates the expression and activity of eNOS and iNOS in pulmonary vascular endothelium (le Cras et al., 1996; Xue et al., 1994). Yet expression of eNOS on immunostaining is reportedly diffuse in pulmonary vessels of patients without pulmonary hypertension and nearly absent in patients with pulmonary arterial hypertension (Giaid et al., 1995). Others have also reported decreased pulmonary endothelial expression of eNOS and increased expression of arginase II, an enzyme that decreases the bioavailability of the NOS cofactor L-arginase, in patients with pulmonary arterial hypertension (Xu et al., 2004). Studies of eNOS knockout mice have not been any more definitive, either, as both elevated and normal pulmonary artery pressures have been found in these mice (Fagan et al., 1999a; Fagan et al., 1999b; Quinlan et al., 2000; Steudel et al., 1997; Steudel et al., 1998).
Howsoever NOS dysfunction contributes to pulmonary hypertension, it is clear from studies of sGCα deficient mice that sGCα1 is essential for NO-mediated pulmonary vasodilation and limits chronic-hypoxia induced pulmonary vascular remodeling (Vermeersch et al., 2007). Thus while the mechanism by which abnormal NO-cGMP signaling leads to pathologic pulmonary hypertension remains unclear, the consensus has arisen that activating the NO-cGMP pathway in pulmonary hypertension can reduce pulmonary artery pressure and reverse pulmonary vascular remodeling. Inhaled nitric oxide and PDE5A inhibitors are used in the treatment of patients with pulmonary hypertension. Several groups are also investigating the therapeutic potential of enhancers of eNOS and activators of soluble guanylyl cyclase (Coggins et al., 2007; Dumitrascu et al., 2006; Evgenov et al., 2007; Tzao et al., 2001). The pre-clinical and clinical studies that support the use of these NO-cGMP modulating interventions are well summarized in a review centered on NO-cGMP signaling in pulmonary hypertension (Klinger, 2007).
4.4 Cardiac hypertrophy and ventricular remodeling
The heart responds to the physiological stresses of pressure- and volume-overload by increasing the heart wall thickness, a compensatory mechanism that initially serves to preserve cardiac function but ultimately leads to functional decline and clinical heart failure. Pressure-and/or volume-overload occur with a variety of cardiovascular diseases, from hypertension to myocardial infarction to valvular disease to cardiomyopathy. This ventricular remodeling, as the morphologic and structural changes are also called, is in fact the common end pathway of many cardiovascular diseases.
As detailed earlier, cGMP-PKG signaling in cardiac myocytes serves to counter the hypertrophic program. Both abnormal NO- and NP-triggered cGMP-PKG signaling have been associated with ventricular hypertrophy and heart failure in animal models and in human patients. Abnormalities range from dysfunctional cGMP production to altered catabolism of cGMP by differential PDE isoform expression and activity. In rodent models of pressure-overload hypertrophy, differential expression of the NOS isoforms and consequent alterations in cGMP signal compartmentation appear to contribute to the pathophysiology of cardiac hypertrophy and heart failure (Loyer et al., 2008). NOS uncoupling is also a significant component of the pathophysiology of hypertrophy. Sustained pressure overload activates reactive oxygen species which decrease the bioavailablity of the NOS cofactor BH4. Exogenous BH4 administration has been shown to recouple NOS and reverse advanced hypertrophy in mice with transaortic constriction induced pressure overload (Moens et al., 2008b). A functional uncoupling of membrane-associated sGC in pressure overload hypertrophy is also under investigation (Tsai et al., 2008) whereby a dissociation of sGC from the plasmalemmal lipid raft domains appears to result in decreased cGMP production upon NO stimulation. Abnormal NP-cGMP signaling is also seen in hypertrophy. A functional deletion mutation of the human NPR-A gene and decreased receptor expression has been associated with ventricular hypertrophy in a Japanese population (Nakayama et al., 2000). While this has not born out in other populations, it is one example of the significance of reduced NP signaling in hypertrophy.
The activity of myocardial PDEs has been shown to change in hypertrophy (Osadchii, 2007). In Dahl salt-sensitive hypertensive rats, pressure overload hypertrophy and heart failure are associated with increased myocardial expression and activity of PDE3 and reduced cAMP levels (Takahashi et al., 2002). Similarly, increasing cAMP-hydrolysis by cytosolic PDE1 and PDE2 have been found in hypertrophied ventricles of rats with aortic constriction (Yanaka et al., 2003). Myocardial PDE5 expression and activity also increases in pressure overload hypertrophy in mice (Takimoto et al., 2005a; Takimoto et al., 2005b; Zhang et al., 2008a).
4.5 Myocardial ischemia
Myocardial injury associated with ischemia and reperfusion can be limited by ischemic pre-conditioning, the process by which brief, sublethal episodes of ischemia stimulate a protective response against subsequent, more severe ischemia that would otherwise cause myocardial cell death. Ischemic pre-conditioning has been described in many tissues, including the heart, brain, and liver, and can be divided into two phases--- an early, rapid phase and a delayed, longer phase (Downey et al., 2007). The NO-cGMP-PKG pathway has been implicated in both phases of ischemic preconditioning as well as in ischemic post-conditioning, by which reperfusion is briefly interrupted initially, shortly after the end of ischemia (Costa et al., 2008; Javadov et al., 2003; Tsang et al., 2005; Wang et al., 2005; Yellon et al., 2003). The molecular mechanism underlying NO-cGMP-PKG signaling in ischemic preconditioning is not fully understood, but ischemia-reperfusion injury and cardiac ischemic pre-conditioning appear to depend on iNOS, rather than nNOS or eNOS (Guo et al., 2005; Jones et al., 2006). PKG-Iα is thought to exert its protective effect by opening inner membrane mitochondrial ATP-sensitive K+ (mitoKATP) channels. How PKG-Iα opens mitoKATP is not fully understood but it is thought to phosphorylate a serine or threonine of an unknown mitochondrial outer membrane protein. Opening of mitoKATP causes swelling of the matrix and generation of ROS. Mitochondrial ROS activates cytosolic PKC through redox signalling, triggering PKC phosphorylation of Akt, ERK, JNK, and glycogen synthase kinase 3β, and increasing the expression of NOS and Bcl-2 (Das et al., 2006; Das et al., 2008). All play essential roles in the protective anti-necrotic and anti-apoptotic effect of PKGIα following ischemia-reperfusion injury in cardiac myocytes.
NO-cGMP-PKG signaling also plays an important role in post-infarction cardiac remodeling, a process that involves myocyte hypertrophy, chamber dilation, and interstitial fibrosis. As discussed in earlier sections, uncoupled eNOS, reduced NO bioavailability, reduced BH4 bioavailability, and altered PDE isoform activity mediate the pathophysiology of cardiac hypertrophy and thus post-infarction remodeling.
4.6 Mitochondrial metabolism
Over the past few years, cGMP-PKG signaling in mitrochondria has received increasing attention for its role in ischemic pre-conditioning and anti-oxidant cardioprotection. Several groups have demonstrated that NO acts as an endogenous opener of the inner membrane mitochondrial ATP-sensitive K+ (mitoKATP) channels by activating sGC production of cGMP with subsequent PKG activation (Costa et al., 2005; Quinlan et al., 2008; Sasaki et al., 2000). PKG is thought to phosphorylate an as yet unidentified target protein which transmits the cardioprotective signal from the cytosol to the mitochondrial membrane via PKCε, a novel isoform of PKC that is calcium-independent but phospholipids- and diacylglycerol- dependent. PKCε then directly interacts with mitoKATP, opening the channel, thereby blunting mitochondrial calcium overload and conferring cardioprotection (Costa et al., 2005). Furthermore, NO has been shown to mobilize intracellular Zn2+ via a cGMP-PKG-dependent signal pathway in rat cardiac myocytes, preventing the mitochondrial death pathway via Zn2+(Jang et al., 2007).
4.7 Dystrophy related cardiomyopathies
The far-reaching impact of dysregulated cGMP signaling is also demonstrated by a subset of muscular dystrophy that is accompanied by cardiomyopathy. The underlying genetic defect, mutations in the gene encoding the plasma membrane protein dystrophin, affects both skeletal muscle and cardiac muscle. Dystrophin forms a protein complex at the plasma membrane (dystrophin glycoprotein complex, DGC) that functions as a mechano-signal transducer and stabilizes the interaction of cardiac and skeletal myocytes with the extracellular matrix. DGC is known to interact with NOS. Abnormalities of dystrophin have been implicated in both inherited and acquired cardiomyopathies. In Duchenes muscular dystrophy (DMD), dystrophin deficiency leads to a dilated cardiomyopathy, which can develop independent of the skeletal and vascular pathology and accounts for significant mortality in DMD patients. While the exact function of dystrophin in the myocardium remains incompletely understood, several studies have suggested that NO-cGMP signaling is impaired in dystrophin cardiomyopathy.
The most widely used animal model of DMD, the mdx mouse, lacks dystrophin and develops a wide range of cardiac defects, including impaired conduction, arrhhythmias, left ventricular dysfunction, and dilated cardiomyopathy (Quinlan et al., 2004). Early studies of mdx mice revealed significantly decreased nNOS activity (Bia et al., 1999), suggesting that increasing NO production may have therapeutic benefits. Transgenic myocardial expression of nNOS in dystrophin-deficient mdx mice in fact did prevent cardiac fibrosis and mitigated cardiac autonomic dysfunction and arrhythmogenicity (Wehling-Henricks et al., 2005). Though the exact relationship between nNOS activity and cardiomyopathy is not fully understood, subsequent studies reinforce a link between dysfunctional NO-cGMP signaling and cardiac dysfunction in mdx mice. For example, transgenic cardiac-specific overexpression of sGC in dystrophin-deficient mdx mice improved cardiac contractile performance, myocardial metabolic status, and sarcolemmal integrity (Khairallah et al., 2008), and this group recently reported similar benefits in mdx mice treated with sildenafil (Khairallah et al., 2008).
Another model of muscular dystrophy and cardiomyopathy, the sarcoglycan mutant mice, is also characterized by NOS dysfunction. In this model, nNOS expression is normal. However, mislocalization of myocardial eNOS is associated with pathologic NO gradients and regions of focal damage (Heydemann et al., 2004), and and NOS inhibition with L-NAME improved cardiac autonomic dysfunction and arrhythmogencity. These findings are at odds with mdx model and may be particular to the sarcoglycan mutation, as overexpression of eNOS alone does not generate cardiomyopathy (Brunner et al., 2001).
4.8 Heart failure
Disease progression of the hypertrophied or remodeled heart inevitably leads to heart failure. As such, the above discussion of dysregulated cGMP signaling in hypertrophy and remodeling also applies in heart failure. Moreover, natriuretic peptide signaling gains greater prominence in heart failure as plasma natriuretic peptide concentrations become markedly elevated (Burnett, Jr. et al., 1986; Sugawara et al., 1988; Tikkanen et al., 1985). Vasodilatory and diuretic responses to ANP are blunted in heart failure animal models (Drexler et al., 1987; Riegger et al., 1988) and in heart failure patients (Cody et al., 1986; Hirooka et al., 1990; Tsutamoto et al., 1993). The blunted response has been attributed by some to a reduction in NPR-A expression, as seen in the pulmonary vasculature of heart failure patients (Tsutamoto et al., 1992) and in the systemic vasculature of rats models of high-output heart failure (Garcia et al., 1992). Others have noted a decreased responsiveness of NPR-A to ANP without significant change in NPR-A receptor expression levels (Kuhn et al., 2004). An upregulation of an NP clearance receptor (NPR-C) has also been suggested as the mechanism by which natriuretic peptide receptor responsiveness is diminished (Andreassi et al., 2001; Kuhn et al., 2004). Similarly a reduction in the bioavailability of NO in heart failure is felt to be related to the decreased expression and/or activity of eNOS in the failing myocardium of patients (Drexler et al., 1998; Heymes et al., 1999) and animal models (Balligand et al., 1997; Bauersachs et al., 1998; Crabos et al., 1997; Wiemer et al., 1997).
5 Pharmacologic modulation of cGMP signaling in cardiovascular disease
Given that cGMP signaling is dysregulated in a wide spectrum of cardiovascular diseases and can be altered at multiple levels, from upstream triggering events to downstream effectors and regulatory molecules, each step of the cascade becomes a promising target for pharmacologic therapy (Table 1).
Table 1. Novel cardiovascular pharmacotherapies that target the cGMP signaling cascade.
| Drug | Structure | Target Molecule | Action | Disease Therapy |
|---|---|---|---|---|
| Folate | 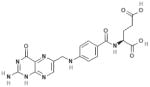 |
eNOS | Active metabolite 5-methyltetrahydro-folate (5-MTHF) is structurally similar to BH4 ↑eNOS coupling ↑BH4 binding affinity to eNOS ↑electron transfer ↑Regeneration of BH4 from inactive form BH2 |
Familial hypercholesterolemia DM CAD Ischemia/reperfusion* |
| BH4 | 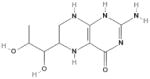 |
eNOS | ↑Electron transfer from eNOS reductase domain Maintain ferrous heme moiety in redox active form ↑Homodimerization of active eNOS |
Ventricular remodeling* HTN PAH CAD Intermittent claudication |
| NCX4016 (NO-NSAID) |
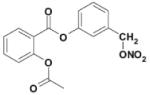 |
sGC | Release nitric oxide | CTEPH Myocardial ischemia* Vascular injury* |
| NCX 6550 (NO-pravastatin) NCX 6553 (NO-fluvastatin) |
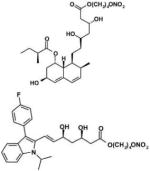 |
sGC | Release nitric oxide ↓Vascular smooth muscle cell proliferation ↓Inflammatory effects on macrophages/monocytes ↓Platelet activation ↓Thrombotic activity |
Atherosclerosis* Peripheral vascular disease* |
| BNP (Nesiritide) |
 |
NPR-A (pGC) |
Vasodilatation Natriuresis Diuresis |
CHF |
| CD-NP |  |
NPR-A, NPR-B | Venodilation Natriuresis Diuresis Renin inhibition Cardiac unloading ↓Proliferation |
CHF |
| sGC stimulators (i.e. YC-1, BAY 60-4552) | 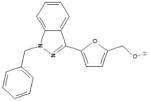 |
sGC | Activates sGC in its reduced form (heme-dependent) Activates sGC synergistically with NO and CO Vasodilation |
CTEPH PAH CHF |
| sGC activators (i.e. BAY 58-2667) | 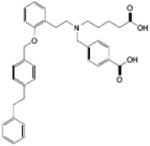 |
sGC | Activates sGC in a heme-independent fashion (heme-deficient or heme-oxidized) Inhibit platelet aggregation and thrombus formation Arterial and venous vasodilation Diuresis Natriuresis Cardiorenal protection |
CHF |
5.1 Enhanced cGMP production
Upregulation of cGMP production can be approached by either NO or NP triggered pathways with direct activation of the respective guanylyl cyclases. With the NO-cGMP pathway, other strategies for recoupling NOS and enhancing NO bioavailability have also been pursued.
5.1.1 NOS coupling and activation
Both folate and tetrahydrobiopterin are being explored for their therapeutic potential in reversing endothelial and myocardial dysfunction in cardiovascular diseases. Folate, the synthetic form of the water soluble B vitamin folic acid, is metabolized into the active form 5-methyltetrahydrofolate (5-MTHF). 5-MTHF is structurally similar to tetrahydrobiopterin (BH4), the essential co-factor of endothelial nitric oxide synthase (eNOS). As previously discussed, BH4 facilitates the electron transfer from eNOS reductase domain, thereby maintaining the ferrous heme moiety in its redox active form and promoting the homodimerization of active eNOS. Diminished bioavailability of BH4 with subsequent uncoupling of eNOS, decreased NO formation, and increased production of reactive oxygen species have been associated with various cardiovascular disorders.
Folate can restore the bioavailability of BH4 through several mechanisms. 5-MTHF can enhance the efficacy of BH4 on eNOS coupling by improving the binding affinity of BH4 for eNOS or facilitating the electron transfer oxidation of BH4 into its radical form(Stroes et al., 2000). 5-MTHF can also promote regeneration of BH4 from its inactive form BH2 (Kaufman, 1991), further stabilize BH4 itself, and possibly interact with eNOS directly (Hyndman et al., 2002).
Clinical studies of folate have demonstrated improvement in endothelial function with folate supplementation in patients with familial hypercholesterolemia (Verhaar et al., 1998; Verhaar et al., 1999), diabetes (van Etten et al., 2002), and coronary artery disease (Doshi et al., 2001; Doshi et al., 2002; Shirodaria et al., 2007). Most recently, a study of folic acid pretreatment in rats demonstrated a protective effect of folate against myocardial dysfunction in acute ischemia and mitigated post-reperfusion injury (Moens et al., 2008a). The beneficial effect of folic acid was associated with the preservation of high-energy phosphates and reductions in ROS generation, eNOS uncoupling, and post-reperfusion cell death.
Similarly, BH4 has been found in animal studies to blunt ventricular remodeling in models of ischemic injury (Masano et al., 2008) and pressure overload (Moens et al., 2008b). An intriguing aspect of this pressure-overload study was that while BH4 treatment re-coupled NOS and resulted in a marked decline in oxidative stress in the myocardium, its efficacy in reversing hypertrophy/fibrosis and improving chamber function did not appear attributable to an anti-oxidant effect alone. A similar protocol performed with the anti-oxidant Tempol failed to mimic the effects of NOS-recoupling. Furthermore, despite recoupled NOS and enhanced NO-generation compared to the untreated pressure-overload controls, PKG activity was not increased by BH4 therapy. Thus, its effects do not appear to depend upon a cGMP-trigger, but more on re-balancing NO/ROS. Chronic oral therapy with BH4 has also been shown to reverse endothelial dysfunction and oxidative stress in patients with hypercholesterolemia (Cosentino et al., 2008). Whether these effects are cGMP/PKG dependent or not remains to be elucidated. Several phase II clinical trials of BH4 and a synthetic version of BH4 called 6R-BH4 are underway to test the safety and efficacy of these therapies for the treatment of systemic hypertension (ClinicalTrials.gov NCT00325962, NCT00208780), pulmonary hypertension (Clinical Trials.gov NCT00435331), intermittent claudication (Clinical Trials.gov NCT00403494), and coronary artery disease (Clinical Trials.gov NCT00423280).
5.1.2 Activation of guanylyl cyclases
A more direct approach to increasing cGMP production is activation of either guanylyl cyclase via ligand stimulation (e.g. natriuretic peptides, organonitrates) or synthetic activators (e.g. sGC activators)
5.1.2.1 Nitrates and natriuretic peptides
Organic nitrates have long been used as short term treatments for unstable angina, acute myocardial infarction, decompensated heart failure, and hypertensive crisis. Their chronic use, however, is limited by tolerance and cross-tolerance, phenomena whereby impaired response to nitrates and other endothelium-dependent and –independent vasodilators develop respectively. At present, nitrate-free periods are recommended in conjunction with sustained chronic nitrate therapy to help reduce tolerance. However, the limitation remains and has given rise to a novel class of NO-releasing drugs.
Nitro-non-steroidal anti-inflammatory drugs (NO-NSAIDs) were developed by the grafting of an organic NO-releasing moiety onto a well-established non-steroidal anti-inflamatory drug (NSAID) (Keeble et al., 2002). NO is slowly released upon catabolism of the NO-NSAID by esterases. The beneficial effects of NO on gastric mucosa (Takeuchi et al., 1998a; Takeuchi et al., 1998b) improves the gastrointestinal safety profile of NO-NSAIDS compared to conventional NSAIDs. NO-releasing aspirin (NCX 4016) has been studied in animal models of pulmonary thromboembolism (Momi et al., 2000), myocardial ischemia (Rossoni et al., 2000; Yamamoto et al., 2000), and vascular injury (Napoli et al., 2001), and each study demonstrated protective and beneficial effects with this drug.
Another novel NO-releasing drug class of particular interest is the NO-releasing derivatives of inhibitors of 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase. NCX 6550 and NCX 6553 are based upon the structures of pravastatin and fluvastatin, respectively. In in vitro studies with vascular smooth muscle cells and monocyte/macrophage cell line (Ongini et al., 2004) and in vivo mouse models of atherosclerosis (Dever et al., 2007; Dever et al., 2008), NCX 6550 and NCX 6553 demonstrated enhanced antiproliferative and anti-inflammatory effects beyond the pleiotropic actions observed with their parent statins. Like the NO-NSAIDs, NO-releasing statin derivatives also exert antiplatelet and antithrombotic activity (Rossiello et al., 2005). Interestingly, NO-releasing statins also promote reparative neovascularization in diabetic mouse model of peripheral limb ischemia, suggesting a role for these hybrid drugs in the treatment of peripheral vascular disease (Emanueli et al., 2007). In vitro cell culture studies have shown that the NO-releasing statins produce dose-dependent increases in cGMP levels (Ongini et al., 2004).
Synthetic recombinant B-type natriuretic peptide (nesiritide) was shown in several studies to have beneficial hemodynamic effects, including vasodilatation, natriuresis, and diuresis, in patients with congestive heart failure (Abraham et al., 1998; Holmes et al., 1993; Marcus et al., 1996; Mills et al., 1999). Outpatient ‘tune-ups’ with intravenous nesiritide infusión became popular earlier in the past decade based in part on results of a trial that suggested the treatment could be a valuable addition to the pharmacologic armamentarium in treating acute decompensated heart failure (Colucci et al., 2000). However, follow-up data of these studies raised concerns regarding an increase in 30-day mortality and worsening renal function with nesiritide treatment (Sackner-Bernstein et al., 2005a; Sackner-Bernstein et al., 2005b). Given the lack of sufficient hard data to support this type of use, nesiritide has since fallen out of favor. Development of other novel synthetic natriuretic peptides has nonetheless continued.
The more recent development in the NP field is the generation of a novel chimeric peptide composed of C-type natriuretic peptide (CNP) fused to the carboxyl terminal tail of Dendroaspis natriuretic peptide (DNP) was shown in pre-clinical in vitro cell culture assays and in vivo animal models to have venodilating, natriuretic, diuretic, renin inhibiting, cardiac unloading, and antiproliferative effects, all while enhancing glomerular filtration rate (Dickey et al., 2008; Lisy et al., 2008). That CD-NP has the hemodynamic benefits of BNP but not the adverse effects of profound hypotension or renal dysfunction has positioned this novel chimeric drug as potentially quite useful in the clinical heart failure therapy. Furthermore, CD-NP can activate NPR-B in addition to NPR-A, which suggests an added potential benefit of preventing or inhibiting cardiac hypertrophy and remodeling (Langenickel et al., 2006; Soeki et al., 2005). A Phase I human physiologic study to evaluate the renal, neurohumoral and hemodynamic effects of CD-NP in chronic stable NYHA Class II-III heart failure patients is being sponsored by the Mayo Clinic, though enrollment has yet to begin.
5.1.2.2 Synthetic activators and stimulators of sGC
NO-induced activation of sGC requires the presence of the reduced Fe2+ heme moiety. Removal or oxidation of the haem moiety renders sGC insensitive to NO, and can occur under conditions of oxidative stress. Two novel drug classes, sGC stimulators and sGC activators, can overcome these obstacles respectively by enhancing the sensitivity of the reduced enzyme to low levels of NO or by directly activating sGC (Evgenov et al., 2006). sGC stimulators then are heme-dependent drugs, whereas sGC activators are heme-independent drugs which can activate NO-unresponsive, heme-oxidized or heme-free sGC.
On the whole, the heme-dependent sGC stimulators (YC-1, BAY 41-2272, BAY 41-8543, BAY 63-2521, CFM-1571, and A-350619) directly activate sGC but do so synergistically with NO, potentiating the effect of low levels of NO and increasing the maximal catalytic rate of sGC. Many of the sGC stimulators, except for A-350619, are structurally based on YC-1. The potentiation effect of sGC stimulators are attributed in part to the stabilization of the nitrosyl-heme complex and the transformation of the NO-activated enzyme from a low- to a high-output activation state. The exact nature of the interaction between YC-1 and other sGC stimulators is not known. Binding of the compound to the sGC catalytic domain has been suggested (Yazawa et al., 2006). However, YC-1 induced effects have also been observed in heme-containing fragments of the β1 subunit lacking the catalytic domain (Denninger et al., 2000). YC-1 additionally inhibits PDE5, thereby further amplifying the downstream effects of its production of cGMP.
Heme-independent sGC activators (BAY 58-2667, HMR-1766) can activate heme-deficient and heme-oxidized sGC and thus exert an additive effect with NO. BAY 58-2667 in particular is the most potent NO-independent sGC activator and its action is further potentiated by the oxidation or removal of the prosthetic heme group of sGC. Reduced sGC however is virtually unresponsive to BAY 58-2667. This selective activation of heme-free or NO-insensitive, oxidized state of sGC by BAY 58-2667 makes it a novel enzyme specific and redox specific drug. sGC activators can mimic the spatial structure of the sGC porphyrin ligand and thus exert their effect either by binding to the unoccupied heme-binding pocket or by replacing the weakly bound oxidized heme.
sGC stimulators and activators were initially noted for their vasodilatory property and considered promising alternatives to nitrovasodilators. However, additional effects on platelet aggregation and overall hemodynamics in in vitro and in vivo animal studies suggest greater potential for sGC stimulators and activators as therapy for a broad range of cardiovascular diseases. Heme-independent, NO-independent sGC activator BAY 58-2667 potently inhibited platelet aggregation induced by thromboxane A2 mimic U46619, collagen, and ADP in human platelet rich plasma, independent of a thrombin-mediated pathway (Stasch et al., 2002). The antiplatelet effects of BAY 58-2667 were confirmed in vivo by prolonged rat tail bleeding times and the reduction of thrombus formation in FeCl3 thrombosis rat model (Stasch et al., 2002). BAY 58-2667 has also been shown to have a hemodynamic profile similar to nitroglycerin; it causes a dose-dependent, long-lasting decrease in blood pressure and reflex increase in heart rate by causing both arterial and venous vasorelaxation in anesthetized dogs (Stasch et al., 2002). The pharmacokinetics of BAY 58-2667 differ however in that it has lower clearance and longer half-life. The redox selectivity of BAY 58-2667 also becomes clinically significant as vascular dysfunction, ROS formation, heme oxidation, and ultimately heme loss are demonstrated in various pathophysiological animal models and human cardiovascular diseases. In fact, BAY 58-2667 was demonstrated to induce selective vasodilation of diseased blood vessels in both humans and animals (Stasch et al., 2006).
Pre-clinical studies of BAY 58-2667 in a dog model of tachypacing-induced heart failure demonstrated that the sGC activator potently unloaded the heart, reducing mean arterial, right atrial, pulmonary artery, and pulmonary capillary wedge pressures in a dose-dependent fashion. It increased cardiac output and renal blood flow, while preserving glomerular filtration rate and sodium and water excretion without further neurohumoral activation (Boerrigter et al., 2007). Similar hemodynamic and cardiorenal benefits had been shown with the sGC stimulator BAY 41-2272 as well (Boerrigter et al., 2003).
A phase I clinical trial has already assessed the safety, tolerability, pharmacokinetics, and pharmacodynamics of BAY 58-2667 in normal healthy volunteers (Frey et al., 2008). Intravenous administration of BAY 58-2667 potently reduced both preload and afterload, suggesting a potential role for acute administration of the drug for the management of decompensated heart failure. Currently a double-blind, placebo-controlled, multicenter, randomized Phase II clinical trial is underway to study the efficacy and tolerability of BAY 58-2667 in patients with decompensated chronic heart failure (ClinicalTrials.gov NCT00559650). The primary outcome measure is the change of the pulmonary capillary wedge pressure over 8 hours, with the secondary outcome measures as the quality of life and need for rehospitalization at 30 days follow-up.
An oral formulation of a sGC stimulator, BAY 60-4552, is being studied in a Phase I clinical trial as a proof of concept study to investigate the safety, tolerability, pharmacokinetics, and hemodynamic effects of the single dose medication in patients with biventricular heart failure and pulmonary hypertension (ClinicalTrials.gov NCT00565565).
Both sGC stimulators and activators have been studied extensively in models of systemic hypertension (Rothermund et al., 2000; Ruetten et al., 1999; Zanfolin et al., 2006), pulmonary hypertension (Deruelle et al., 2006; Deruelle et al., 2005a; Deruelle et al., 2005b; Dumitrascu et al., 2006; Evgenov et al., 2004), and vascular injury (Pan et al., 2004; Tulis et al., 2002) as well. In a rat model of hypertension, chronic treatment with the NOS inhibitor N-nitro-L-arginine methyl ester (L-NAME) caused marked and sustained arterial hypertension (Erley et al., 1995; Ribeiro et al., 1992), as well as cardiac hypertrophy, focal necrosis, and fibrosis (Laflamme et al., 1998; Navarro-Cid et al., 1996). Concomitant treatment of L-NAME-induced hypertensive rats with sGC stimulator BAY 41-2272 inhibited hypertension and protected against cardiac hypertrophy and fibrosis (Frey et al., 2008; Zanfolin et al., 2006). Pulmonary vascular remodeling and right heart hypertrophy have likewise been reversed or blunted by treatment with BAY 41-2272 and BAY 58-2667 in experimental models of pulmonary hypertension in NOS3-/- mice and monocrotaline-injected rats (Dumitrascu et al., 2006).
The oral sGC stimulator BAY 63-2521 is actively being studied in multiple clinical trials for the treatment of pulmonary hypertension of various etiologies--- chronic obstructive pulmonary disease, chronic thromboembolic disease, pulmonary arterial hypertension, and interstitial lung disease (ClinicalTrials.gov NCT00640315, NCT00454558, NCT00694850).
5.2 Enhanced activation of downstream effectors
While cGMP-dependent protein kinases execute the physiologic effects of cGMP in the cardiovascular system, direct small molecular activators of PKG have yet to be developed as a drug therapy. Much still remains to be elucidated about the various protein targets for PKG and mechanisms of intracellular modulation, regulation, and compartmentalization of PKG activity. PKG activators based on cGMP analogues are used in cell-systems but cannot be used clinically. “Hydrolysis-resistant” PKG activators (i.e. 8-pCPT-cGMP and 8-Br-PET-cGMP) can be degraded by distinct PDEs, and certain “specific” PKG inhibitors (i.e. Rp-8-pCPT-cGMPS and Rp-8-Br-PET-cGMPS) also inhibit certain PDEs. Moreover, Rp inhibitors can chemically convert into PKG activators. Thus the prevailing strategies for modulating cGMP levels are by enhancing its production through guanylyl cyclase stimulation and by halting its degradation through phosphodiesterase inhibition.
5.3 Modifying cGMP catabolism
Over the last twenty five years, inhibition of PDE5 has evolved as a therapeutic target for angina and systemic hypertension to erectile dysfunction to pulmonary arterial hypertension and now chronic heart failure. Sildenafil was the first of the PDE5 inhibitors to be developed, followed by vardenafil and then tadalafil. Initial clinical studies of sildenafil focused on its potential as an anti-angina drug. While sildenafil did not appear to have any significant benefit in the treatment of angina, a major side effect of the medication propelled a seismic change in its subsequent clinical development and marketing as the pharmacotherapy of erectile dysfunction was born. All three PDE5 inhibitors are similar in chemical structure, though vary somewhat with respect to pharmacokinetics (tadalafil having the longest t1/2), potency (vardenafil has the lowest IC50), and selectivity (tadalafil is arguably the most selective). Newer PDE5 inhibitors are under development with longer durations of action.
Given the high concentration of PDE5 in the lungs, a role for sildenafil in the treatment of primary pulmonary hypertension was also hypothesized. Several studies demonstrated a benefit of sildenafil in primary pulmonary hypertension with respect to exercise capacity, hemodynamics, functional status, and quality of life (Bhatia et al., 2003; Galie et al., 2005; Michelakis et al., 2002; Michelakis et al., 2003; Prasad et al., 2000; Rossi et al., 2008; Sastry et al., 2004). In 2005, sildenafil was ultimately approved for the treatment of pulmonary arterial hypertension. Comparison of the three PDE5 inhibitors in patients with pulmonary arterial hypertension revealed significant differences in pharmacokinetics, selectivity, and outcome(Ghofrani et al., 2004). Vardenafil exerts the most rapid effect on pulmonary vasorelaxation; however, only sildenafil and tadalafil are selective for the pulmonary vasculature. Sildenafil, alone, improved arterial oxygenation. However, cases have been reported in which tadalafil does improve arterial oxygenation in primary pulmonary arterial hypertension(Ghofrani et al., 2004; Palmieri et al., 2004). Presently, tadalafil is in Phase 3 clinical trials for pulmonary arterial hypertension. PDE5 inhibitors are also being studied in conjunction with upstream activators of the cGMP signaling cascade, such as natriuretic peptides, for a synergistic approach to pulmonary hypertension therapy (Baliga et al., 2008). Combination therapy with endothelin receptor antagonists (Wrishko et al., 2008) or prostacyclin (Bendayan et al., 2008) is another strategy for management of pulmonary arterial hypertension.
More recently, several groups have been studying the benefits of PDE5 inhibition in chronic heart failure. Preclinical studies of sildenafil in animal models of pressure-overload induced cardiac hypertrophy and heart failure have established that PDE5 inhibition indeed suppresses pathologic cardiac hypertrophy and fibrosis and improves cardiac function (Dumitrascu et al., 2006; Nagayama et al., 2008; Takimoto et al., 2005b), and is coupled with depressed activation of calcium-calmodulin dependent serine/threonine phosphatase calcineurin (Cn) and nuclear factor of activated T-cells (NFAT) (Fiedler et al., 2002; Takimoto et al., 2005b). In vivo studies recently demonstrated that PDE5 inhibition with sildenafil and subsequent PKG activation blunted cardiac hypertrophy and improved cardiac function even in Cn-/- mice subjected to transaortic constriction induced chronic pressure overload, suggesting that cGMP-PKG activation targets multiple downstream pathways and not only Cn/NFAT (Hsu et al., 2008). Even as the downstream signaling effects of PDE5 inhibition are being elucidated, several studies have already examined the clinical benefits of sildenafil in chronic heart failure.
Chronic sildenafil therapy has been shown in small clinical studies to improve functional exercise capacity (Guazzi et al., 2007b; Lewis et al., 2007b), exercise hemodynamics, and oxygen uptake (Bussotti et al., 2008; Lewis et al., 2007a) in chronic heart failure patients. The benefits of sildenafil in heart failure are derived from multiple effects, not just pulmonary vasodilation. Symptomatic improvement and increased exercise capacity was achieved by chronic sildenafil treatment largely through improved nitric oxide-mediated vasodilation, tempered the peripheral stimulus to hyperventilation, and heightened ventilatory efficiency and exercise performance (Guazzi et al., 2007).
Multiple clinical trials continue to investigate the efficacy of sildenafil in the treatment of heart failure. A Phase IV efficacy clinical trial of sildenafil in the management of moderate congestive heart failure is scheduled to begin shortly (ClinicalTrials.gov NCT00793338). The primary outcome of the study is change in 6-minute walk distance with secondary outcome measures including change in peak oxygen consumption, neurohormone levels, and quality of life. Sildenafil is also being examined in the RELAX study (ClinicalTrails.gov NCT00763867) for its potential in managing diastolic heart failure, with a primary outcome measure of change in exercise capacity as determined by peak oxygen uptake.
6 Conclusions and perspectives
The central role of cGMP in mediating various physiological processes within the cardiovascular system presents this second messenger as a prime target for pharmacologic modulation. Many different approaches have been undertaken to enhance the cGMP signal. Supplementation with essential co-factors for eNOS can restore the bioavailability of NO in diseased states. NO-donor drugs and synthetic chimeric natriuretic peptides can trigger cGMP production through their respective guanylyl cyclases. Synthetic stimulators and activators of soluble guanylyl cyclase further enhance and potentiate sGC activity. There are no other bioengineered direct activators of particulate guanylyl cyclases aside from synthetic natriuretic peptides. cGMP analogues are limited even as research tools. PDE5 inhibitors amplify the cGMP signal by blocking its degradation. Of the different cGMP modulating drugs discussed, PDE5 inhibitors are the only class approved for clinical use and remains to be approved for cardiovascular disease treatment. Novel PDE5 inhibitors are also in development which can offer improved pharmacokinetics and selectivity. Overall, the various drugs in clinical trial offer great promise in expanding our pharmacologic armamentarium for the treatment of cardiovascular diseases, ranging from hypertension to decompensated heart failure, and without the adverse side effects associated with current therapies, particularly as they relate to the cardiorenal syndrome.
The greatest challenge in modulating the cGMP signal perhaps lies in our limited understanding of the compartmentalization of cGMP and its functional implications. As we unravel the details of cGMP compartmentalization, we may find ourselves developing novel drugs that target not only a singular step of the cGMP signaling cascade but also a particular subcellular compartment wherein cGMP acts.
Abbreviations
- BH4
tetrahydrobiopterin
- cGMP
cyclic guanosine 3′,5′-monophosphate
- cAMP
cyclic adenosine 3′,5′-monophosphate
- DEA/NO
diethylamine NONOate
- DGC
dystrophin glycoprotein complex
- NO
nitric oxide
- NOS
nitric oxide synthase
- NP
natriuretic peptide
- NPR
natriuretic peptide receptor
- PDE
phosphodiesterase
- pGC
particulate guanylyl cyclase
- PKG
protein kinase G (cGMP-dependent protein kinase)
- sGC
soluble guanylyl cyclase
- VSMC
vascular smooth muscle cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham WT, Lowes BD, Ferguson DA, Odom J, Kim JK, Robertson AD, Bristow MR, Schrier RW. Systemic hemodynamic, neurohormonal, and renal effects of a steady-state infusion of human brain natriuretic peptide in patients with hemodynamically decompensated heart failure. J Card Fail. 1998;4:37–44. doi: 10.1016/s1071-9164(98)90506-1. [DOI] [PubMed] [Google Scholar]
- Ackermann U, Irizawa TG, Milojevic S, Sonnenberg H. Cardiovascular effects of atrial extracts in anesthetized rats. Can J Physiol Pharmacol. 1984;62:819–826. doi: 10.1139/y84-135. [DOI] [PubMed] [Google Scholar]
- Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammendola A, Geiselhoringer A, Hofmann F, Schlossmann J. Molecular determinants of the interaction between the inositol 1,4,5-trisphosphate receptor-associated cGMP kinase substrate (IRAG) and cGMP kinase Ibeta. J Biol Chem. 2001;276:24153–24159. doi: 10.1074/jbc.M101530200. [DOI] [PubMed] [Google Scholar]
- Andreassi MG, Del Ry S, Palmieri C, Clerico A, Biagini A, Giannessi D. Up-regulation of ‘clearance’ receptors in patients with chronic heart failure: a possible explanation for the resistance to biological effects of cardiac natriuretic hormones. Eur J Heart Fail. 2001;3:407–414. doi: 10.1016/s1388-9842(01)00161-1. [DOI] [PubMed] [Google Scholar]
- Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1994;91:7583–7587. doi: 10.1073/pnas.91.16.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci U S A. 1977;74:3203–3207. doi: 10.1073/pnas.74.8.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni FI, Hislop AA, Pollock JS, Haworth SG, Mitchell JA. Birth upregulates nitric oxide synthase activity in the porcine lung. Life Sci. 2002;70:1609–1620. doi: 10.1016/s0024-3205(02)01471-6. [DOI] [PubMed] [Google Scholar]
- Arstall MA, Sawyer DB, Fukazawa R, Kelly RA. Cytokine-mediated apoptosis in cardiac myocytes: the role of inducible nitric oxide synthase induction and peroxynitrite generation. Circ Res. 1999;85:829–840. doi: 10.1161/01.res.85.9.829. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Mulrow PJ, Franco-Saenz R, Snajdar R, Rapp J. Inhibition of aldosterone production by an atrial extract. Science. 1984;224:992–994. doi: 10.1126/science.6326267. [DOI] [PubMed] [Google Scholar]
- Atlas SA, Kleinert HD, Camargo MJ, Januszewicz A, Sealey JE, Laragh JH, Schilling JW, Lewicki JA, Johnson LK, Maack T. Purification, sequencing and synthesis of natriuretic and vasoactive rat atrial peptide. Nature. 1984;309:717–719. doi: 10.1038/309717a0. [DOI] [PubMed] [Google Scholar]
- Attina TM, Malatino LS, Maxwell SR, Padfield PL, Webb DJ. Phosphodiesterase type 5 inhibition reverses impaired forearm exercise-induced vasodilatation in hypertensive patients. J Hypertens. 2008;26:501–507. doi: 10.1097/HJH.0b013e3282f382ff. [DOI] [PubMed] [Google Scholar]
- Baliga RS, Zhao L, Madhani M, Lopez-Torondel B, Visintin C, Selwood D, Wilkins MR, MacAllister RJ, Hobbs AJ. Synergy between natriuretic peptides and phosphodiesterase 5 inhibitors ameliorates pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:861–869. doi: 10.1164/rccm.200801-121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balligand JL, Cannon PJ. Nitric oxide synthases and cardiac muscle. Autocrine and paracrine influences. Arterioscler Thromb Vasc Biol. 1997;17:1846–1858. doi: 10.1161/01.atv.17.10.1846. [DOI] [PubMed] [Google Scholar]
- Balligand JL, Ungureanu-Longrois D, Simmons WW, Kobzik L, Lowenstein CJ, Lamas S, Kelly RA, Smith TW, Michel T. Induction of NO synthase in rat cardiac microvascular endothelial cells by IL-1 beta and IFN-gamma. Am J Physiol. 1995;268:H1293–H1303. doi: 10.1152/ajpheart.1995.268.3.H1293. [DOI] [PubMed] [Google Scholar]
- Bauersachs J, Bouloumie A, Mulsch A, Wiemer G, Fleming I, Busse R. Vasodilator dysfunction in aged spontaneously hypertensive rats: changes in NO synthase III and soluble guanylyl cyclase expression, and in superoxide anion production. Cardiovasc Res. 1998;37:772–779. doi: 10.1016/s0008-6363(97)00250-2. [DOI] [PubMed] [Google Scholar]
- Beall AC, Kato K, Goldenring JR, Rasmussen H, Brophy CM. Cyclic nucleotide-dependent vasorelaxation is associated with the phosphorylation of a small heat shock-related protein. J Biol Chem. 1997;272:11283–11287. doi: 10.1074/jbc.272.17.11283. [DOI] [PubMed] [Google Scholar]
- Behrends S, Harteneck C, Schultz G, Koesling D. A variant of the alpha 2 subunit of soluble guanylyl cyclase contains an insert homologous to a region within adenylyl cyclases and functions as a dominant negative protein. J Biol Chem. 1995;270:21109–21113. doi: 10.1074/jbc.270.36.21109. [DOI] [PubMed] [Google Scholar]
- Bendayan D, Shitrit D, Kramer MR. Combination therapy with prostacyclin and tadalafil for severe pulmonary arterial hypertension: a pilot study. Respirology. 2008;13:916–918. doi: 10.1111/j.1440-1843.2007.01176.x. [DOI] [PubMed] [Google Scholar]
- Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- Bender AT, Ostenson CL, Wang EH, Beavo JA. Selective up-regulation of PDE1B2 upon monocyte-to-macrophage differentiation. Proc Natl Acad Sci U S A. 2005;102:497–502. doi: 10.1073/pnas.0408535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethke T, Eschenhagen T, Klimkiewicz A, Kohl C, von der LH, Mehl H, Mende U, Meyer W, Neumann J, Rosswag S. Phosphodiesterase inhibition by enoximone in preparations from nonfailing and failing human hearts. Arzneimittelforschung. 1992;42:437–445. [PubMed] [Google Scholar]
- Bethke T, Meyer W, Schmitz W, Scholz H, Stein B, Thomas K, Wenzlaff H. Phosphodiesterase inhibition in ventricular cardiomyocytes from guinea-pig hearts. Br J Pharmacol. 1992;107:127–133. doi: 10.1111/j.1476-5381.1992.tb14474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia S, Frantz RP, Severson CJ, Durst LA, McGoon MD. Immediate and long-term hemodynamic and clinical effects of sildenafil in patients with pulmonary arterial hypertension receiving vasodilator therapy. Mayo Clin Proc. 2003;78:1207–1213. doi: 10.4065/78.10.1207. [DOI] [PubMed] [Google Scholar]
- Bia BL, Cassidy PJ, Young ME, Rafael JA, Leighton B, Davies KE, Radda GK, Clarke K. Decreased myocardial nNOS, increased iNOS and abnormal ECGs in mouse models of Duchenne muscular dystrophy. J Mol Cell Cardiol. 1999;31:1857–1862. doi: 10.1006/jmcc.1999.1018. [DOI] [PubMed] [Google Scholar]
- Birschmann I, Walter U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3′,5′-cyclic monophosphate-dependent protein kinase. Acta Biochim Pol. 2004;51:397–404. [PubMed] [Google Scholar]
- Birukova AA, Zagranichnaya T, Alekseeva E, Bokoch GM, Birukov KG. Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protection. J Cell Physiol. 2008;215:715–724. doi: 10.1002/jcp.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch KD, Scott JA, Zisfein JB, Fallon JT, Margolies MN, Seidman CE, Matsueda GR, Homcy CJ, Graham RM, Seidman JG. Biosynthesis and secretion of proatrial natriuretic factor by cultured rat cardiocytes. Science. 1985;230:1168–1171. doi: 10.1126/science.2933808. [DOI] [PubMed] [Google Scholar]
- Blumenthal DK, Stull JT, Gill GN. Phosphorylation of cardiac troponin by guanosine 3′:5′-monophosphate-dependent protein kinase. J Biol Chem. 1978;253:324–326. [PubMed] [Google Scholar]
- Boerrigter G, Costello-Boerrigter LC, Cataliotti A, Lapp H, Stasch JP, Burnett JC., Jr Targeting heme-oxidized soluble guanylate cyclase in experimental heart failure. Hypertension. 2007;49:1128–1133. doi: 10.1161/HYPERTENSIONAHA.106.083832. [DOI] [PubMed] [Google Scholar]
- Boerrigter G, Costello-Boerrigter LC, Cataliotti A, Tsuruda T, Harty GJ, Lapp H, Stasch JP, Burnett JC., Jr Cardiorenal and humoral properties of a novel direct soluble guanylate cyclase stimulator BAY 41-2272 in experimental congestive heart failure. Circulation. 2003;107:686–689. doi: 10.1161/01.cir.0000055737.15443.f8. [DOI] [PubMed] [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- Boolell M, Gepi-Attee S, Gingell JC, Allen MJ. Sildenafil, a novel effective oral therapy for male erectile dysfunction. Br J Urol. 1996;78:257–261. doi: 10.1046/j.1464-410x.1996.10220.x. [DOI] [PubMed] [Google Scholar]
- Borlaug BA, Melenovsky V, Marhin T, Fitzgerald P, Kass DA. Sildenafil inhibits beta-adrenergic-stimulated cardiac contractility in humans. Circulation. 2005;112:2642–2649. doi: 10.1161/CIRCULATIONAHA.105.540500. [DOI] [PubMed] [Google Scholar]
- Bottiger BW, Motsch J, Dorsam J, Mieck U, Gries A, Weimann J, Martin E. Inhaled nitric oxide selectively decreases pulmonary artery pressure and pulmonary vascular resistance following acute massive pulmonary microembolism in piglets. Chest. 1996;110:1041–1047. doi: 10.1378/chest.110.4.1041. [DOI] [PubMed] [Google Scholar]
- Brovkovych V, Gao XP, Ong E, Brovkovych S, Brennan ML, Su X, Hazen SL, Malik AB, Skidgel RA. Augmented inducible nitric oxide synthase expression and increased NO production reduce sepsis-induced lung injury and mortality in myeloperoxidase-null mice. Am J Physiol Lung Cell Mol Physiol. 2008;295:L96–103. doi: 10.1152/ajplung.00450.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune B, Schmidt KU, Ullrich V. Activation of soluble guanylate cyclase by carbon monoxide and inhibition by superoxide anion. Eur J Biochem. 1990;192:683–688. doi: 10.1111/j.1432-1033.1990.tb19276.x. [DOI] [PubMed] [Google Scholar]
- Brunner F, Andrew P, Wolkart G, Zechner R, Mayer B. Myocardial contractile function and heart rate in mice with myocyte-specific overexpression of endothelial nitric oxide synthase. Circulation. 2001;104:3097–3102. doi: 10.1161/hc5001.101966. [DOI] [PubMed] [Google Scholar]
- Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schroder E, Browning DD, Eaton P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317:1393–1397. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- Burnett JC, Jr, Kao PC, Hu DC, Heser DW, Heublein D, Granger JP, Opgenorth TJ, Reeder GS. Atrial natriuretic peptide elevation in congestive heart failure in the human. Science. 1986;231:1145–1147. doi: 10.1126/science.2935937. [DOI] [PubMed] [Google Scholar]
- Bussolati B, Dunk C, Grohman M, Kontos CD, Mason J, Ahmed A. Vascular endothelial growth factor receptor-1 modulates vascular endothelial growth factor-mediated angiogenesis via nitric oxide. Am J Pathol. 2001;159:993–1008. doi: 10.1016/S0002-9440(10)61775-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussotti M, Montorsi P, Amato M, Magini A, Baldassarre D, Tantardini F, Veglia F, Agostoni P. Sildenafil improves the alveolar-capillary function in heart failure patients. Int J Cardiol. 2008;126:68–72. doi: 10.1016/j.ijcard.2007.03.118. [DOI] [PubMed] [Google Scholar]
- Butt E, Bernhardt M, Smolenski A, Kotsonis P, Frohlich LG, Sickmann A, Meyer HE, Lohmann SM, Schmidt HH. Endothelial nitric-oxide synthase (type III) is activated and becomes calcium independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J Biol Chem. 2000;275:5179–5187. doi: 10.1074/jbc.275.7.5179. [DOI] [PubMed] [Google Scholar]
- Carrier GO, Fuchs LC, Winecoff AP, Giulumian AD, White RE. Nitrovasodilators relax mesenteric microvessels by cGMP-induced stimulation of Ca-activated K channels. Am J Physiol. 1997;273:H76–H84. doi: 10.1152/ajpheart.1997.273.1.H76. [DOI] [PubMed] [Google Scholar]
- Cary SP, Winger JA, Marletta MA. Tonic and acute nitric oxide signaling through soluble guanylate cyclase is mediated by nonheme nitric oxide, ATP, and GTP. Proc Natl Acad Sci U S A. 2005;102:13064–13069. doi: 10.1073/pnas.0506289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro LR, Verde I, Cooper DM, Fischmeister R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation. 2006;113:2221–2228. doi: 10.1161/CIRCULATIONAHA.105.599241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Levine YC, Golan DE, Michel T, Lin AJ. Atrial natriuretic peptide-initiated cGMP pathways regulate vasodilator-stimulated phosphoprotein phosphorylation and angiogenesis in vascular endothelium. J Biol Chem. 2008;283:4439–4447. doi: 10.1074/jbc.M709439200. [DOI] [PubMed] [Google Scholar]
- Cherry PD, Furchgott RF, Zawadzki JV, Jothianandan D. Role of endothelial cells in relaxation of isolated arteries by bradykinin. Proc Natl Acad Sci U S A. 1982;79:2106–2110. doi: 10.1073/pnas.79.6.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiche JD, Schlutsmeyer SM, Bloch DB, de la Monte SM, Roberts JD, Jr, Filippov G, Janssens SP, Rosenzweig A, Bloch KD. Adenovirus-mediated gene transfer of cGMP-dependent protein kinase increases the sensitivity of cultured vascular smooth muscle cells to the antiproliferative and pro-apoptotic effects of nitric oxide/cGMP. J Biol Chem. 1998;273:34263–34271. doi: 10.1074/jbc.273.51.34263. [DOI] [PubMed] [Google Scholar]
- Clarkson PB, Lim PO, MacDonald TM. Influence of basal nitric oxide secretion on cardiac function in man. British Journal of Clinical Pharmacology. 1995;40:299–305. doi: 10.1111/j.1365-2125.1995.tb04550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerk A, Cullingford TE, Fuller SJ, Giraldo A, Markou T, Pikkarainen S, Sugden PH. Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. J Cell Physiol. 2007;212:311–322. doi: 10.1002/jcp.21094. [DOI] [PubMed] [Google Scholar]
- Cody RJ, Atlas SA, Laragh JH, Kubo SH, Covit AB, Ryman KS, Shaknovich A, Pondolfino K, Clark M, Camargo MJ. Atrial natriuretic factor in normal subjects and heart failure patients. Plasma levels and renal, hormonal, and hemodynamic responses to peptide infusion. J Clin Invest. 1986;78:1362–1374. doi: 10.1172/JCI112723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggins MP, Bloch KD. Nitric oxide in the pulmonary vasculature. Arterioscler Thromb Vasc Biol. 2007;27:1877–1885. doi: 10.1161/ATVBAHA.107.142943. [DOI] [PubMed] [Google Scholar]
- Colucci WS, Elkayam U, Horton DP, Abraham WT, Bourge RC, Johnson AD, Wagoner LE, Givertz MM, Liang CS, Neibaur M, Haught WH, LeJemtel TH. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. N Engl J Med. 2000;343:246–253. doi: 10.1056/NEJM200007273430403. [DOI] [PubMed] [Google Scholar]
- Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- Cornwell TL, Arnold E, Boerth NJ, Lincoln TM. Inhibition of smooth muscle cell growth by nitric oxide and activation of cAMP-dependent protein kinase by cGMP. Am J Physiol. 1994;267:C1405–C1413. doi: 10.1152/ajpcell.1994.267.5.C1405. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Hurlimann D, Delli GC, Chenevard R, Blau N, Alp NJ, Channon KM, Eto M, Lerch P, Enseleit F, Ruschitzka F, Volpe M, Luscher TF, Noll G. Chronic treatment with tetrahydrobiopterin reverses endothelial dysfunction and oxidative stress in hypercholesterolaemia. Heart. 2008;94:487–492. doi: 10.1136/hrt.2007.122184. [DOI] [PubMed] [Google Scholar]
- Costa AD, Garlid KD, West IC, Lincoln TM, Downey JM, Cohen MV, Critz SD. Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ Res. 2005;97:329–336. doi: 10.1161/01.RES.0000178451.08719.5b. [DOI] [PubMed] [Google Scholar]
- Costa AD, Pierre SV, Cohen MV, Downey JM, Garlid KD. cGMP signalling in pre- and post-conditioning: the role of mitochondria. Cardiovasc Res. 2008;77:344–352. doi: 10.1093/cvr/cvm050. [DOI] [PubMed] [Google Scholar]
- Crabos M, Coste P, Paccalin M, Tariosse L, Daret D, Besse P, Bonoron-Adele S. Reduced basal NO-mediated dilation and decreased endothelial NO-synthase expression in coronary vessels of spontaneously hypertensive rats. J Mol Cell Cardiol. 1997;29:55–65. doi: 10.1006/jmcc.1996.0251. [DOI] [PubMed] [Google Scholar]
- Croom KF, Curran MP, Abman SH, Channick RN, Heresi GA, Rubin LJ, Torbicki A. Sildenafil: a review of its use in pulmonary arterial hypertension. Drugs. 2008;68:383–397. doi: 10.2165/00003495-200868030-00009. [DOI] [PubMed] [Google Scholar]
- D'Souza SP, Davis M, Baxter GF. Autocrine and paracrine actions of natriuretic peptides in the heart. Pharmacol Ther. 2004;101:113–129. doi: 10.1016/j.pharmthera.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Das A, Smolenski A, Lohmann SM, Kukreja RC. Cyclic GMP-dependent protein kinase Ialpha attenuates necrosis and apoptosis following ischemia/reoxygenation in adult cardiomyocyte. J Biol Chem. 2006;281:38644–38652. doi: 10.1074/jbc.M606142200. [DOI] [PubMed] [Google Scholar]
- Das A, Xi L, Kukreja RC. Protein kinase G dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem. 2008 doi: 10.1074/jbc.M801547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bold AJ. Atrial natriuretic factor of the rat heart. Studies on isolation and properties. Proc Soc Exp Biol Med. 1982;170:133–138. doi: 10.3181/00379727-170-41408. [DOI] [PubMed] [Google Scholar]
- de Bold AJ. Atrial natriuretic factor: a hormone produced by the heart. Science. 1985;230:767–770. doi: 10.1126/science.2932797. [DOI] [PubMed] [Google Scholar]
- de Bold AJ, Ma KK, Zhang Y, de Bold ML, Bensimon M, Khoshbaten A. The physiological and pathophysiological modulation of the endocrine function of the heart. Can J Physiol Pharmacol. 2001;79:705–714. [PubMed] [Google Scholar]
- DeMeester SL, Qiu Y, Buchman TG, Hotchkiss RS, Dunnigan K, Karl IE, Cobb JP. Nitric oxide inhibits stress-induced endothelial cell apoptosis. Crit Care Med. 1998;26:1500–1509. doi: 10.1097/00003246-199809000-00016. [DOI] [PubMed] [Google Scholar]
- Denninger JW, Schelvis JP, Brandish PE, Zhao Y, Babcock GT, Marletta MA. Interaction of soluble guanylate cyclase with YC-1: kinetic and resonance Raman studies. Biochemistry. 2000;39:4191–4198. doi: 10.1021/bi992332q. [DOI] [PubMed] [Google Scholar]
- Deruelle P, Balasubramaniam V, Kunig AM, Seedorf GJ, Markham NE, Abman SH. BAY 41-2272, a direct activator of soluble guanylate cyclase, reduces right ventricular hypertrophy and prevents pulmonary vascular remodeling during chronic hypoxia in neonatal rats. Biol Neonate. 2006;90:135–144. doi: 10.1159/000092518. [DOI] [PubMed] [Google Scholar]
- Deruelle P, Grover TR, Abman SH. Pulmonary vascular effects of nitric oxide-cGMP augmentation in a model of chronic pulmonary hypertension in fetal and neonatal sheep. Am J Physiol Lung Cell Mol Physiol. 2005;289:L798–L806. doi: 10.1152/ajplung.00119.2005. [DOI] [PubMed] [Google Scholar]
- Deruelle P, Grover TR, Storme L, Abman SH. Effects of BAY 41-2272, a soluble guanylate cyclase activator, on pulmonary vascular reactivity in the ovine fetus. Am J Physiol Lung Cell Mol Physiol. 2005;288:L727–L733. doi: 10.1152/ajplung.00409.2004. [DOI] [PubMed] [Google Scholar]
- Dever G, Spickett CM, Kennedy S, Rush C, Tennant G, Monopoli A, Wainwright CL. The nitric oxide-donating pravastatin derivative, NCX 6550 [(1S-[1alpha(betaS*, deltaS*), 2alpha, 6alpha, 8beta-(R*), 8a alpha]]-1,2,6,7,8,8a-Hexahydro-beta, delta, 6-trihydroxy-2-methyl-8-(2-methyl-1-oxobutoxy)-1-naphtalene-heptanoic acid 4-(nitrooxy)butyl ester)], reduces splenocyte adhesion and reactive oxygen species generation in normal and atherosclerotic mice. J Pharmacol Exp Ther. 2007;320:419–426. doi: 10.1124/jpet.106.109298. [DOI] [PubMed] [Google Scholar]
- Dever GJ, Benson R, Wainwright CL, Kennedy S, Spickett CM. Phospholipid chlorohydrin induces leukocyte adhesion to ApoE-/- mouse arteries via upregulation of P-selectin. Free Radic Biol Med. 2008;44:452–463. doi: 10.1016/j.freeradbiomed.2007.10.038. [DOI] [PubMed] [Google Scholar]
- Dickey DM, Burnett JC, Jr, Potter LR. Novel bifunctional natriuretic peptides as potential therapeutics. J Biol Chem. 2008 doi: 10.1074/jbc.M804538200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi SN, McDowell IF, Moat SJ, Lang D, Newcombe RG, Kredan MB, Lewis MJ, Goodfellow J. Folate improves endothelial function in coronary artery disease: an effect mediated by reduction of intracellular superoxide? Arterioscler Thromb Vasc Biol. 2001;21:1196–1202. doi: 10.1161/hq0701.092000. [DOI] [PubMed] [Google Scholar]
- Doshi SN, McDowell IF, Moat SJ, Payne N, Durrant HJ, Lewis MJ, Goodfellow J. Folic acid improves endothelial function in coronary artery disease via mechanisms largely independent of homocysteine lowering. Circulation. 2002;105:22–26. doi: 10.1161/hc0102.101388. [DOI] [PubMed] [Google Scholar]
- Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemic preconditioning. Heart Fail Rev. 2007;12:181–188. doi: 10.1007/s10741-007-9025-2. [DOI] [PubMed] [Google Scholar]
- Doyle DD, Upshaw-Earley J, Bell EL, Palfrey HC. Natriuretic peptide receptor-B in adult rat ventricle is predominantly confined to the nonmyocyte population. Am J Physiol Heart Circ Physiol. 2002;282:H2117–H2123. doi: 10.1152/ajpheart.00988.2001. [DOI] [PubMed] [Google Scholar]
- Draijer R, Atsma DE, van der LA, van Hinsbergh VW. cGMP and nitric oxide modulate thrombin-induced endothelial permeability. Regulation via different pathways in human aortic and umbilical vein endothelial cells. Circ Res. 1995;76:199–208. doi: 10.1161/01.res.76.2.199. [DOI] [PubMed] [Google Scholar]
- Draijer R, Vaandrager AB, Nolte C, de Jonge HR, Walter U, van Hinsbergh VW. Expression of cGMP-dependent protein kinase I and phosphorylation of its substrate, vasodilator-stimulated phosphoprotein, in human endothelial cells of different origin. Circ Res. 1995;77:897–905. doi: 10.1161/01.res.77.5.897. [DOI] [PubMed] [Google Scholar]
- Drexler H, Finkh M, Hoing S, Toth M, Just H, Lang RE. Systemic and regional vascular effects of atrial natriuretic peptide in a rat model of chronic heart failure. Basic Res Cardiol. 1987;82:517–529. doi: 10.1007/BF01907221. [DOI] [PubMed] [Google Scholar]
- Drexler H, Kastner S, Strobel A, Studer R, Brodde OE, Hasenfuss G. Expression, activity and functional significance of inducible nitric oxide synthase in the failing human heart. J Am Coll Cardiol. 1998;32:955–963. doi: 10.1016/s0735-1097(98)00336-2. [DOI] [PubMed] [Google Scholar]
- Driscoll JA, Chakinala MM. Medical therapy for pulmonary arterial hypertension. Expert Opin Pharmacother. 2008;9:65–81. doi: 10.1517/14656566.9.1.65. [DOI] [PubMed] [Google Scholar]
- Dumitrascu R, Weissmann N, Ghofrani HA, Dony E, Beuerlein K, Schmidt H, Stasch JP, Gnoth MJ, Seeger W, Grimminger F, Schermuly RT. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation. 2006;113:286–295. doi: 10.1161/CIRCULATIONAHA.105.581405. [DOI] [PubMed] [Google Scholar]
- Dunkerley HA, Tilley DG, Palmer D, Liu H, Jimmo SL, Maurice DH. Reduced phosphodiesterase 3 activity and phosphodiesterase 3A level in synthetic vascular smooth muscle cells: implications for use of phosphodiesterase 3 inhibitors in cardiovascular tissues. Mol Pharmacol. 2002;61:1033–1040. doi: 10.1124/mol.61.5.1033. [DOI] [PubMed] [Google Scholar]
- Eddahibi S, Raffestin B, Le Monnier de Gouville AC, Adnot S. Effect of DMPPO, a phosphodiesterase type 5 inhibitor, on hypoxic pulmonary hypertension in rats. Br J Pharmacol. 1998;125:681–688. doi: 10.1038/sj.bjp.0702124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont JA, Vrolix M, Wuytack F, Raeymaekers L, Casteels R. The (Ca2+-Mg2+)-ATPases of the plasma membrane and of the endoplasmic reticulum in smooth muscle cells and their regulation. J Cardiovasc Pharmacol. 1988;12 5:S51–S55. [PubMed] [Google Scholar]
- Emanueli C, Monopoli A, Kraenkel N, Meloni M, Gadau S, Campesi I, Ongini E, Madeddu P. Nitropravastatin stimulates reparative neovascularisation and improves recovery from limb Ischaemia in type-1 diabetic mice. Br J Pharmacol. 2007;150:873–882. doi: 10.1038/sj.bjp.0707142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erley CM, Rebmann S, Strobel U, Schmidt T, Wehrmann M, Osswald H, Risler T. Effects of antihypertensive therapy on blood pressure and renal function in rats with hypertension due to chronic blockade of nitric oxide synthesis. Exp Nephrol. 1995;3:293–299. [PubMed] [Google Scholar]
- Evgenov OV, Ichinose F, Evgenov NV, Gnoth MJ, Falkowski GE, Chang Y, Bloch KD, Zapol WM. Soluble guanylate cyclase activator reverses acute pulmonary hypertension and augments the pulmonary vasodilator response to inhaled nitric oxide in awake lambs. Circulation. 2004;110:2253–2259. doi: 10.1161/01.CIR.0000144469.01521.8A. [DOI] [PubMed] [Google Scholar]
- Evgenov OV, Kohane DS, Bloch KD, Stasch JP, Volpato GP, Bellas E, Evgenov NV, Buys ES, Gnoth MJ, Graveline AR, Liu R, Hess DR, Langer R, Zapol WM. Inhaled agonists of soluble guanylate cyclase induce selective pulmonary vasodilation. Am J Respir Crit Care Med. 2007;176:1138–1145. doi: 10.1164/rccm.200707-1121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5:755–768. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan KA, Fouty BW, Tyler RC, Morris KG, Jr, Hepler LK, Sato K, LeCras TD, Abman SH, Weinberger HD, Huang PL, McMurtry IF, Rodman DM. The pulmonary circulation of homozygous or heterozygous eNOS-null mice is hyperresponsive to mild hypoxia. J Clin Invest. 1999;103:291–299. doi: 10.1172/JCI3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan KA, Tyler RC, Sato K, Fouty BW, Morris KG, Jr, Huang PL, McMurtry IF, Rodman DM. Relative contributions of endothelial, inducible, and neuronal NOS to tone in the murine pulmonary circulation. Am J Physiol. 1999;277:L472–L478. doi: 10.1152/ajplung.1999.277.3.L472. [DOI] [PubMed] [Google Scholar]
- Fiedler B, Lohmann SM, Smolenski A, Linnemuller S, Pieske B, Schroder F, Molkentin JD, Drexler H, Wollert KC. Inhibition of calcineurin-NFAT hypertrophy signaling by cGMP-dependent protein kinase type I in cardiac myocytes. Proc Natl Acad Sci U S A. 2002;99:11363–11368. doi: 10.1073/pnas.162100799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineman JR, Wong J, Morin FC, III, Wild LM, Soifer SJ. Chronic nitric oxide inhibition in utero produces persistent pulmonary hypertension in newborn lambs. J Clin Invest. 1994;93:2675–2683. doi: 10.1172/JCI117281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischmeister R, Castro L, Abi-Gerges A, Rochais F, Vandecasteele G. Species-and tissue-dependent effects of NO and cyclic GMP on cardiac ion channels. Comp Biochem Physiol A Mol Integr Physiol. 2005;142:136–143. doi: 10.1016/j.cbpb.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Fischmeister R, Castro LR, Abi-Gerges A, Rochais F, Jurevicius J, Leroy J, Vandecasteele G. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res. 2006;99:816–828. doi: 10.1161/01.RES.0000246118.98832.04. [DOI] [PubMed] [Google Scholar]
- Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. 2005;111:1601–1610. doi: 10.1161/01.CIR.0000160359.49478.C2. [DOI] [PubMed] [Google Scholar]
- Flaherty MP, Brown M, Grupp IL, Schultz JE, Murphree SS, Jones WK. eNOS deficient mice develop progressive cardiac hypertrophy with altered cytokine and calcium handling protein expression. Cardiovasc Toxicol. 2007;7:165–177. doi: 10.1007/s12012-007-0028-y. [DOI] [PubMed] [Google Scholar]
- Frey R, Muck W, Unger S, Artmeier-Brandt U, Weimann G, Wensing G. Pharmacokinetics, Pharmacodynamics, Tolerability and Safety of the Soluble Guanylate Cyclase Activator Cinaciguat (BAY 58-2667) in Healthy Male Volunteers. J Clin Pharmacol. 2008 doi: 10.1177/0091270008322906. [DOI] [PubMed] [Google Scholar]
- Frostell CG, Blomqvist H, Hedenstierna G, Lundberg J, Zapol WM. Inhaled nitric oxide selectively reverses human hypoxic pulmonary vasoconstriction without causing systemic vasodilation. Anesthesiology. 1993;78:427–435. doi: 10.1097/00000542-199303000-00005. [DOI] [PubMed] [Google Scholar]
- Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL, Jain RK. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A. 2001;98:2604–2609. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuo K, Hata S, Suhara T, Nakahashi T, Shinto Y, Tsujimoto Y, Morimoto S, Ogihara T. Nitric oxide induces upregulation of Fas and apoptosis in vascular smooth muscle. Hypertension. 1996;27:823–826. doi: 10.1161/01.hyp.27.3.823. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Khan MT, Jothianandan D. Comparison of Endothelium-Dependent Relaxation and Nitric Oxide-Induced Relaxation in Rabbit Aorta. Federation Proceedings. 1987;46:385–385. [Google Scholar]
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- Furst R, Bubik MF, Bihari P, Mayer BA, Khandoga AG, Hoffmann F, Rehberg M, Krombach F, Zahler S, Vollmar AM. Atrial natriuretic peptide protects against histamine-induced endothelial barrier dysfunction in vivo. Mol Pharmacol. 2008;74:1–8. doi: 10.1124/mol.108.045773. [DOI] [PubMed] [Google Scholar]
- Gaide MS, Fitterman WS, Wiggins JR, Myerburg RJ, Cameron JS, Bassett AL. Amrinone relaxes potassium-induced contracture of failing right ventricular muscle of cats. J Cardiovasc Pharmacol. 1983;5:335–340. doi: 10.1097/00005344-198303000-00028. [DOI] [PubMed] [Google Scholar]
- Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, Fleming T, Parpia T, Burgess G, Branzi A, Grimminger F, Kurzyna M, Simonneau G. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- Garcia R, Bonhomme MC, Schiffrin EL. Divergent regulation of atrial natriuretic factor receptors in high-output heart failure. Am J Physiol. 1992;263:H1790–H1797. doi: 10.1152/ajpheart.1992.263.6.H1790. [DOI] [PubMed] [Google Scholar]
- Ghofrani HA, Voswinckel R, Reichenberger F, Olschewski H, Haredza P, Karadas B, Schermuly RT, Weissmann N, Seeger W, Grimminger F. Differences in hemodynamic and oxygenation responses to three different phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension: a randomized prospective study. J Am Coll Cardiol. 2004;44:1488–1496. doi: 10.1016/j.jacc.2004.06.060. [DOI] [PubMed] [Google Scholar]
- Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–221. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- Gillies HC, Roblin D, Jackson G. Coronary and systemic hemodynamic effects of sildenafil citrate: from basic science to clinical studies in patients with cardiovascular disease. Int J Cardiol. 2002;86:131–141. doi: 10.1016/s0167-5273(02)00421-7. [DOI] [PubMed] [Google Scholar]
- Grunfeld S, Hamilton CA, Mesaros S, McClain SW, Dominiczak AF, Bohr DF, Malinski T. Role of superoxide in the depressed nitric oxide production by the endothelium of genetically hypertensive rats. Hypertension. 1995;26:854–857. doi: 10.1161/01.hyp.26.6.854. [DOI] [PubMed] [Google Scholar]
- Guazzi M, Samaja M. The role of PDE5-inhibitors in cardiopulmonary disorders: from basic evidence to clinical development. Curr Med Chem. 2007;14:2181–2191. doi: 10.2174/092986707781389619. [DOI] [PubMed] [Google Scholar]
- Guazzi M, Samaja M, Arena R, Vicenzi M, Guazzi MD. Long-term use of sildenafil in the therapeutic management of heart failure. J Am Coll Cardiol. 2007;50:2136–2144. doi: 10.1016/j.jacc.2007.07.078. [DOI] [PubMed] [Google Scholar]
- Guo Y, Stein AB, Wu WJ, Zhu X, Tan W, Li Q, Bolli R. Late preconditioning induced by NO donors, adenosine A1 receptor agonists, and delta1-opioid receptor agonists is mediated by iNOS. Am J Physiol Heart Circ Physiol. 2005;289:H2251–H2257. doi: 10.1152/ajpheart.00341.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwathmey JK, Morgan JP. The effects of milrinone and piroximone on intracellular calcium handling in working myocardium from the ferret. Br J Pharmacol. 1985;85:97–108. doi: 10.1111/j.1476-5381.1985.tb08835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyurko R, Kuhlencordt P, Fishman MC, Huang PL. Modulation of mouse cardiac function in vivo by eNOS and ANP. Am J Physiol Heart Circ Physiol. 2000;278:H971–H981. doi: 10.1152/ajpheart.2000.278.3.H971. [DOI] [PubMed] [Google Scholar]
- Han B, Fixler R, Beeri R, Wang Y, Bachrach U, Hasin Y. The opposing effects of endothelin-1 and C-type natriuretic peptide on apoptosis of neonatal rat cardiac myocytes. Eur J Pharmacol. 2003;474:15–20. doi: 10.1016/s0014-2999(03)01995-2. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Charbonneau H, Beavo JA. Purification of calmodulin-stimulated cyclic nucleotide phosphodiesterase by monoclonal antibody affinity chromatography. Methods Enzymol. 1988;159:543–557. doi: 10.1016/0076-6879(88)59053-5. [DOI] [PubMed] [Google Scholar]
- Harteneck C, Wedel B, Koesling D, Malkewitz J, Bohme E, Schultz G. Molecular cloning and expression of a new alpha-subunit of soluble guanylyl cyclase. Interchangeability of the alpha-subunits of the enzyme. FEBS Lett. 1991;292:217–222. doi: 10.1016/0014-5793(91)80871-y. [DOI] [PubMed] [Google Scholar]
- Hathaway CA, Heistad DD, Piegors DJ, Miller FJ., Jr Regression of atherosclerosis in monkeys reduces vascular superoxide levels. Circ Res. 2002;90:277–283. doi: 10.1161/hh0302.104724. [DOI] [PubMed] [Google Scholar]
- He L, Chen J, Liu X, Dinger B, Fidone S. Enhanced nitric oxide-mediated chemoreceptor inhibition and altered cyclic GMP signaling in rat carotid body following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1463–L1468. doi: 10.1152/ajplung.00249.2007. [DOI] [PubMed] [Google Scholar]
- Heineke J, Kempf T, Kraft T, Hilfiker A, Morawietz H, Scheubel RJ, Caroni P, Lohmann SM, Drexler H, Wollert KC. Downregulation of cytoskeletal muscle LIM protein by nitric oxide: impact on cardiac myocyte hypertrophy. Circulation. 2003;107:1424–1432. doi: 10.1161/01.cir.0000055319.94801.fc. [DOI] [PubMed] [Google Scholar]
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- Herget S, Lohse MJ, Nikolaev VO. Real-time monitoring of phosphodiesterase inhibition in intact cells. Cell Signal. 2008;20:1423–1431. doi: 10.1016/j.cellsig.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Heydemann A, Huber JM, Kakkar R, Wheeler MT, McNally EM. Functional nitric oxide synthase mislocalization in cardiomyopathy. J Mol Cell Cardiol. 2004;36:213–223. doi: 10.1016/j.yjmcc.2003.09.020. [DOI] [PubMed] [Google Scholar]
- Heymes C, Vanderheyden M, Bronzwaer JG, Shah AM, Paulus WJ. Endomyocardial nitric oxide synthase and left ventricular preload reserve in dilated cardiomyopathy. Circulation. 1999;99:3009–3016. doi: 10.1161/01.cir.99.23.3009. [DOI] [PubMed] [Google Scholar]
- Hirooka Y, Takeshita A, Imaizumi T, Suzuki S, Yoshida M, Ando S, Nakamura M. Attenuated forearm vasodilative response to intra-arterial atrial natriuretic peptide in patients with heart failure. Circulation. 1990;82:147–153. doi: 10.1161/01.cir.82.1.147. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- Holmes SJ, Espiner EA, Richards AM, Yandle TG, Frampton C. Renal, endocrine, and hemodynamic effects of human brain natriuretic peptide in normal man. J Clin Endocrinol Metab. 1993;76:91–96. doi: 10.1210/jcem.76.1.8380606. [DOI] [PubMed] [Google Scholar]
- Holschermann H, Noll T, Hempel A, Piper HM. Dual role of cGMP in modulation of macromolecule permeability of aortic endothelial cells. Am J Physiol. 1997;272:H91–H98. doi: 10.1152/ajpheart.1997.272.1.H91. [DOI] [PubMed] [Google Scholar]
- Holtwick R, van Eickels M, Skryabin BV, Baba HA, Bubikat A, Begrow F, Schneider MD, Garbers DL, Kuhn M. Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J Clin Invest. 2003;111:1399–1407. doi: 10.1172/JCI17061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda A, Adams SR, Sawyer CL, Lev-Ram V, Tsien RY, Dostmann WR. Spatiotemporal dynamics of guanosine 3′,5′-cyclic monophosphate revealed by a genetically encoded, fluorescent indicator. Proc Natl Acad Sci U S A. 2001;98:2437–2442. doi: 10.1073/pnas.051631298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood J, Granger HJ. Protein kinase G mediates vascular endothelial growth factor-induced Raf-1 activation and proliferation in human endothelial cells. J Biol Chem. 1998;273:23504–23508. doi: 10.1074/jbc.273.36.23504. [DOI] [PubMed] [Google Scholar]
- Horio T, Nishikimi T, Yoshihara F, Matsuo H, Takishita S, Kangawa K. Inhibitory regulation of hypertrophy by endogenous atrial natriuretic peptide in cultured cardiac myocytes. Hypertension. 2000;35:19–24. doi: 10.1161/01.hyp.35.1.19. [DOI] [PubMed] [Google Scholar]
- Hsu S, Nagayama T, Koitabashi N, Zhang M, Zhou L, Bedja D, Gabrielson KL, Molkentin JD, Kass DA, Takimoto E. Phosphodiesterase 5 Inhibition Blocks Pressure Overload-Induced Cardiac Hypertrophy Independent of the Calcineurin Pathway. Cardiovasc Res. 2008 doi: 10.1093/cvr/cvn324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson HG, Trindade PT, Cunanan DB, Wu CF, Pratt RE. Mechanisms of natriuretic-peptide-induced growth inhibition of vascular smooth muscle cells. Cardiovasc Res. 1997;35:158–167. doi: 10.1016/s0008-6363(97)00086-2. [DOI] [PubMed] [Google Scholar]
- Hyndman ME, Verma S, Rosenfeld RJ, Anderson TJ, Parsons HG. Interaction of 5-methyltetrahydrofolate and tetrahydrobiopterin on endothelial function. Am J Physiol Heart Circ Physiol. 2002;282:H2167–H2172. doi: 10.1152/ajpheart.00935.2001. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ, Byrns RE, Buga GM, Wood KS. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ Res. 1987;61:866–879. doi: 10.1161/01.res.61.6.866. [DOI] [PubMed] [Google Scholar]
- Irwin DC, Rhodes J, Baker DC, Nelson SE, Tucker A. Atrial natriuretic peptide blockade exacerbates high altitude pulmonary edema in endotoxin-primed rats. High Alt Med Biol. 2001;2:349–360. doi: 10.1089/15270290152608525. [DOI] [PubMed] [Google Scholar]
- Irwin DC, Tissot van Patot MC, Tucker A, Bowen R. Direct ANP inhibition of hypoxia-induced inflammatory pathways in pulmonary microvascular and macrovascular endothelial monolayers. Am J Physiol Lung Cell Mol Physiol. 2005;288:L849–L859. doi: 10.1152/ajplung.00294.2004. [DOI] [PubMed] [Google Scholar]
- Jang Y, Wang H, Xi J, Mueller RA, Norfleet EA, Xu Z. NO mobilizes intracellular Zn2+ via cGMP/PKG signaling pathway and prevents mitochondrial oxidant damage in cardiomyocytes. Cardiovasc Res. 2007;75:426–433. doi: 10.1016/j.cardiores.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javadov SA, Clarke S, Das M, Griffiths EJ, Lim KH, Halestrap AP. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol. 2003;549:513–524. doi: 10.1113/jphysiol.2003.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang BH, Maruyama J, Yokochi A, Amano H, Mitani Y, Maruyama K. Correlation of inhaled nitric-oxide induced reduction of pulmonary artery pressure and vascular changes. Eur Respir J. 2002;20:52–58. doi: 10.1183/09031936.02.00249302. [DOI] [PubMed] [Google Scholar]
- John SW, Krege JH, Oliver PM, Hagaman JR, Hodgin JB, Pang SC, Flynn TG, Smithies O. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science. 1995;267:679–681. doi: 10.1126/science.7839143. [DOI] [PubMed] [Google Scholar]
- Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Jurevicius J, Skeberdis VA, Fischmeister R. Role of cyclic nucleotide phosphodiesterase isoforms in cAMP compartmentation following beta2-adrenergic stimulation of ICa, L in frog ventricular myocytes. J Physiol. 2003;551:239–252. doi: 10.1113/jphysiol.2003.045211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambayashi Y, Nakao K, Mukoyama M, Saito Y, Ogawa Y, Shiono S, Inouye K, Yoshida N, Imura H. Isolation and sequence determination of human brain natriuretic peptide in human atrium. FEBS Lett. 1990;259:341–345. doi: 10.1016/0014-5793(90)80043-i. [DOI] [PubMed] [Google Scholar]
- Kamisaki Y, Saheki S, Nakane M, Palmieri JA, Kuno T, Chang BY, Waldman SA, Murad F. Soluble guanylate cyclase from rat lung exists as a heterodimer. J Biol Chem. 1986;261:7236–7241. [PubMed] [Google Scholar]
- Kass DA, Takimoto E, Nagayama T, Champion HC. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc Res. 2007;75:303–314. doi: 10.1016/j.cardiores.2007.02.031. [DOI] [PubMed] [Google Scholar]
- Kato T, Sano M, Miyoshi S, Sato T, Hakuno D, Ishida H, Kinoshita-Nakazawa H, Fukuda K, Ogawa S. Calmodulin kinases II and IV and calcineurin are involved in leukemia inhibitory factor-induced cardiac hypertrophy in rats. Circ Res. 2000;87:937–945. doi: 10.1161/01.res.87.10.937. [DOI] [PubMed] [Google Scholar]
- Katsuki S, Arnold WP, Murad F. Effects of sodium nitroprusside, nitroglycerin, and sodium azide on levels of cyclic nucleotides and mechanical activity of various tissues. J Cyclic Nucleotide Res. 1977;3:239–247. [PubMed] [Google Scholar]
- Kaufman S. Some metabolic relationships between biopterin and folate: implications for the “methyl trap hypothesis”. Neurochem Res. 1991;16:1031–1036. doi: 10.1007/BF00965847. [DOI] [PubMed] [Google Scholar]
- Kawai N, Bloch DB, Filippov G, Rabkina D, Suen HC, Losty PD, Janssens SP, Zapol WM, de la MS, Bloch KD. Constitutive endothelial nitric oxide synthase gene expression is regulated during lung development. Am J Physiol. 1995;268:L589–L595. doi: 10.1152/ajplung.1995.268.4.L589. [DOI] [PubMed] [Google Scholar]
- Keeble JE, Moore PK. Pharmacology and potential therapeutic applications of nitric oxide-releasing non-steroidal anti-inflammatory and related nitric oxide-donating drugs. Br J Pharmacol. 2002;137:295–310. doi: 10.1038/sj.bjp.0704876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khairallah M, Khairallah RJ, Young ME, Allen BG, Gillis MA, Danialou G, Deschepper CF, Petrof BJ, Des RC. Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc Natl Acad Sci U S A. 2008;105:7028–7033. doi: 10.1073/pnas.0710595105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SA, Skaf MW, Harrison RW, Lee K, Minhas KM, Kumar A, Fradley M, Shoukas AA, Berkowitz DE, Hare JM. Nitric oxide regulation of myocardial contractility and calcium cycling: independent impact of neuronal and endothelial nitric oxide synthases. Circ Res. 2003;92:1322–1329. doi: 10.1161/01.RES.0000078171.52542.9E. [DOI] [PubMed] [Google Scholar]
- Kinugawa KI, Kohmoto O, Yao A, Serizawa T, Takahashi T. Cardiac inducible nitric oxide synthase negatively modulates myocardial function in cultured rat myocytes. Am J Physiol. 1997;272:H35–H47. doi: 10.1152/ajpheart.1997.272.1.H35. [DOI] [PubMed] [Google Scholar]
- Klinger JR. The nitric oxide/cGMP signaling pathway in pulmonary hypertension. Clin Chest Med. 2007;28:143–67. ix. doi: 10.1016/j.ccm.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Klinger JR, Warburton R, Carino GP, Murray J, Murphy C, Napier M, Harrington EO. Natriuretic peptides differentially attenuate thrombin-induced barrier dysfunction in pulmonary microvascular endothelial cells. Exp Cell Res. 2006;312:401–410. doi: 10.1016/j.yexcr.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Klinger JR, Warburton RR, Pietras L, Hill NS. Brain natriuretic peptide inhibits hypoxic pulmonary hypertension in rats. J Appl Physiol. 1998;84:1646–1652. doi: 10.1152/jappl.1998.84.5.1646. [DOI] [PubMed] [Google Scholar]
- Kloss S, Bouloumie A, Mulsch A. Aging and chronic hypertension decrease expression of rat aortic soluble guanylyl cyclase. Hypertension. 2000;35:43–47. [PubMed] [Google Scholar]
- Koeppen M, Feil R, Siegl D, Feil S, Hofmann F, Pohl U, de Wit C. cGMP-dependent protein kinase mediates NO- but not acetylcholine-induced dilations in resistance vessels in vivo. Hypertension. 2004;44:952–955. doi: 10.1161/01.HYP.0000147661.80059.ca. [DOI] [PubMed] [Google Scholar]
- Koide M, Kawahara Y, Tsuda T, Yokoyama M. Cytokine-induced expression of an inducible type of nitric oxide synthase gene in cultured vascular smooth muscle cells. FEBS Lett. 1993;318:213–217. doi: 10.1016/0014-5793(93)80514-u. [DOI] [PubMed] [Google Scholar]
- Koizumi T, Gupta R, Banerjee M, Newman JH. Changes in pulmonary vascular tone during exercise. Effects of nitric oxide (NO) synthase inhibition, L-arginine infusion, and NO inhalation. J Clin Invest. 1994;94:2275–2282. doi: 10.1172/JCI117590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojda G, Kottenberg K. Regulation of basal myocardial function by NO. Cardiovasc Res. 1999;41:514–523. doi: 10.1016/s0008-6363(98)00314-9. [DOI] [PubMed] [Google Scholar]
- Kojda G, Kottenberg K, Nix P, Schluter KD, Piper HM, Noack E. Low increase in cGMP induced by organic nitrates and nitrovasodilators improves contractile response of rat ventricular myocytes. Circ Res. 1996;78:91–101. doi: 10.1161/01.res.78.1.91. [DOI] [PubMed] [Google Scholar]
- Kook H, Itoh H, Choi BS, Sawada N, Doi K, Hwang TJ, Kim KK, Arai H, Baik YH, Nakao K. Physiological concentration of atrial natriuretic peptide induces endothelial regeneration in vitro. Am J Physiol Heart Circ Physiol. 2003;284:H1388–H1397. doi: 10.1152/ajpheart.00414.2002. [DOI] [PubMed] [Google Scholar]
- Kuhn M, Holtwick R, Baba HA, Perriard JC, Schmitz W, Ehler E. Progressive cardiac hypertrophy and dysfunction in atrial natriuretic peptide receptor (GC-A) deficient mice. Heart. 2002;87:368–374. doi: 10.1136/heart.87.4.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M, Voss M, Mitko D, Stypmann J, Schmid C, Kawaguchi N, Grabellus F, Baba HA. Left ventricular assist device support reverses altered cardiac expression and function of natriuretic peptides and receptors in end-stage heart failure. Cardiovasc Res. 2004;64:308–314. doi: 10.1016/j.cardiores.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Kurihara N, Alfie ME, Sigmon DH, Rhaleb NE, Shesely EG, Carretero OA. Role of nNOS in blood pressure regulation in eNOS null mutant mice. Hypertension. 1998;32:856–861. doi: 10.1161/01.hyp.32.5.856. [DOI] [PubMed] [Google Scholar]
- Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem. 2003;278:22546–22554. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- Laber U, Kober T, Schmitz V, Schrammel A, Meyer W, Mayer B, Weber M, Kojda G. Effect of hypercholesterolemia on expression and function of vascular soluble guanylyl cyclase. Circulation. 2002;105:855–860. doi: 10.1161/hc0702.103975. [DOI] [PubMed] [Google Scholar]
- Laflamme A, Foucart S, Moreau P, Lambert C, Cardinal R, de Champlain J. Sympathetic functions in NG-nitro-L-arginine-methyl-ester-induced hypertension: modulation by the renin-angiotensin system. J Hypertens. 1998;16:63–76. doi: 10.1097/00004872-199816010-00011. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenickel TH, Buttgereit J, Pagel-Langenickel I, Lindner M, Monti J, Beuerlein K, Al Saadi N, Plehm R, Popova E, Tank J, Dietz R, Willenbrock R, Bader M. Cardiac hypertrophy in transgenic rats expressing a dominant-negative mutant of the natriuretic peptide receptor B. Proc Natl Acad Sci U S A. 2006;103:4735–4740. doi: 10.1073/pnas.0510019103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, Tarpey M, Fukai T, Harrison DG. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–1288. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- Lavi S, McConnell JP, Rihal CS, Prasad A, Mathew V, Lerman LO, Lerman A. Local production of lipoprotein-associated phospholipase A2 and lysophosphatidylcholine in the coronary circulation: association with early coronary atherosclerosis and endothelial dysfunction in humans. Circulation. 2007;115:2715–2721. doi: 10.1161/CIRCULATIONAHA.106.671420. [DOI] [PubMed] [Google Scholar]
- Lavi S, Yang EH, Prasad A, Mathew V, Barsness GW, Rihal CS, Lerman LO, Lerman A. The interaction between coronary endothelial dysfunction, local oxidative stress, and endogenous nitric oxide in humans. Hypertension. 2008;51:127–133. doi: 10.1161/HYPERTENSIONAHA.107.099986. [DOI] [PubMed] [Google Scholar]
- Layland J, Li JM, Shah AM. Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol. 2002;540:457–467. doi: 10.1113/jphysiol.2001.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Cras TD, Xue C, Rengasamy A, Johns RA. Chronic hypoxia upregulates endothelial and inducible NO synthase gene and protein expression in rat lung. Am J Physiol. 1996;270:L164–L170. doi: 10.1152/ajplung.1996.270.1.L164. [DOI] [PubMed] [Google Scholar]
- Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechene P, Mazet JL, Conti M, Fischmeister R, Vandecasteele G. Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ Res. 2008;102:1091–1100. doi: 10.1161/CIRCRESAHA.107.167817. [DOI] [PubMed] [Google Scholar]
- Lewis GD, Lachmann J, Camuso J, Lepore JJ, Shin J, Martinovic ME, Systrom DM, Bloch KD, Semigran MJ. Sildenafil improves exercise hemodynamics and oxygen uptake in patients with systolic heart failure. Circulation. 2007;115:59–66. doi: 10.1161/CIRCULATIONAHA.106.626226. [DOI] [PubMed] [Google Scholar]
- Lewis GD, Shah R, Shahzad K, Camuso JM, Pappagianopoulos PP, Hung J, Tawakol A, Gerszten RE, Systrom DM, Bloch KD, Semigran MJ. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation. 2007;116:1555–1562. doi: 10.1161/CIRCULATIONAHA.107.716373. [DOI] [PubMed] [Google Scholar]
- Li D, Zhou N, Johns RA. Soluble guanylate cyclase gene expression and localization in rat lung after exposure to hypoxia. Am J Physiol. 1999;277:L841–L847. doi: 10.1152/ajplung.1999.277.4.L841. [DOI] [PubMed] [Google Scholar]
- Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X, Novak L, Renfrow MB, Chen YF. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ Res. 2008;102:185–192. doi: 10.1161/CIRCRESAHA.107.157677. [DOI] [PubMed] [Google Scholar]
- Li W, Mital S, Ojaimi C, Csiszar A, Kaley G, Hintze TH. Premature death and age-related cardiac dysfunction in male eNOS-knockout mice. J Mol Cell Cardiol. 2004;37:671–680. doi: 10.1016/j.yjmcc.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Lincoln TM, Wu X, Sellak H, Dey N, Choi CS. Regulation of vascular smooth muscle cell phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. Front Biosci. 2006;11:356–367. doi: 10.2741/1803. [DOI] [PubMed] [Google Scholar]
- Linder AE, McCluskey LP, Cole KR, III, Lanning KM, Webb RC. Dynamic association of nitric oxide downstream signaling molecules with endothelial caveolin-1 in rat aorta. J Pharmacol Exp Ther. 2005;314:9–15. doi: 10.1124/jpet.105.083634. [DOI] [PubMed] [Google Scholar]
- Lisy O, Huntley BK, McCormick DJ, Kurlansky PA, Burnett JC., Jr Design, synthesis, and actions of a novel chimeric natriuretic peptide: CD-NP. J Am Coll Cardiol. 2008;52:60–68. doi: 10.1016/j.jacc.2008.02.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann SM, Fischmeister R, Walter U. Signal transduction by cGMP in heart. Basic Res Cardiol. 1991;86:503–514. doi: 10.1007/BF02190700. [DOI] [PubMed] [Google Scholar]
- Lopez MJ, Wong SK, Kishimoto I, Dubois S, Mach V, Friesen J, Garbers DL, Beuve A. Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature. 1995;378:65–68. doi: 10.1038/378065a0. [DOI] [PubMed] [Google Scholar]
- Lorenowicz MJ, Fernandez-Borja M, Kooistra MR, Bos JL, Hordijk PL. PKA and Epac1 regulate endothelial integrity and migration through parallel and independent pathways. Eur J Cell Biol. 2008;87:779–792. doi: 10.1016/j.ejcb.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Loyer X, Heymes C, Samuel JL. Constitutive nitric oxide synthases in the heart from hypertrophy to failure. Clin Exp Pharmacol Physiol. 2008;35:483–488. doi: 10.1111/j.1440-1681.2008.04901.x. [DOI] [PubMed] [Google Scholar]
- Lugnier C, Keravis T, Eckly-Michel A. Cross talk between NO and cyclic nucleotide phosphodiesterases in the modulation of signal transduction in blood vessel. J Physiol Pharmacol. 1999;50:639–652. [PubMed] [Google Scholar]
- MacNaul KL, Hutchinson NI. Differential expression of iNOS and cNOS mRNA in human vascular smooth muscle cells and endothelial cells under normal and inflammatory conditions. Biochem Biophys Res Commun. 1993;196:1330–1334. doi: 10.1006/bbrc.1993.2398. [DOI] [PubMed] [Google Scholar]
- Malecot CO, Bers DM, Katzung BG. Biphasic contractions induced by milrinone at low temperature in ferret ventricular muscle: role of the sarcoplasmic reticulum and transmembrane calcium influx. Circ Res. 1986;59:151–162. doi: 10.1161/01.res.59.2.151. [DOI] [PubMed] [Google Scholar]
- Marcantoni A, Levi RC, Gallo MP, Hirsch E, Alloatti G. Phosphoinositide 3-kinasegamma (PI3Kgamma) controls L-type calcium current (ICa, L) through its positive modulation of type-3 phosphodiesterase (PDE3) J Cell Physiol. 2006;206:329–336. doi: 10.1002/jcp.20467. [DOI] [PubMed] [Google Scholar]
- Marcus LS, Hart D, Packer M, Yushak M, Medina N, Danziger RS, Heitjan DF, Katz SD. Hemodynamic and renal excretory effects of human brain natriuretic peptide infusion in patients with congestive heart failure. A double-blind, placebo-controlled, randomized crossover trial. Circulation. 1996;94:3184–3189. doi: 10.1161/01.cir.94.12.3184. [DOI] [PubMed] [Google Scholar]
- Martin SR, Emanuel K, Sears CE, Zhang YH, Casadei B. Are myocardial eNOS and nNOS involved in the beta-adrenergic and muscarinic regulation of inotropy? A systematic investigation. Cardiovasc Res. 2006;70:97–106. doi: 10.1016/j.cardiores.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Martins TJ, Mumby MC, Beavo JA. Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J Biol Chem. 1982;257:1973–1979. [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Masano T, Kawashima S, Toh R, Satomi-Kobayashi S, Shinohara M, Takaya T, Sasaki N, Takeda M, Tawa H, Yamashita T, Yokoyama M, Hirata K. Beneficial effects of exogenous tetrahydrobiopterin on left ventricular remodeling after myocardial infarction in rats: the possible role of oxidative stress caused by uncoupled endothelial nitric oxide synthase. Circ J. 2008;72:1512–1519. doi: 10.1253/circj.cj-08-0072. [DOI] [PubMed] [Google Scholar]
- Massion PB, Balligand JL. Modulation of cardiac contraction, relaxation and rate by the endothelial nitric oxide synthase (eNOS): lessons from genetically modified mice. J Physiol. 2003;546:63–75. doi: 10.1113/jphysiol.2002.025973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice DH. Cyclic nucleotide phosphodiesterase-mediated integration of cGMP and cAMP signaling in cells of the cardiovascular system. Front Biosci. 2005;10:1221–1228. doi: 10.2741/1614. [DOI] [PubMed] [Google Scholar]
- Maurice DH, Palmer D, Tilley DG, Dunkerley HA, Netherton SJ, Raymond DR, Elbatarny HS, Jimmo SL. Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol Pharmacol. 2003;64:533–546. doi: 10.1124/mol.64.3.533. [DOI] [PubMed] [Google Scholar]
- McLemore EC, Tessier DJ, Thresher J, Komalavilas P, Brophy CM. Role of the small heat shock proteins in regulating vascular smooth muscle tone. J Am Coll Surg. 2005;201:30–36. doi: 10.1016/j.jamcollsurg.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- Mery PF, Pavoine C, Pecker F, Fischmeister R. Erythro-9-(2-hydroxy-3-nonyl)adenine inhibits cyclic GMP-stimulated phosphodiesterase in isolated cardiac myocytes. Mol Pharmacol. 1995;48:121–130. [PubMed] [Google Scholar]
- Michelakis E, Tymchak W, Lien D, Webster L, Hashimoto K, Archer S. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation. 2002;105:2398–2403. doi: 10.1161/01.cir.0000016641.12984.dc. [DOI] [PubMed] [Google Scholar]
- Michelakis ED, Tymchak W, Noga M, Webster L, Wu XC, Lien D, Wang SH, Modry D, Archer SL. Long-term treatment with oral sildenafil is safe and improves functional capacity and hemodynamics in patients with pulmonary arterial hypertension. Circulation. 2003;108:2066–2069. doi: 10.1161/01.CIR.0000099502.17776.C2. [DOI] [PubMed] [Google Scholar]
- Mills RM, LeJemtel TH, Horton DP, Liang C, Lang R, Silver MA, Lui C, Chatterjee K. Sustained hemodynamic effects of an infusion of nesiritide (human b-type natriuretic peptide) in heart failure: a randomized, double-blind, placebo-controlled clinical trial. Natrecor Study Group. J Am Coll Cardiol. 1999;34:155–162. doi: 10.1016/s0735-1097(99)00184-9. [DOI] [PubMed] [Google Scholar]
- Mingone CJ, Gupte SA, Ali N, Oeckler RA, Wolin MS. Thiol oxidation inhibits nitric oxide-mediated pulmonary artery relaxation and guanylate cyclase stimulation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L549–L557. doi: 10.1152/ajplung.00331.2005. [DOI] [PubMed] [Google Scholar]
- Mitaka C, Hirata Y, Nagura T, Tsunoda Y, Amaha K. Beneficial effect of atrial natriuretic peptide on pulmonary gas exchange in patients with acute lung injury. Chest. 1998;114:223–228. doi: 10.1378/chest.114.1.223. [DOI] [PubMed] [Google Scholar]
- Moens AL, Champion HC, Claeys MJ, Tavazzi B, Kaminski PM, Wolin MS, Borgonjon DJ, Van Nassauw L, Haile A, Zviman M, Bedja D, Wuyts FL, Elsaesser RS, Cos P, Gabrielson KL, Lazzarino G, Paolocci N, Timmermans JP, Vrints CJ, Kass DA. High-dose folic acid pretreatment blunts cardiac dysfunction during ischemia coupled to maintenance of high-energy phosphates and reduces postreperfusion injury. Circulation. 2008;117:1810–1819. doi: 10.1161/CIRCULATIONAHA.107.725481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G, Ketner EA, Majmudar M, Gabrielson K, Halushka MK, Mitchell JB, Biswal S, Channon KM, Wolin MS, Alp NJ, Paolocci N, Champion HC, Kass DA. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation. 2008;117:2626–2636. doi: 10.1161/CIRCULATIONAHA.107.737031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan P, Brutsaert DL, Paulus WJ, Sys SU. Myocardial contractile response to nitric oxide and cGMP. Circulation. 1996;93:1223–1229. doi: 10.1161/01.cir.93.6.1223. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollnau H, Schulz E, Daiber A, Baldus S, Oelze M, August M, Wendt M, Walter U, Geiger C, Agrawal R, Kleschyov AL, Meinertz T, Munzel T. Nebivolol prevents vascular NOS III uncoupling in experimental hyperlipidemia and inhibits NADPH oxidase activity in inflammatory cells. Arterioscler Thromb Vasc Biol. 2003;23:615–621. doi: 10.1161/01.ATV.0000065234.70518.26. [DOI] [PubMed] [Google Scholar]
- Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, Tsilimingas N, Walter U, Forstermann U, Meinertz T, Griendling K, Munzel T. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002;90:E58–E65. doi: 10.1161/01.res.0000012569.55432.02. [DOI] [PubMed] [Google Scholar]
- Momi S, Emerson M, Paul W, Leone M, Mezzasoma AM, Del Soldato P, Page CP, Gresele P. Prevention of pulmonary thromboembolism by NCX 4016, a nitric oxide-releasing aspirin. Eur J Pharmacol. 2000;397:177–185. doi: 10.1016/s0014-2999(00)00223-5. [DOI] [PubMed] [Google Scholar]
- Mongillo M, Tocchetti CG, Terrin A, Lissandron V, Cheung YF, Dostmann WR, Pozzan T, Kass DA, Paolocci N, Houslay MD, Zaccolo M. Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ Res. 2006;98:226–234. doi: 10.1161/01.RES.0000200178.34179.93. [DOI] [PubMed] [Google Scholar]
- Mope L, McClellan GB, Winegrad S. Calcium sensitivity of the contractile system and phosphorylation of troponin in hyperpermeable cardiac cells. J Gen Physiol. 1980;75:271–282. doi: 10.1085/jgp.75.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–928. doi: 10.1038/nature06798. [DOI] [PubMed] [Google Scholar]
- Mukoyama M, Nakao K, Saito Y, Ogawa Y, Hosoda K, Suga S, Shirakami G, Jougasaki M, Imura H. Human brain natriuretic peptide, a novel cardiac hormone. Lancet. 1990;335:801–802. doi: 10.1016/0140-6736(90)90925-u. [DOI] [PubMed] [Google Scholar]
- Munzel T, Feil R, Mulsch A, Lohmann SM, Hofmann F, Walter U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3′,5′-cyclic monophosphate-dependent protein kinase [corrected] Circulation. 2003;108:2172–2183. doi: 10.1161/01.CIR.0000094403.78467.C3. [DOI] [PubMed] [Google Scholar]
- Murad F, Rapoport RM, Fiscus R. Role of cyclic-GMP in relaxations of vascular smooth muscle. J Cardiovasc Pharmacol. 1985;7 3:S111–S118. [PubMed] [Google Scholar]
- Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray F, MacLean MR, Pyne NJ. Increased expression of the cGMP-inhibited cAMP-specific (PDE3) and cGMP binding cGMP-specific (PDE5) phosphodiesterases in models of pulmonary hypertension. Br J Pharmacol. 2002;137:1187–1194. doi: 10.1038/sj.bjp.0704984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama T, Zhang M, Hsu S, Takimoto E, Kass DA. Sustained soluble guanylate cyclase stimulation offsets nitric-oxide synthase inhibition to restore acute cardiac modulation by sildenafil. J Pharmacol Exp Ther. 2008;326:380–387. doi: 10.1124/jpet.108.137422. [DOI] [PubMed] [Google Scholar]
- Nagel DJ, Aizawa T, Jeon KI, Liu W, Mohan A, Wei H, Miano JM, Florio VA, Gao P, Korshunov VA, Berk BC, Yan C. Role of nuclear Ca2+/calmodulin-stimulated phosphodiesterase 1A in vascular smooth muscle cell growth and survival. Circ Res. 2006;98:777–784. doi: 10.1161/01.RES.0000215576.27615.fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Ichikawa K, Ito M, Yamamori B, Okinaka T, Isaka N, Yoshida Y, Fujita S, Nakano T. Effects of the phosphorylation of myosin phosphatase by cyclic GMP-dependent protein kinase. Cell Signal. 1999;11:671–676. doi: 10.1016/s0898-6568(99)00036-4. [DOI] [PubMed] [Google Scholar]
- Nakao K, Itoh H, Kambayashi Y, Hosoda K, Saito Y, Yamada T, Mukoyama M, Arai H, Shirakami G, Suga S. Rat brain natriuretic peptide. Isolation from rat heart and tissue distribution. Hypertension. 1990;15:774–778. doi: 10.1161/01.hyp.15.6.774. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Soma M, Takahashi Y, Rehemudula D, Kanmatsuse K, Furuya K. Functional deletion mutation of the 5′-flanking region of type A human natriuretic peptide receptor gene and its association with essential hypertension and left ventricular hypertrophy in the Japanese. Circ Res. 2000;86:841–845. doi: 10.1161/01.res.86.8.841. [DOI] [PubMed] [Google Scholar]
- Napoli C, Cirino G, Del Soldato P, Sorrentino R, Sica V, Condorelli M, Pinto A, Ignarro LJ. Effects of nitric oxide-releasing aspirin versus aspirin on restenosis in hypercholesterolemic mice. Proc Natl Acad Sci U S A. 2001;98:2860–2864. doi: 10.1073/pnas.041602898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nausch LW, Ledoux J, Bonev AD, Nelson MT, Dostmann WR. Differential patterning of cGMP in vascular smooth muscle cells revealed by single GFP-linked biosensors. Proc Natl Acad Sci U S A. 2008;105:365–370. doi: 10.1073/pnas.0710387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Cid J, Maeso R, Rodrigo E, Munoz-Garcia R, Ruilope LM, Lahera V, Cachofeiro V. Renal and vascular consequences of the chronic nitric oxide synthase inhibition. Effects of antihypertensive drugs. Am J Hypertens. 1996;9:1077–1083. doi: 10.1016/0895-7061(96)00195-1. [DOI] [PubMed] [Google Scholar]
- Netherton SJ, Maurice DH. Vascular endothelial cell cyclic nucleotide phosphodiesterases and regulated cell migration: implications in angiogenesis. Mol Pharmacol. 2005;67:263–272. doi: 10.1124/mol.104.004853. [DOI] [PubMed] [Google Scholar]
- Nichol CA, Lee CL, Edelstein MP, Chao JY, Duch DS. Biosynthesis of tetrahydrobiopterin by de novo and salvage pathways in adrenal medulla extracts, mammalian cell cultures, and rat brain in vivo. Proc Natl Acad Sci U S A. 1983;80:1546–1550. doi: 10.1073/pnas.80.6.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North AJ, Star RA, Brannon TS, Ujiie K, Wells LB, Lowenstein CJ, Snyder SH, Shaul PW. Nitric oxide synthase type I and type III gene expression are developmentally regulated in rat lung. Am J Physiol. 1994;266:L635–L641. doi: 10.1152/ajplung.1994.266.6.L635. [DOI] [PubMed] [Google Scholar]
- Oelze M, Mollnau H, Hoffmann N, Warnholtz A, Bodenschatz M, Smolenski A, Walter U, Skatchkov M, Meinertz T, Munzel T. Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ Res. 2000;87:999–1005. doi: 10.1161/01.res.87.11.999. [DOI] [PubMed] [Google Scholar]
- Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest. 1993;91:2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, Reddick RL, Pandey KN, Milgram SL, Smithies O, Maeda N. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci U S A. 1997;94:14730–14735. doi: 10.1073/pnas.94.26.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongini E, Impagnatiello F, Bonazzi A, Guzzetta M, Govoni M, Monopoli A, Del Soldato P, Ignarro LJ. Nitric oxide (NO)-releasing statin derivatives, a class of drugs showing enhanced antiproliferative and antiinflammatory properties. Proc Natl Acad Sci U S A. 2004;101:8497–8502. doi: 10.1073/pnas.0401996101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osadchii OE. Myocardial phosphodiesterases and regulation of cardiac contractility in health and cardiac disease. Cardiovasc Drugs Ther. 2007;21:171–194. doi: 10.1007/s10557-007-6014-6. [DOI] [PubMed] [Google Scholar]
- Palmieri EA, Affuso F, Fazio S, Lembo D. Tadalafil in primary pulmonary arterial hypertension. Ann Intern Med. 2004;141:743–744. doi: 10.7326/0003-4819-141-9-200411020-00033. [DOI] [PubMed] [Google Scholar]
- Pan SL, Guh JH, Chang YL, Kuo SC, Lee FY, Teng CM. YC-1 prevents sodium nitroprusside-mediated apoptosis in vascular smooth muscle cells. Cardiovasc Res. 2004;61:152–158. doi: 10.1016/j.cardiores.2003.09.013. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos A, Desai KM, Rudic RD, Mayer B, Zhang R, Ruiz-Torres MP, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide synthase inhibitors attenuate transforming-growth-factor-beta 1-stimulated capillary organization in vitro. Am J Pathol. 1997;150:1835–1844. [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti A, Morbidelli L, Cui XL, Douglas JG, Hood JD, Granger HJ, Ledda F, Ziche M. Nitric oxide is an upstream signal of vascular endothelial growth factor-induced extracellular signal-regulated kinase1/2 activation in postcapillary endothelium. J Biol Chem. 1998;273:4220–4226. doi: 10.1074/jbc.273.7.4220. [DOI] [PubMed] [Google Scholar]
- Park MH. Advances in diagnosis and treatment in patients with pulmonary arterial hypertension. Catheter Cardiovasc Interv. 2008;71:205–213. doi: 10.1002/ccd.21389. [DOI] [PubMed] [Google Scholar]
- Parker TA, le Cras TD, Kinsella JP, Abman SH. Developmental changes in endothelial nitric oxide synthase expression and activity in ovine fetal lung. Am J Physiol Lung Cell Mol Physiol. 2000;278:L202–L208. doi: 10.1152/ajplung.2000.278.1.L202. [DOI] [PubMed] [Google Scholar]
- Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin SD, Silengo L, Altruda F, Wetzker R, Wymann MP, Lembo G, Hirsch E. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell. 2004;118:375–387. doi: 10.1016/j.cell.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Pauvert O, Salvail D, Rousseau E, Lugnier C, Marthan R, Savineau JP. Characterisation of cyclic nucleotide phosphodiesterase isoforms in the media layer of the main pulmonary artery. Biochem Pharmacol. 2002;63:1763–1772. doi: 10.1016/s0006-2952(02)00919-x. [DOI] [PubMed] [Google Scholar]
- Peng W, Hoidal JR, Farrukh IS. Regulation of Ca(2+)-activated K+ channels in pulmonary vascular smooth muscle cells: role of nitric oxide. J Appl Physiol. 1996;81:1264–1272. doi: 10.1152/jappl.1996.81.3.1264. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, Hirneiss C, Wang GX, Korth M, Aszodi A, Andersson KE, Krombach F, Mayerhofer A, Ruth P, Fassler R, Hofmann F. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998;17:3045–3051. doi: 10.1093/emboj/17.11.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfitzer G, Ruegg JC, Flockerzi V, Hofmann F. cGMP-dependent protein kinase decreases calcium sensitivity of skinned cardiac fibers. FEBS Lett. 1982;149:171–175. doi: 10.1016/0014-5793(82)81095-8. [DOI] [PubMed] [Google Scholar]
- Phillips PG, Long L, Wilkins MR, Morrell NW. cAMP phosphodiesterase inhibitors potentiate effects of prostacyclin analogs in hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2005;288:L103–L115. doi: 10.1152/ajplung.00095.2004. [DOI] [PubMed] [Google Scholar]
- Piggott LA, Hassell KA, Berkova Z, Morris AP, Silberbach M, Rich TC. Natriuretic peptides and nitric oxide stimulate cGMP synthesis in different cellular compartments. J Gen Physiol. 2006;128:3–14. doi: 10.1085/jgp.200509403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilz RB, Casteel DE. Regulation of gene expression by cyclic GMP. Circ Res. 2003;93:1034–1046. doi: 10.1161/01.RES.0000103311.52853.48. [DOI] [PubMed] [Google Scholar]
- Pison U, Lopez FA, Heidelmeyer CF, Rossaint R, Falke KJ. Inhaled nitric oxide reverses hypoxic pulmonary vasoconstriction without impairing gas exchange. J Appl Physiol. 1993;74:1287–1292. doi: 10.1152/jappl.1993.74.3.1287. [DOI] [PubMed] [Google Scholar]
- Pollman MJ, Yamada T, Horiuchi M, Gibbons GH. Vasoactive substances regulate vascular smooth muscle cell apoptosis. Countervailing influences of nitric oxide and angiotensin II. Circ Res. 1996;79:748–756. doi: 10.1161/01.res.79.4.748. [DOI] [PubMed] [Google Scholar]
- Prasad S, Wilkinson J, Gatzoulis MA. Sildenafil in primary pulmonary hypertension. N Engl J Med. 2000;343:1342. doi: 10.1056/NEJM200011023431814. [DOI] [PubMed] [Google Scholar]
- Price CJ, Brindle NP. Vasodilator-stimulated phosphoprotein is involved in stress-fiber and membrane ruffle formation in endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:2051–2056. doi: 10.1161/01.atv.20.9.2051. [DOI] [PubMed] [Google Scholar]
- Quinlan CL, Costa AD, Costa CL, Pierre SV, Dos SP, Garlid KD. Conditioning the heart induces formation of signalosomes that interact with mitochondria to open mitoKATP channels. Am J Physiol Heart Circ Physiol. 2008;295:H953–H961. doi: 10.1152/ajpheart.00520.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord. 2004;14:491–496. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Quinlan TR, Li D, Laubach VE, Shesely EG, Zhou N, Johns RA. eNOS-deficient mice show reduced pulmonary vascular proliferation and remodeling to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2000;279:L641–L650. doi: 10.1152/ajplung.2000.279.4.L641. [DOI] [PubMed] [Google Scholar]
- Raeymaekers L, Hofmann F, Casteels R. Cyclic GMP-dependent protein kinase phosphorylates phospholamban in isolated sarcoplasmic reticulum from cardiac and smooth muscle. Biochem J. 1988;252:269–273. doi: 10.1042/bj2520269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport RM, Draznin MB, Murad F. Endothelium-dependent relaxation in rat aorta may be mediated through cyclic GMP-dependent protein phosphorylation. Nature. 1983;306:174–176. doi: 10.1038/306174a0. [DOI] [PubMed] [Google Scholar]
- Rapoport RM, Murad F. Agonist-induced endothelium-dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circ Res. 1983;52:352–357. doi: 10.1161/01.res.52.3.352. [DOI] [PubMed] [Google Scholar]
- Rastaldo R, Pagliaro P, Cappello S, Penna C, Mancardi D, Westerhof N, Losano G. Nitric oxide and cardiac function. Life Sci. 2007;81:779–793. doi: 10.1016/j.lfs.2007.07.019. [DOI] [PubMed] [Google Scholar]
- Reffelmann T, Kloner RA. Therapeutic potential of phosphodiesterase 5 inhibition for cardiovascular disease. Circulation. 2003;108:239–244. doi: 10.1161/01.CIR.0000081166.87607.E2. [DOI] [PubMed] [Google Scholar]
- Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch BM, Walter U. The proline-rich focal adhesion and microfilament protein VASP is a ligand for profilins. EMBO J. 1995;14:1583–1589. doi: 10.1002/j.1460-2075.1995.tb07146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembold CM, Foster DB, Strauss JD, Wingard CJ, Eyk JE. cGMP-mediated phosphorylation of heat shock protein 20 may cause smooth muscle relaxation without myosin light chain dephosphorylation in swine carotid artery. J Physiol. 2000;524(Pt 3):865–878. doi: 10.1111/j.1469-7793.2000.00865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembold CM, Kaufman E. Heat induced HSP20 phosphorylation without increased cyclic nucleotide levels in swine carotid media. BMC Physiol 3. 2003:3. doi: 10.1186/1472-6793-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentsendorj O, Mirzapoiazova T, Adyshev D, Servinsky LE, Renne T, Verin AD, Pearse DB. Role of vasodilator-stimulated phosphoprotein in cGMP-mediated protection of human pulmonary artery endothelial barrier function. Am J Physiol Lung Cell Mol Physiol. 2008;294:L686–L697. doi: 10.1152/ajplung.00417.2007. [DOI] [PubMed] [Google Scholar]
- Ribeiro MO, Antunes E, de Nucci G, Lovisolo SM, Zatz R. Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension. Hypertension. 1992;20:298–303. doi: 10.1161/01.hyp.20.3.298. [DOI] [PubMed] [Google Scholar]
- Rich GF, Roos CM, Anderson SM, Urich DC, Daugherty MO, Johns RA. Inhaled nitric oxide: dose response and the effects of blood in the isolated rat lung. J Appl Physiol. 1993;75:1278–1284. doi: 10.1152/jappl.1993.75.3.1278. [DOI] [PubMed] [Google Scholar]
- Riegger GA, Elsner D, Kromer EP, Daffner C, Forssmann WG, Muders F, Pascher EW, Kochsiek K. Atrial natriuretic peptide in congestive heart failure in the dog: plasma levels, cyclic guanosine monophosphate, ultrastructure of atrial myoendocrine cells, and hemodynamic, hormonal, and renal effects. Circulation. 1988;77:398–406. doi: 10.1161/01.cir.77.2.398. [DOI] [PubMed] [Google Scholar]
- Rizzoni D, Porteri E, Castellano M, Bettoni G, Muiesan ML, Tiberio G, Giulini SM, Rossi G, Bernini G, Agabiti-Rosei E. Endothelial dysfunction in hypertension is independent from the etiology and from vascular structure. Hypertension. 1998;31:335–341. doi: 10.1161/01.hyp.31.1.335. [DOI] [PubMed] [Google Scholar]
- Rochais F, Abi-Gerges A, Horner K, Lefebvre F, Cooper DM, Conti M, Fischmeister R, Vandecasteele G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res. 2006;98:1081–1088. doi: 10.1161/01.RES.0000218493.09370.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen GM, Tsai P, Weaver J, Porasuphatana S, Roman LJ, Starkov AA, Fiskum G, Pou S. The role of tetrahydrobiopterin in the regulation of neuronal nitric-oxide synthase-generated superoxide. J Biol Chem. 2002;277:40275–40280. doi: 10.1074/jbc.M200853200. [DOI] [PubMed] [Google Scholar]
- Rosen RC, Kostis JB. Overview of phosphodiesterase 5 inhibition in erectile dysfunction. Am J Cardiol. 2003;92:9M–18M. doi: 10.1016/s0002-9149(03)00824-5. [DOI] [PubMed] [Google Scholar]
- Rosenkranz AC, Woods RL, Dusting GJ, Ritchie RH. Antihypertrophic actions of the natriuretic peptides in adult rat cardiomyocytes: importance of cyclic GMP. Cardiovasc Res. 2003;57:515–522. doi: 10.1016/s0008-6363(02)00667-3. [DOI] [PubMed] [Google Scholar]
- Rossaint R, Falke KJ, Lopez F, Slama K, Pison U, Zapol WM. Inhaled nitric oxide for the adult respiratory distress syndrome. N Engl J Med. 1993;328:399–405. doi: 10.1056/NEJM199302113280605. [DOI] [PubMed] [Google Scholar]
- Rossi R, Nuzzo A, Lattanzi A, Coppi F, Modena MG. Sildenafil improves endothelial function in patients with pulmonary hypertension. Pulm Pharmacol Ther. 2008;21:172–177. doi: 10.1016/j.pupt.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Rossiello MR, Momi S, Caracchini R, Giannini S, Guglielmini G, Monopoli A, Ongini E, Semeraro N, Colucci M, Gresele P. A novel nitric oxide-releasing statin derivative exerts an antiplatelet/antithrombotic activity and inhibits tissue factor expression. J Thromb Haemost. 2005;3:2554–2562. doi: 10.1111/j.1538-7836.2005.01605.x. [DOI] [PubMed] [Google Scholar]
- Rossoni G, Berti M, Colonna VD, Bernareggi M, Del Soldato P, Berti F. Myocardial protection by the nitroderivative of aspirin, NCX 4016: in vitro and in vivo experiments in the rabbit. Ital Heart J. 2000;1:146–155. [PubMed] [Google Scholar]
- Rothermund L, Friebe A, Paul M, Koesling D, Kreutz R. Acute blood pressure effects of YC-1-induced activation of soluble guanylyl cyclase in normotensive and hypertensive rats. Br J Pharmacol. 2000;130:205–208. doi: 10.1038/sj.bjp.0703320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruetten H, Dimmeler S, Gehring D, Ihling C, Zeiher AM. Concentric left ventricular remodeling in endothelial nitric oxide synthase knockout mice by chronic pressure overload. Cardiovasc Res. 2005;66:444–453. doi: 10.1016/j.cardiores.2005.01.021. [DOI] [PubMed] [Google Scholar]
- Ruetten H, Zabel U, Linz W, Schmidt HH. Downregulation of soluble guanylyl cyclase in young and aging spontaneously hypertensive rats. Circ Res. 1999;85:534–541. doi: 10.1161/01.res.85.6.534. [DOI] [PubMed] [Google Scholar]
- Russwurm M, Koesling D. NO activation of guanylyl cyclase. EMBO J. 2004;23:4443–4450. doi: 10.1038/sj.emboj.7600422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russwurm M, Wittau N, Koesling D. Guanylyl cyclase/PSD-95 interaction: targeting of the nitric oxide-sensitive alpha2beta1 guanylyl cyclase to synaptic membranes. J Biol Chem. 2001;276:44647–44652. doi: 10.1074/jbc.M105587200. [DOI] [PubMed] [Google Scholar]
- Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res. 2003;93:280–291. doi: 10.1161/01.RES.0000087541.15600.2B. [DOI] [PubMed] [Google Scholar]
- Sabrane K, Kruse MN, Fabritz L, Zetsche B, Mitko D, Skryabin BV, Zwiener M, Baba HA, Yanagisawa M, Kuhn M. Vascular endothelium is critically involved in the hypotensive and hypovolemic actions of atrial natriuretic peptide. J Clin Invest. 2005;115:1666–1674. doi: 10.1172/JCI23360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K. Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA. 2005;293:1900–1905. doi: 10.1001/jama.293.15.1900. [DOI] [PubMed] [Google Scholar]
- Sackner-Bernstein JD, Skopicki HA, Aaronson KD. Risk of worsening renal function with nesiritide in patients with acutely decompensated heart failure. Circulation. 2005;111:1487–1491. doi: 10.1161/01.CIR.0000159340.93220.E4. [DOI] [PubMed] [Google Scholar]
- Sadhu K, Hensley K, Florio VA, Wolda SL. Differential expression of the cyclic GMP-stimulated phosphodiesterase PDE2A in human venous and capillary endothelial cells. J Histochem Cytochem. 1999;47:895–906. doi: 10.1177/002215549904700707. [DOI] [PubMed] [Google Scholar]
- Sakuma I, Togashi H, Yoshioka M, Saito H, Yanagida M, Tamura M, Kobayashi T, Yasuda H, Gross SS, Levi R. NG-methyl-L-arginine, an inhibitor of L-arginine-derived nitric oxide synthesis, stimulates renal sympathetic nerve activity in vivo. A role for nitric oxide in the central regulation of sympathetic tone? Circ Res. 1992;70:607–611. doi: 10.1161/01.res.70.3.607. [DOI] [PubMed] [Google Scholar]
- Salloum FN, Abbate A, Das A, Houser JE, Mudrick CA, Qureshi IZ, Hoke NN, Roy SK, Brown WR, Prabhakar S, Kukreja RC. Sildenafil (Viagra) attenuates ischemic cardiomyopathy and improves left ventricular function in mice. Am J Physiol Heart Circ Physiol. 2008;294:H1398–H1406. doi: 10.1152/ajpheart.91438.2007. [DOI] [PubMed] [Google Scholar]
- Saraiva RM, Hare JM. Nitric oxide signaling in the cardiovascular system: implications for heart failure. Curr Opin Cardiol. 2006;21:221–228. doi: 10.1097/01.hco.0000221584.56372.dc. [DOI] [PubMed] [Google Scholar]
- Sasaki N, Sato T, Ohler A, O'Rourke B, Marban E. Activation of mitochondrial ATP-dependent potassium channels by nitric oxide. Circulation. 2000;101:439–445. doi: 10.1161/01.cir.101.4.439. [DOI] [PubMed] [Google Scholar]
- Sastry BK, Narasimhan C, Reddy NK, Raju BS. Clinical efficacy of sildenafil in primary pulmonary hypertension: a randomized, placebo-controlled, double-blind, crossover study. J Am Coll Cardiol. 2004;43:1149–1153. doi: 10.1016/j.jacc.2003.10.056. [DOI] [PubMed] [Google Scholar]
- Sausbier M, Schubert R, Voigt V, Hirneiss C, Pfeifer A, Korth M, Kleppisch T, Ruth P, Hofmann F. Mechanisms of NO/cGMP-dependent vasorelaxation. Circ Res. 2000;87:825–830. doi: 10.1161/01.res.87.9.825. [DOI] [PubMed] [Google Scholar]
- Sayed N, Kim DD, Fioramonti X, Iwahashi T, Duran WN, Beuve A. Nitroglycerin-induced S-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ Res. 2008;103:606–614. doi: 10.1161/CIRCRESAHA.108.175133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer A, Burkhardt M, Vollkommer T, Bauersachs J, Munzel T, Walter U, Smolenski A. Endothelium-dependent and -independent relaxation and VASP serines 157/239 phosphorylation by cyclic nucleotide-elevating vasodilators in rat aorta. Biochem Pharmacol. 2003;65:397–405. doi: 10.1016/s0006-2952(02)01523-x. [DOI] [PubMed] [Google Scholar]
- Scherer-Oppliger T, Leimbacher W, Blau N, Thony B. Serine 19 of human 6-pyruvoyltetrahydropterin synthase is phosphorylated by cGMP protein kinase II. J Biol Chem. 1999;274:31341–31348. doi: 10.1074/jbc.274.44.31341. [DOI] [PubMed] [Google Scholar]
- Schlossmann J, Ammendola A, Ashman K, Zong X, Huber A, Neubauer G, Wang GX, Allescher HD, Korth M, Wilm M, Hofmann F, Ruth P. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature. 2000;404:197–201. doi: 10.1038/35004606. [DOI] [PubMed] [Google Scholar]
- Schoser BG, Behrends S. Soluble guanylyl cyclase is localized at the neuromuscular junction in human skeletal muscle. Neuroreport. 2001;12:979–981. doi: 10.1097/00001756-200104170-00023. [DOI] [PubMed] [Google Scholar]
- Schultz K, Schultz K, Schultz G. Sodium nitroprusside and other smooth muscle-relaxants increase cyclic GMP levels in rat ductus deferens. Nature. 1977;265:750–751. doi: 10.1038/265750a0. [DOI] [PubMed] [Google Scholar]
- Schulz E, Jansen T, Wenzel P, Daiber A, Munzel T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid Redox Signal. 2008;10:1115–1126. doi: 10.1089/ars.2007.1989. [DOI] [PubMed] [Google Scholar]
- Sears CE, Bryant SM, Ashley EA, Lygate CA, Rakovic S, Wallis HL, Neubauer S, Terrar DA, Casadei B. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res. 2003;92:e52–e59. doi: 10.1161/01.RES.0000064585.95749.6D. [DOI] [PubMed] [Google Scholar]
- Senzaki H, Smith CJ, Juang GJ, Isoda T, Mayer SP, Ohler A, Paolocci N, Tomaselli GF, Hare JM, Kass DA. Cardiac phosphodiesterase 5 (cGMP-specific) modulates beta-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J. 2001;15:1718–1726. doi: 10.1096/fj.00-0538com. [DOI] [PubMed] [Google Scholar]
- Shah AM. Paracrine modulation of heart cell function by endothelial cells. Cardiovasc Res. 1996;31:847–867. [PubMed] [Google Scholar]
- Shah AM, MacCarthy PA. Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacol Ther. 2000;86:49–86. doi: 10.1016/s0163-7258(99)00072-8. [DOI] [PubMed] [Google Scholar]
- Shah AM, Spurgeon HA, Sollott SJ, Talo A, Lakatta EG. 8-bromo-cGMP reduces the myofilament response to Ca2+ in intact cardiac myocytes. Circ Res. 1994;74:970–978. doi: 10.1161/01.res.74.5.970. [DOI] [PubMed] [Google Scholar]
- Shakur Y, Holst LS, Landstrom TR, Movsesian M, Degerman E, Manganiello V. Regulation and function of the cyclic nucleotide phosphodiesterase (PDE3) gene family. Prog Nucleic Acid Res Mol Biol. 2001;66:241–277. doi: 10.1016/s0079-6603(00)66031-2. [DOI] [PubMed] [Google Scholar]
- Sharma RK, Adachi AM, Adachi K, Wang JH. Demonstration of bovine brain calmodulin-dependent cyclic nucleotide phosphodiesterase isozymes by monoclonal antibodies. J Biol Chem. 1984;259:9248–9254. [PubMed] [Google Scholar]
- Sharma RK, Wang JH. Calmodulin and Ca2+-dependent phosphorylation and dephosphorylation of 63-kDa subunit-containing bovine brain calmodulin-stimulated cyclic nucleotide phosphodiesterase isozyme. J Biol Chem. 1986;261:1322–1328. [PubMed] [Google Scholar]
- Shaul PW, Afshar S, Gibson LL, Sherman TS, Kerecman JD, Grubb PH, Yoder BA, McCurnin DC. Developmental changes in nitric oxide synthase isoform expression and nitric oxide production in fetal baboon lung. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1192–L1199. doi: 10.1152/ajplung.00112.2002. [DOI] [PubMed] [Google Scholar]
- Shaul PW, Yuhanna IS, German Z, Chen Z, Steinhorn RH, Morin FC., III Pulmonary endothelial NO synthase gene expression is decreased in fetal lambs with pulmonary hypertension. Am J Physiol. 1997;272:L1005–L1012. doi: 10.1152/ajplung.1997.272.5.L1005. [DOI] [PubMed] [Google Scholar]
- Shirodaria C, Antoniades C, Lee J, Jackson CE, Robson MD, Francis JM, Moat SJ, Ratnatunga C, Pillai R, Refsum H, Neubauer S, Channon KM. Global improvement of vascular function and redox state with low-dose folic acid: implications for folate therapy in patients with coronary artery disease. Circulation. 2007;115:2262–2270. doi: 10.1161/CIRCULATIONAHA.106.679084. [DOI] [PubMed] [Google Scholar]
- Silberbach M, Roberts CT., Jr Natriuretic peptide signalling: molecular and cellular pathways to growth regulation. Cell Signal. 2001;13:221–231. doi: 10.1016/s0898-6568(01)00139-5. [DOI] [PubMed] [Google Scholar]
- Smolenski A, Poller W, Walter U, Lohmann SM. Regulation of human endothelial cell focal adhesion sites and migration by cGMP-dependent protein kinase I. J Biol Chem. 2000;275:25723–25732. doi: 10.1074/jbc.M909632199. [DOI] [PubMed] [Google Scholar]
- Snyder PB, Florio VA, Ferguson K, Loughney K. Isolation, expression and analysis of splice variants of a human Ca2+/calmodulin-stimulated phosphodiesterase (PDE1A) Cell Signal. 1999;11:535–544. doi: 10.1016/s0898-6568(99)00027-3. [DOI] [PubMed] [Google Scholar]
- Soeki T, Kishimoto I, Okumura H, Tokudome T, Horio T, Mori K, Kangawa K. C-type natriuretic peptide, a novel antifibrotic and antihypertrophic agent, prevents cardiac remodeling after myocardial infarction. J Am Coll Cardiol. 2005;45:608–616. doi: 10.1016/j.jacc.2004.10.067. [DOI] [PubMed] [Google Scholar]
- Sonnenburg WK, Seger D, Kwak KS, Huang J, Charbonneau H, Beavo JA. Identification of inhibitory and calmodulin-binding domains of the PDE1A1 and PDE1A2 calmodulin-stimulated cyclic nucleotide phosphodiesterases. J Biol Chem. 1995;270:30989–31000. doi: 10.1074/jbc.270.52.30989. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Loh E, Roddy MA, Currie KE, Creager MA. Nitric oxide regulates basal systemic and pulmonary vascular resistance in healthy humans. Circulation. 1994;89:2035–2040. doi: 10.1161/01.cir.89.5.2035. [DOI] [PubMed] [Google Scholar]
- Stasch JP, Schmidt P, Alonso-Alija C, Apeler H, Dembowsky K, Haerter M, Heil M, Minuth T, Perzborn E, Pleiss U, Schramm M, Schroeder W, Schroder H, Stahl E, Steinke W, Wunder F. NO- and haem-independent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of a new pharmacological principle. Br J Pharmacol. 2002;136:773–783. doi: 10.1038/sj.bjp.0704778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, AK HS, Meurer S, Deile M, Taye A, Knorr A, Lapp H, Muller H, Turgay Y, Rothkegel C, Tersteegen A, Kemp-Harper B, Muller-Esterl W, Schmidt HH. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116:2552–2561. doi: 10.1172/JCI28371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauss HM, Godecke A, Mrowka R, Schrader J, Persson PB. Enhanced blood pressure variability in eNOS knockout mice. Hypertension. 1999;33:1359–1363. doi: 10.1161/01.hyp.33.6.1359. [DOI] [PubMed] [Google Scholar]
- Steudel W, Ichinose F, Huang PL, Hurford WE, Jones RC, Bevan JA, Fishman MC, Zapol WM. Pulmonary vasoconstriction and hypertension in mice with targeted disruption of the endothelial nitric oxide synthase (NOS 3) gene. Circ Res. 1997;81:34–41. doi: 10.1161/01.res.81.1.34. [DOI] [PubMed] [Google Scholar]
- Steudel W, Scherrer-Crosbie M, Bloch KD, Weimann J, Huang PL, Jones RC, Picard MH, Zapol WM. Sustained pulmonary hypertension and right ventricular hypertrophy after chronic hypoxia in mice with congenital deficiency of nitric oxide synthase 3. J Clin Invest. 1998;101:2468–2477. doi: 10.1172/JCI2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroes ES, van Faassen EE, Yo M, Martasek P, Boer P, Govers R, Rabelink TJ. Folic acid reverts dysfunction of endothelial nitric oxide synthase. Circ Res. 2000;86:1129–1134. doi: 10.1161/01.res.86.11.1129. [DOI] [PubMed] [Google Scholar]
- Suenobu N, Shichiri M, Iwashina M, Marumo F, Hirata Y. Natriuretic peptides and nitric oxide induce endothelial apoptosis via a cGMP-dependent mechanism. Arterioscler Thromb Vasc Biol. 1999;19:140–146. doi: 10.1161/01.atv.19.1.140. [DOI] [PubMed] [Google Scholar]
- Sugawara A, Nakao K, Morii N, Yamada T, Itoh H, Shiono S, Saito Y, Mukoyama M, Arai H, Nishimura K. Synthesis of atrial natriuretic polypeptide in human failing hearts. Evidence for altered processing of atrial natriuretic polypeptide precursor and augmented synthesis of beta-human ANP. J Clin Invest. 1988;81:1962–1970. doi: 10.1172/JCI113544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surapisitchat J, Jeon KI, Yan C, Beavo JA. Differential regulation of endothelial cell permeability by cGMP via phosphodiesterases 2 and 3. Circ Res. 2007;101:811–818. doi: 10.1161/CIRCRESAHA.107.154229. [DOI] [PubMed] [Google Scholar]
- Surks HK, Mendelsohn ME. Dimerization of cGMP-dependent protein kinase 1alpha and the myosin-binding subunit of myosin phosphatase: role of leucine zipper domains. Cell Signal. 2003;15:937–944. doi: 10.1016/s0898-6568(03)00057-3. [DOI] [PubMed] [Google Scholar]
- Taigen T, De Windt LJ, Lim HW, Molkentin JD. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2000;97:1196–1201. doi: 10.1073/pnas.97.3.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taimor G, Hofstaetter B, Piper HM. Apoptosis induction by nitric oxide in adult cardiomyocytes via cGMP-signaling and its impairment after simulated ischemia. Cardiovasc Res. 2000;45:588–594. doi: 10.1016/s0008-6363(99)00272-2. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Osanai T, Nakano T, Wakui M, Okumura K. Enhanced activities and gene expression of phosphodiesterase types 3 and 4 in pressure-induced congestive heart failure. Heart Vessels. 2002;16:249–256. doi: 10.1007/s003800200032. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Suzuki K, Yamamoto H, Araki H, Mizoguchi H, Ukawa H. Cyclooxygenase-2 selective and nitric oxide-releasing nonsteroidal anti-inflammatory drugs and gastric mucosal responses. J Physiol Pharmacol. 1998;49:501–513. [PubMed] [Google Scholar]
- Takeuchi K, Ukawa H, Konaka A, Kitamura M, Sugawa Y. Effect of nitric oxide-releasing aspirin derivative on gastric functional and ulcerogenic responses in rats: comparison with plain aspirin. J Pharmacol Exp Ther. 1998;286:115–121. [PubMed] [Google Scholar]
- Takimoto E, Belardi D, Tocchetti CG, Vahebi S, Cormaci G, Ketner EA, Moens AL, Champion HC, Kass DA. Compartmentalization of cardiac beta-adrenergic inotropy modulation by phosphodiesterase type 5. Circulation. 2007;115:2159–2167. doi: 10.1161/CIRCULATIONAHA.106.643536. [DOI] [PubMed] [Google Scholar]
- Takimoto E, Champion HC, Belardi D, Moslehi J, Mongillo M, Mergia E, Montrose DC, Isoda T, Aufiero K, Zaccolo M, Dostmann WR, Smith CJ, Kass DA. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res. 2005;96:100–109. doi: 10.1161/01.RES.0000152262.22968.72. [DOI] [PubMed] [Google Scholar]
- Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Ueda M, Endo M, Kitajima M. The effect of atrial natriuretic peptide on pulmonary acid injury in a pig model. Am J Respir Crit Care Med. 1996;154:1351–1356. doi: 10.1164/ajrccm.154.5.8912747. [DOI] [PubMed] [Google Scholar]
- Tatsumi T, Matoba S, Kawahara A, Keira N, Shiraishi J, Akashi K, Kobara M, Tanaka T, Katamura M, Nakagawa C, Ohta B, Shirayama T, Takeda K, Asayama J, Fliss H, Nakagawa M. Cytokine-induced nitric oxide production inhibits mitochondrial energy production and impairs contractile function in rat cardiac myocytes. J Am Coll Cardiol. 2000;35:1338–1346. doi: 10.1016/s0735-1097(00)00526-x. [DOI] [PubMed] [Google Scholar]
- Thompson WJ, Ashikaga T, Kelly JJ, Liu L, Zhu B, Vemavarapu L, Strada SJ. Regulation of cyclic AMP in rat pulmonary microvascular endothelial cells by rolipram-sensitive cyclic AMP phosphodiesterase (PDE4) Biochem Pharmacol. 2002;63:797–807. doi: 10.1016/s0006-2952(01)00914-5. [DOI] [PubMed] [Google Scholar]
- Tikkanen I, Fyhrquist F, Metsarinne K, Leidenius R. Plasma atrial natriuretic peptide in cardiac disease and during infusion in healthy volunteers. Lancet. 1985;2:66–69. doi: 10.1016/s0140-6736(85)90178-3. [DOI] [PubMed] [Google Scholar]
- Tokudome T, Horio T, Soeki T, Mori K, Kishimoto I, Suga S, Yoshihara F, Kawano Y, Kohno M, Kangawa K. Inhibitory effect of C-type natriuretic peptide (CNP) on cultured cardiac myocyte hypertrophy: interference between CNP and endothelin-1 signaling pathways. Endocrinology. 2004;145:2131–2140. doi: 10.1210/en.2003-1260. [DOI] [PubMed] [Google Scholar]
- Tsai EJ, Nagayama T, Friebe A, Kass DA, Takimoto E. A Nitric Oxide-Soluble Guanylyl Cyclase-Cyclic Guanosine Monophosphate Signaling Lipid Raft Microdomain in Cardiac Myocytes is Altered by Pressure-Overload and Restored with Chronic Sildenafil Treatment. Circ Res. 2008;103:5–5. [Google Scholar]
- Tsang A, Hausenloy DJ, Mocanu MM, Carr RD, Yellon DM. Preconditioning the diabetic heart: the importance of Akt phosphorylation. Diabetes. 2005;54:2360–2364. doi: 10.2337/diabetes.54.8.2360. [DOI] [PubMed] [Google Scholar]
- Tsoukias NM, Kavdia M, Popel AS. A theoretical model of nitric oxide transport in arterioles: frequency- vs. amplitude-dependent control of cGMP formation. Am J Physiol Heart Circ Physiol. 2004;286:H1043–H1056. doi: 10.1152/ajpheart.00525.2003. [DOI] [PubMed] [Google Scholar]
- Tsutamoto T, Kanamori T, Morigami N, Sugimoto Y, Yamaoka O, Kinoshita M. Possibility of downregulation of atrial natriuretic peptide receptor coupled to guanylate cyclase in peripheral vascular beds of patients with chronic severe heart failure. Circulation. 1993;87:70–75. doi: 10.1161/01.cir.87.1.70. [DOI] [PubMed] [Google Scholar]
- Tsutamoto T, Kanamori T, Wada A, Kinoshita M. Uncoupling of atrial natriuretic peptide extraction and cyclic guanosine monophosphate production in the pulmonary circulation in patients with severe heart failure. J Am Coll Cardiol. 1992;20:541–546. doi: 10.1016/0735-1097(92)90005-8. [DOI] [PubMed] [Google Scholar]
- Tulis DA, Bohl Masters KS, Lipke EA, Schiesser RL, Evans AJ, Peyton KJ, Durante W, West JL, Schafer AI. YC-1-mediated vascular protection through inhibition of smooth muscle cell proliferation and platelet function. Biochem Biophys Res Commun. 2002;291:1014–1021. doi: 10.1006/bbrc.2002.6552. [DOI] [PubMed] [Google Scholar]
- Tzao C, Nickerson PA, Russell JA, Gugino SF, Steinhorn RH. Pulmonary hypertension alters soluble guanylate cyclase activity and expression in pulmonary arteries isolated from fetal lambs. Pediatr Pulmonol. 2001;31:97–105. doi: 10.1002/1099-0496(200102)31:2<97::aid-ppul1016>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Vaandrager AB, de Jonge HR. Signalling by cGMP-dependent protein kinases. Mol Cell Biochem. 1996;157:23–30. doi: 10.1007/BF00227877. [DOI] [PubMed] [Google Scholar]
- Vaandrager AB, Ehlert EM, Jarchau T, Lohmann SM, de Jonge HR. N-terminal myristoylation is required for membrane localization of cGMP-dependent protein kinase type II. J Biol Chem. 1996;271:7025–7029. doi: 10.1074/jbc.271.12.7025. [DOI] [PubMed] [Google Scholar]
- van Etten RW, de Koning EJ, Verhaar MC, Gaillard CA, Rabelink TJ. Impaired NO-dependent vasodilation in patients with Type II (non-insulin-dependent) diabetes mellitus is restored by acute administration of folate. Diabetologia. 2002;45:1004–1010. doi: 10.1007/s00125-002-0862-1. [DOI] [PubMed] [Google Scholar]
- Vandecasteele G, Verde I, Rucker-Martin C, Donzeau-Gouge P, Fischmeister R. Cyclic GMP regulation of the L-type Ca(2+) channel current in human atrial myocytes. J Physiol. 2001;533:329–340. doi: 10.1111/j.1469-7793.2001.0329a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeput F, Wolda SL, Krall J, Hambleton R, Uher L, McCaw KN, Radwanski PB, Florio V, Movsesian MA. Cyclic nucleotide phosphodiesterase PDE1C1 in human cardiac myocytes. J Biol Chem. 2007;282:32749–32757. doi: 10.1074/jbc.M703173200. [DOI] [PubMed] [Google Scholar]
- Vasquez-Vivar J, Martasek P, Whitsett J, Joseph J, Kalyanaraman B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: an EPR spin trapping study. Biochem J. 2002;362:733–739. doi: 10.1042/0264-6021:3620733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verde I, Vandecasteele G, Lezoualc'h F, Fischmeister R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br J Pharmacol. 1999;127:65–74. doi: 10.1038/sj.bjp.0702506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaar MC, Wever RM, Kastelein JJ, van Dam T, Koomans HA, Rabelink TJ. 5-methyltetrahydrofolate, the active form of folic acid, restores endothelial function in familial hypercholesterolemia. Circulation. 1998;97:237–241. doi: 10.1161/01.cir.97.3.237. [DOI] [PubMed] [Google Scholar]
- Verhaar MC, Wever RM, Kastelein JJ, van Loon D, Milstien S, Koomans HA, Rabelink TJ. Effects of oral folic acid supplementation on endothelial function in familial hypercholesterolemia. A randomized placebo-controlled trial. Circulation. 1999;100:335–338. doi: 10.1161/01.cir.100.4.335. [DOI] [PubMed] [Google Scholar]
- Vermeersch P, Buys E, Pokreisz P, Marsboom G, Ichinose F, Sips P, Pellens M, Gillijns H, Swinnen M, Graveline A, Collen D, Dewerchin M, Brouckaert P, Bloch KD, Janssens S. Soluble guanylate cyclase-alpha1 deficiency selectively inhibits the pulmonary vasodilator response to nitric oxide and increases the pulmonary vascular remodeling response to chronic hypoxia. Circulation. 2007;116:936–943. doi: 10.1161/CIRCULATIONAHA.106.677245. [DOI] [PubMed] [Google Scholar]
- Vila-Petroff MG, Younes A, Egan J, Lakatta EG, Sollott SJ. Activation of distinct cAMP-dependent and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circ Res. 1999;84:1020–1031. doi: 10.1161/01.res.84.9.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosatka RJ, Kashyap S, Trifiletti RR. Arginine deficiency accompanies persistent pulmonary hypertension of the newborn. Biol Neonate. 1994;66:65–70. doi: 10.1159/000244091. [DOI] [PubMed] [Google Scholar]
- Wang G, Liem DA, Vondriska TM, Honda HM, Korge P, Pantaleon DM, Qiao X, Wang Y, Weiss JN, Ping P. Nitric oxide donors protect murine myocardium against infarction via modulation of mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 2005;288:H1290–H1295. doi: 10.1152/ajpheart.00796.2004. [DOI] [PubMed] [Google Scholar]
- Weber M, Lauer N, Mulsch A, Kojda G. The effect of peroxynitrite on the catalytic activity of soluble guanylyl cyclase. Free Radic Biol Med. 2001;31:1360–1367. doi: 10.1016/s0891-5849(01)00706-7. [DOI] [PubMed] [Google Scholar]
- Wegener JW, Nawrath H, Wolfsgruber W, Kuhbandner S, Werner C, Hofmann F, Feil R. cGMP-dependent protein kinase I mediates the negative inotropic effect of cGMP in the murine myocardium. Circ Res. 2002;90:18–20. doi: 10.1161/hh0102.103222. [DOI] [PubMed] [Google Scholar]
- Wehling-Henricks M, Jordan MC, Roos KP, Deng B, Tidball JG. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum Mol Genet. 2005;14:1921–1933. doi: 10.1093/hmg/ddi197. [DOI] [PubMed] [Google Scholar]
- Weitzberg E, Rudehill A, Lundberg JM. Nitric oxide inhalation attenuates pulmonary hypertension and improves gas exchange in endotoxin shock. Eur J Pharmacol. 1993;233:85–94. doi: 10.1016/0014-2999(93)90352-i. [DOI] [PubMed] [Google Scholar]
- Wenzel S, Rohde C, Wingerning S, Roth J, Kojda G, Schluter KD. Lack of endothelial nitric oxide synthase-derived nitric oxide formation favors hypertrophy in adult ventricular cardiomyocytes. Hypertension. 2007;49:193–200. doi: 10.1161/01.HYP.0000250468.02084.ce. [DOI] [PubMed] [Google Scholar]
- White RE, Lee AB, Shcherbatko AD, Lincoln TM, Schonbrunn A, Armstrong DL. Potassium channel stimulation by natriuretic peptides through cGMP-dependent dephosphorylation. Nature. 1993;361:263–266. doi: 10.1038/361263a0. [DOI] [PubMed] [Google Scholar]
- Wiemer G, Linz W, Hatrik S, Scholkens BA, Malinski T. Angiotensin-converting enzyme inhibition alters nitric oxide and superoxide release in normotensive and hypertensive rats. Hypertension. 1997;30:1183–1190. doi: 10.1161/01.hyp.30.5.1183. [DOI] [PubMed] [Google Scholar]
- Williams JM, White CR, Chang MM, Injeti ER, Zhang L, Pearce WJ. Chronic hypoxic decreases in soluble guanylate cyclase protein and enzyme activity are age dependent in fetal and adult ovine carotid arteries. J Appl Physiol. 2006;100:1857–1866. doi: 10.1152/japplphysiol.00662.2005. [DOI] [PubMed] [Google Scholar]
- Wollert KC, Fiedler B, Gambaryan S, Smolenski A, Heineke J, Butt E, Trautwein C, Lohmann SM, Drexler H. Gene transfer of cGMP-dependent protein kinase I enhances the antihypertrophic effects of nitric oxide in cardiomyocytes. Hypertension. 2002;39:87–92. doi: 10.1161/hy1201.097292. [DOI] [PubMed] [Google Scholar]
- Woodard GE, Rosado JA. Natriuretic peptides in vascular physiology and pathology. Int Rev Cell Mol Biol. 2008;268:59–93. doi: 10.1016/S1937-6448(08)00803-4. [DOI] [PubMed] [Google Scholar]
- Wrishko RE, Dingemanse J, Yu A, Darstein C, Phillips DL, Mitchell MI. Pharmacokinetic interaction between tadalafil and bosentan in healthy male subjects. J Clin Pharmacol. 2008;48:610–618. doi: 10.1177/0091270008315315. [DOI] [PubMed] [Google Scholar]
- Wu CF, Bishopric NH, Pratt RE. Atrial natriuretic peptide induces apoptosis in neonatal rat cardiac myocytes. J Biol Chem. 1997;272:14860–14866. doi: 10.1074/jbc.272.23.14860. [DOI] [PubMed] [Google Scholar]
- Wu P, Wang P. Per-Arnt-Sim domain-dependent association of cAMP-phosphodiesterase 8A1 with IkappaB proteins. Proc Natl Acad Sci U S A. 2004;101:17634–17639. doi: 10.1073/pnas.0407649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci U S A. 1996;93:6770–6774. doi: 10.1073/pnas.93.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci U S A. 1999;96:657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Kaneko FT, Zheng S, Comhair SA, Janocha AJ, Goggans T, Thunnissen FB, Farver C, Hazen SL, Jennings C, Dweik RA, Arroliga AC, Erzurum SC. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004;18:1746–1748. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- Xue C, Rengasamy A, le Cras TD, Koberna PA, Dailey GC, Johns RA. Distribution of NOS in normoxic vs. hypoxic rat lung: upregulation of NOS by chronic hypoxia. Am J Physiol. 1994;267:L667–L678. doi: 10.1152/ajplung.1994.267.6.L667. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Kakar NR, Vina ER, Johnson PE, Bing RJ. The effect of aspirin and two nitric oxide donors on the infarcted heart in situ. Life Sci. 2000;67:839–846. doi: 10.1016/s0024-3205(00)00678-0. [DOI] [PubMed] [Google Scholar]
- Yan C, Miller CL, Abe J. Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart. Circ Res. 2007;100:489–501. doi: 10.1161/01.RES.0000258451.44949.d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanaka N, Kurosawa Y, Minami K, Kawai E, Omori K. cGMP-phosphodiesterase activity is up-regulated in response to pressure overload of rat ventricles. Biosci Biotechnol Biochem. 2003;67:973–979. doi: 10.1271/bbb.67.973. [DOI] [PubMed] [Google Scholar]
- Yang L, Liu G, Zakharov SI, Bellinger AM, Mongillo M, Marx SO. Protein kinase G phosphorylates Cav1.2 alpha1c and beta2 subunits. Circ Res. 2007;101:465–474. doi: 10.1161/CIRCRESAHA.107.156976. [DOI] [PubMed] [Google Scholar]
- Yang XP, Liu YH, Shesely EG, Bulagannawar M, Liu F, Carretero OA. Endothelial nitric oxide gene knockout mice: cardiac phenotypes and the effect of angiotensin-converting enzyme inhibitor on myocardial ischemia/reperfusion injury. Hypertension. 1999;34:24–30. doi: 10.1161/01.hyp.34.1.24. [DOI] [PubMed] [Google Scholar]
- Yano M, Kohno M, Ohkusa T, Mochizuki M, Yamada J, Kohno M, Hisaoka T, Ono K, Tanigawa T, Kobayashi S, Matsuzaki M. Effect of milrinone on left ventricular relaxation and Ca(2+) uptake function of cardiac sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol. 2000;279:H1898–H1905. doi: 10.1152/ajpheart.2000.279.4.H1898. [DOI] [PubMed] [Google Scholar]
- Yazawa S, Tsuchiya H, Hori H, Makino R. Functional characterization of two nucleotide-binding sites in soluble guanylate cyclase. J Biol Chem. 2006;281:21763–21770. doi: 10.1074/jbc.M508983200. [DOI] [PubMed] [Google Scholar]
- Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1113–1151. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- Yin J, Hoffmann J, Kaestle SM, Neye N, Wang L, Baeurle J, Liedtke W, Wu S, Kuppe H, Pries AR, Kuebler WM. Negative-feedback loop attenuates hydrostatic lung edema via a cGMP-dependent regulation of transient receptor potential vanilloid 4. Circ Res. 2008;102:966–974. doi: 10.1161/CIRCRESAHA.107.168724. [DOI] [PubMed] [Google Scholar]
- Yuen PS, Potter LR, Garbers DL. A new form of guanylyl cyclase is preferentially expressed in rat kidney. Biochemistry. 1990;29:10872–10878. doi: 10.1021/bi00501a002. [DOI] [PubMed] [Google Scholar]
- Zabel U, Kleinschnitz C, Oh P, Nedvetsky P, Smolenski A, Muller H, Kronich P, Kugler P, Walter U, Schnitzer JE, Schmidt HH. Calcium-dependent membrane association sensitizes soluble guanylyl cyclase to nitric oxide. Nat Cell Biol. 2002;4:307–311. doi: 10.1038/ncb775. [DOI] [PubMed] [Google Scholar]
- Zanfolin M, Faro R, Araujo EG, Guaraldo AM, Antunes E, de Nucci G. Protective effects of BAY 41-2272 (sGC stimulator) on hypertension, heart, and cardiomyocyte hypertrophy induced by chronic L-NAME treatment in rats. J Cardiovasc Pharmacol. 2006;47:391–395. [PubMed] [Google Scholar]
- Zaragoza C, Soria E, Lopez E, Browning D, Balbin M, Lopez-Otin C, Lamas S. Activation of the mitogen activated protein kinase extracellular signal-regulated kinase 1 and 2 by the nitric oxide-cGMP-cGMP-dependent protein kinase axis regulates the expression of matrix metalloproteinase 13 in vascular endothelial cells. Mol Pharmacol. 2002;62:927–935. doi: 10.1124/mol.62.4.927. [DOI] [PubMed] [Google Scholar]
- Zhang M, Koitabashi N, Nagayama T, Rambaran R, Feng N, Takimoto E, Koenke T, O'Rourke B, Champion HC, Crow MT, Kass DA. Expression, activity, and pro-hypertrophic effects of PDE5A in cardiac myocytes. Cell Signal. 2008 doi: 10.1016/j.cellsig.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Moalem J, Tse J, Scholz PM, Weiss HR. Effects of natriuretic peptides on ventricular myocyte contraction and role of cyclic GMP signaling. Eur J Pharmacol. 2005;510:209–215. doi: 10.1016/j.ejphar.2005.01.031. [DOI] [PubMed] [Google Scholar]
- Zhang R, Wang L, Zhang L, Chen J, Zhu Z, Zhang Z, Chopp M. Nitric oxide enhances angiogenesis via the synthesis of vascular endothelial growth factor and cGMP after stroke in the rat. Circ Res. 2003;92:308–313. doi: 10.1161/01.res.0000056757.93432.8c. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Zhang MH, Sears CE, Emanuel K, Redwood C, El Armouche A, Kranias EG, Casadei B. Reduced phospholamban phosphorylation is associated with impaired relaxation in left ventricular myocytes from neuronal NO synthase-deficient mice. Circ Res. 2008;102:242–249. doi: 10.1161/CIRCRESAHA.107.164798. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Brandish PE, Ballou DP, Marletta MA. A molecular basis for nitric oxide sensing by soluble guanylate cyclase. Proc Natl Acad Sci U S A. 1999;96:14753–14758. doi: 10.1073/pnas.96.26.14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XB, Ruth P, Schlossmann J, Hofmann F, Korth M. Protein phosphatase 2A is essential for the activation of Ca2+-activated K+ currents by cGMP-dependent protein kinase in tracheal smooth muscle and Chinese hamster ovary cells. J Biol Chem. 1996;271:19760–19767. doi: 10.1074/jbc.271.33.19760. [DOI] [PubMed] [Google Scholar]
- Zhu W, Zou Y, Shiojima I, Kudoh S, Aikawa R, Hayashi D, Mizukami M, Toko H, Shibasaki F, Yazaki Y, Nagai R, Komuro I. Ca2+/calmodulin-dependent kinase II and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. J Biol Chem. 2000;275:15239–15245. doi: 10.1074/jbc.275.20.15239. [DOI] [PubMed] [Google Scholar]