

Abstract
Suppression of oncogenic Wnt-mediated signaling holds promise as an anti-cancer therapeutic strategy. We previously reported a novel class of small molecules (IWR-1/2, Inhibitors of Wnt Response) that antagonize Wnt signaling by stabilizing the Axin destruction complex. Herein, we present the results of structure/activity relationship studies of these compounds.
The Wnt signal transduction pathways play a key role in embryogenesis and tissue homeostasis,1 and are frequently mis-activated in cancer.2 Small molecules that antagonize the Wnt signaling pathways therefore hold promise as anti-cancer therapeutics.3,4 We recently reported two small molecules, IWR-1 (1) and IWR-2 (2) (Figure 1) that suppressed Wnt signaling by stabilizing the Axin destruction complex, a negative regulator of canonical Wnt signaling.5 To understand the structure/activity relationship of 1 and 2, we synthesized a series of analogs and compared their ability to suppress Wnt signaling in luciferase-based reporter assays as previously described.5,6 The results of these studies are presented in this letter.
Figure 1.

The structures of IWR-1 (1) and IWR-2 (2).
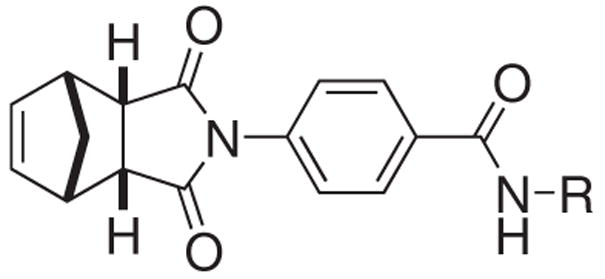
To facilitate our structure/activity relationship analyses, we considered 1 and 2 to be composed of three structural subunits, the norbornyl, spacer and amide regions (Figure 1). The equal potency of 1 and 2 suggested that their amide region may accommodate structural modifications without suffering a significant loss in activity. We therefore focused our efforts on modifying the amide substitution groups (Table 1). Indeed, we found that introduction of a methyl group to the 7-position of the quinoline (3) only slightly decreased activity. However, partial saturation of the quinoline group (4) resulted in considerable reduction of activity. The limited access to 8-aminoquinoline derivatives prompted us to examine the potential of substituting the quinoline with other aromatic groups.
Table 1.
SAR studies on the IWR amide region.
 | ||
|---|---|---|
| Compounds | R | EC50(μM) |
| 3 | 7-methylquinolin-8-yl | 0.8 |
| 4 | 5,6,7,8-tetrahydroquinolin-8-yl | 10 |
| 5 | 2-methoxyphenyl | 1 |
| 6 | phenyl | >25 |
| 7 | 2-fluorophenyl | >25 |
| 8 | 2-chlorophenyl | >25 |
| 9 | 2-bromophenyl | >25 |
| 10 | 3-fluorophenyl | >25 |
| 11 | 3-chlorophenyl | >25 |
| 12 | 3-bromophenyl | 9 |
| 13 | 4-fluorophenyl | 4 |
| 14 | 4-chlorophenyl | 3 |
| 15 | 4-bromophenyl | 1 |
| 16 | 2,3-difluorophenyl | >25 |
| 17 | 2,4-difluorophenyl | 9 |
| 18 | 2,4-dichlorophenyl | 10 |
| 19 | 2,4-dibromophenyl | >25 |
| 20 | 2,5-difluorophenyl | >25 |
| 21 | 2,6-difluorophenyl | >25 |
| 22 | 3,4-difluorophenyl | 3 |
| 23 | 3,4-dichlorophenyl | 5 |
| 24 | 3,4-dibromophenyl | 10 |
| 25 | 3,5-difluorophenyl | >25 |
| 26 | 2-(trifluoromethyl)phenyl | >25 |
| 27 | 3-(trifluoromethyl)phenyl | 20 |
| 28 | 4-(trifluoromethyl)phenyl | >25 |
| 29 | benzyl | 20 |
| 30 | (2-pyridyl)methyl | >25 |
| 31 | (3-pyridyl)methyl | >25 |
| 32 | (4-pyridyl)methyl | 10 |
| 33 | trans-(2-methoxy)cyclohexyl | 2 |
| 34 | trans-(2-hydroxy)cyclohexyl | >25 |
| 35 | cyclohexyl | >25 |
We first sought to simplify the structure of the aromatic amide. We recognized that the anisolyl analog 5 was a less potent hit of the initial screen5 that yielded the discovery of 1 and 2. Interestingly, its parent phenyl analog 6 was inactive at even 50 μM, suggesting that fine-tuning the steric and electronic properties of 6 to be a feasible approach to discover new Wnt inhibitors. We therefore set out to evaluate the activity of a series of halogenated analogs (Table 1).
Significantly, introduction of halogen atoms at the 4-position of the phenyl group (13–15) was found to be effective, with the 4-bromophenyl derivative 15 being the most active compound. Other halogen derivatives (7–11) were found nearly inactive with the exception of modest activity for the 3-bromophenyl derivative 12. We next examined the effects of dihalogen substitutions. These compounds (16–25) were generally less potent than the mono-halogenated derivatives 7–15. Only the 3,4-dihalophenyl derivatives (22–24) showed moderate activity. The trifluoromethylphenyl derivatives 26–28 were also found to be ineffective Wnt inhibitors.
We have also examined the effects of alkyl amides. The benzyl amide 29 was a weak inhibitor and incorporation of a nitrogen atom to the 4-position of the phenyl group (32) slightly improved the activity. Other pyridyl derivatives (30 and 31) were found to be inactive. Saturation of the phenyl group of 5 led to a 2-fold decrease in potency (33). Further truncation of the structure (34 and 35) abolished activity.
To explore the structural requirement of the spacer region of IWR’s, we replaced the central aromatic ring of 1 with five different spacers (Table 2, a–e). Introduction of a substituent group (chloro methyl or methoxyl) at the 2-position of the center aromatic ring significantly twisted the conformation and abolished the activity (36–38). On the other hand, the 3-position of the space tolerates certain modification (39–41). The chloro and methyl derivatives (39 and 40) are moderately active. We also found that extension of the spacer by one nitrogen atom (R=b) to form an aromatic urea linker (42) rendered the compound inactive. Interestingly, saturation of this spacer (R=c) resulted in a slight loss of activity (43–45). Incorporation of a nitrogen atom to the saturated spacer (R=d) to form a urea linker (46) also reduced the activity significantly. Consistent with the observation with 42 (R=b), extension of the saturated spacer by one carbon atom (R=e) led to drastic decrease in activity (47–49). We further found that introduction of an N-alkyl group to the amide abolished the activity. N-Me-IWR (50) was not active even at 50 μM (Figure 2). These results underlined the importance of the amide functional group and the conformation of IWR.
Table 2.
SAR studies on the IWR spacer region.
 | ||||
|---|---|---|---|---|
| Compounds | R | R′ | Ar | EC50 (μM) |
| 36 | a | 2-chloro | quinolin-8-y | >25 |
| 37 | a | 2-methyl | quinolin-8-y | >25 |
| 38 | a | 2-methoxy | quinolin-8-y | >25 |
| 39 | a | 3-chloro | quinolin-8-y | 1 |
| 40 | a | 3-methyl | quinolin-8-y | 2 |
| 41 | a | 3-methoxy | quinolin-8-y | >25 |
| 42 | b | – | quinolin-8-yl | >25 |
| 43 | c | – | quinolin-8-yl | 0.4 |
| 44 | c | – | 4-methylquinolin-8-yl | 1 |
| 45 | c | – | 7-methylquinolin-8-yl | 1 |
| 46 | d | – | quinolin-8-yl | 10 |
| 47 | e | – | quinolin-8-yl | >25 |
| 48 | e | – | 4-methylquinolin-8-yl | >25 |
| 49 | e | – | 7-methylquinolin-8-yl | 10 |
Figure 2.

The structural deviation of 50–52 from 1 and their dose-response curves.
We previously reported that exo-IWR 51 was only active at high concentration.5 Quantitatively, 51 was 25-fold less active than endo-IWR 1 (Figure 2). Interestingly, saturation of the olefin did not affect the activity. Sat-IWR 52 and 1 were equally potent in the in vitro assays (Figure 2). These results indicated that the norbornyl region of 1 could only tolerate subtle steric perturbation.
We have also tested the in vivo activity of IWR’s and found that 1 effectively inhibited zebrafish tail fin regeneration.5 We show herein that the minimum inhibitory concentration of 1 is 0.5 μM (Figure 3). We further demonstrated that the in vivo activity of IWR’s correlated with their in vitro activity. For example, only partial inhibition of fin regeneration was observed with moderate inhibitors 13 and 43. The weak inhibitor 17 only retarded the growth of the tail fin (picture not shown).
Figure 3.

Inhibition of fin regeneration in zebrafish by IWR’s. Arrows indicate the points of resection.
Preliminary metabolic stability studies7 indicated enzymatic cleavage of the amide bond to be the primary degradation pathway of 1 (Figure 4). Loss of the parent compound 1 and subsequent appearance of acid 53 and 8-aminoquinoline (54) were noted when 1 was incubated in murine whole blood. The half-life in blood was 60 min. A similar pattern of cleavage was noted upon incubation of the 1 with murine liver S9 fractions (t½ = 20 min for Phase I); however, a decade of 54 was also noted, suggestive of a secondary metabolism pathway. The half-life of 1 in intact murine hepatocytes was 20 min and those of the less potent inhibitors 5 and 13 were 10 and 60 min respectively.
Figure 4.

The murine whole blood metabolism experiments of 1.
In summary, we have conducted focused structure/activity relationship studies on IWR-1/2 (1 and 2). The norbornyl, spacer and amide regions can all be modified with compromised activity. The in vivo activity of the IWR’s correlated with their in vitro activity. Further investigation will be centered on altering the steric and electronic properties of the norbornyl and spacer regions and modify the amide linkage to improve the activity and pharmacokinetic properties.
Acknowledgments
We thank UT Southwestern Medical Center, NIH (NCI P01-CA095471, NIGMS R01-GM079554, NIGMS R01-GM076398), American Cancer Society (RSG GMC-112251) and Welch Foundation (I-1665) for financial support. L.L. is a Virginia Murchison Linthicum Scholar in Medical Research and C.C. is a Southwestern Medical Foundation Scholar in Biomedical Research. We thank Michael Dodge for critical reading of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Clevers H. Cell. 2006;127:469. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 2.(a) Reya T, Clevers H. Nature. 2005;434:843. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]; (b) Polakis P. Curr Opin Genet Dev. 2007;17:45. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 3.Barker N, Clevers H. Nat Rev Drug Discovery. 2006;5:997. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 4.(a) Lepourcelet M, Chen YNP, France DS, Wang H, Crews P, Petersen F, Bruseo C, Wood AW, Shivdasani RA. Cancer Cell. 2004;5:91. doi: 10.1016/s1535-6108(03)00334-9. [DOI] [PubMed] [Google Scholar]; (b) Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Oh SW, Kim HY, Moon SH, Ha JR, Kahn M. Proc Natl Acad Sci. 2004;101:12682. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu J, Wu X, Mitchell B, Kintner C, Ding S, Schultz PG. Angew Chem Int Ed. 2005;44:1987. doi: 10.1002/anie.200462552. [DOI] [PubMed] [Google Scholar]; (d) Lu D, Cottam HB, Corr M, Carson DA. Proc Natl Acad Sci. 2005;102:18567. doi: 10.1073/pnas.0509316102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Park CH, Hahm ER, Lee JH, Jung KC, Yang CH. Biochem Biophys Res Commun. 2005;331:1222. doi: 10.1016/j.bbrc.2005.03.242. [DOI] [PubMed] [Google Scholar]; (f) Jin G, Lu D, Yao S, Wu CCN, Liu JX, Carson DA, Cottam HB. Bioorg Med Chem Lett. 2009;19:606. doi: 10.1016/j.bmcl.2008.12.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, Wei S, Hao W, Kilgore J, Williams NS, Roth MG, Amatruda JF, Chen C, Lum L. Nat Chem Biol. 2009;5:100. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wnt/β-catenin pathway response was measured in L-cells expressing Wnt3A and harboring control and Wnt-responsive reporters.
- 7.Metabolic stability studies were conducted in murine whole blood by incubating 2 μM of 1 in 150 μL of blood collected from C57BL/6 mice. Stability in the presence of S9 fractions (Celsis/InVitro Technologies) was conducted with 2 μM of 1, 1 mg of S9 protein, and an NADPH-regenerating system (1.7 mg/mL NADP, 7.8 mg/mL glucose-6-phosphate, 6 U/mL glucose-6-phosphate dehydrogenase in 2% w/v NaHCO3/10 mM MgCl2) also containing uridine 5′-diphospho-α-D-glucuronic acid (1.9 mg/mL) and 3′-phosphoadenosine-5′-phosphosulphate (100 mg/mL) for both Phase I and Phase II metabolic reactions. Hepatocyte metabolism was conducted by incubating 2 μM of 1 in the presence of 105 cryopreserved murine CD1 hepatocytes (Celsis/InVitro Technologies). Incubation was terminated at the indicated time points by adding 300 μL of methanol. Reactions conducted in saline or buffer served as controls and no degradation was observed. Parent compounds and fragments were detected by LC/MS/MS.f