

Abstract
In order to directly detect nucleic acid polymers, we have designed biosensors comprising sequence-specific DNA binding proteins tethered to split-reporter proteins, which generate signal upon binding a predetermined nucleic acid target, in an approach termed SEquence-Enabled Reassembly (SEER). Herein we demonstrate that spectroscopically distinct split-fluorescent protein variants, GFPuv, EGFP, Venus, and mCherry, function effectively in the SEER system, providing sensitive DNA detection and the ability to simultaneously detect two target oligonucleotides. Additionally, a methylation-specific SEER-Venus system was generated, which was found to clearly distinguish between methylated versus non-methylated target DNA. These results will aid in refinement of the SEER system for the detection of user defined nucleic acid sequences and their chemical modifications as they relate to human disease.
Specific DNA sequences are typically detected by denaturation of double stranded (ds)DNA followed by hybridization with a labeled oligonucleotide probe, as is the case with Southern blotting, fluorescence in situ hybridization (FISH),1 and DNA microarrays.2 In contrast to these approaches, new methods have been developed to directly detect dsDNA, which include designed systems that rely on sequence-specific recognition in the grooves of the native DNA double helix, including hairpin polyamides3 and triplex forming oligonucleotides (TFOs).4 Currently the utility of these techniques is constrained by limitations on the length of dsDNA that can be targeted and restrictions in the constitution of detectable sequences, respectively.5 In comparison to these design efforts, endogenous sequence-specific DNA binding proteins do not suffer from these drawbacks, while additionally providing a method for nucleic acid detection in its native environment. In our work we have focused on Cys2-His2 zinc fingers (ZFs), which comprise an important class of naturally occurring transcription factors and provide a programmable means of DNA detection.6, 7 In our method derived from split-protein assays also called protein-fragment complementation, fragments of a split-reporter protein are appended to DNA detection domains, and the binding event is monitored by signal generation arising from conditional reassembly of split-protein halves. Numerous proteins and enzymes have been genetically fragmented including ubiquitin,8 dihydrofolate reductase,9 green fluorescent protein (GFP),10 β-lactamase,11 and luciferase.12 GFP is particularly appealing as it singularly lacks any requirement for cofactors or substrates.13 By fusing split-GFP to sequence-specific ZF domains, recognition of target DNA induces GFP reassembly in a method called SEquence-Enabled Reassembly (SEER) (Figure 1).14
Figure 1.
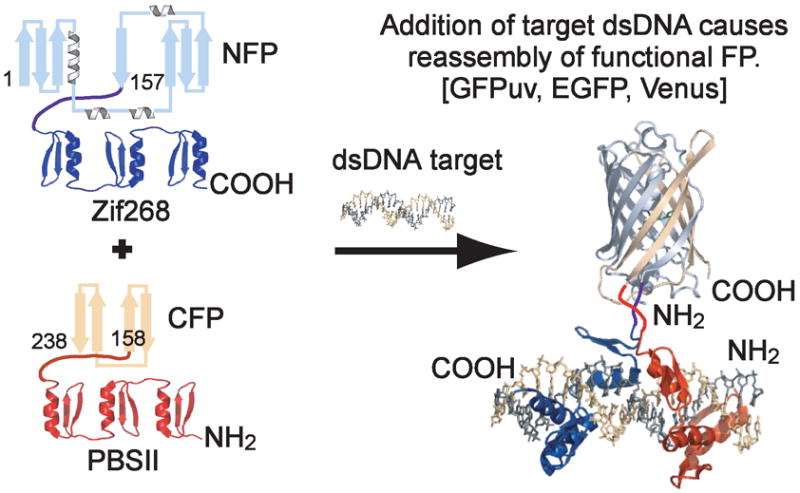
SEquence-Enabled Reassembly. The FP fusion constructs are initially non-fluorescent. The introduction of target DNA results in ZF binding, which induces reassembly of a productively fluorescent FP.
Currently, our reported SEER systems can specifically detect the presence of a single oligonucleotide sequence at a time.14–16 New SEER systems capable of simultaneously detecting multiple targets would be useful for numerous applications, including DNA profiling and ratiometric analysis. The GFP reporter affords the opportunity to accomplish this objective since GFP variants with distinct spectroscopic properties have been extensively studied.17 At present the repertoire of proteins derived from wild type Aequorea victoria GFP includes blue, cyan, and yellow fluorescent proteins (BFP, CFP, and YFP, respectively). Although a red-only emitting FP has not been achieved using the A. victoria scaffold,18 naturally occurring red fluorescent proteins (RFPs) have been identified. The so-called mFruits, monomeric DsRED derivatives, were recently selected and have since been employed as fusion tags in a variety of organisms, offering additional opportunities for protein complementation assays.19, 20 Thus, we constructed SEER systems incorporating five distinct fluorescent protein (FP) variants: a UV-excitable GFP, GFPuv;21 a CFP, Cerulean;22 an enhanced GFP, EGFP;23 a YFP, Venus;24 and a DsRED derived RFP, mCherry19 (Figure 2, left panels). Each N-terminal FP fragment (residues 1–157 for GFP-derived FPs and 1–168 for mCherry) was fused to ZF Zif268, while ZF PBSII was attached to each C-terminal FP fragment (residues 158–238 for GFP-derived FPs and 169–231 for mCherry). In all cases the split-FP domain was connected to the DNA binding domain via a flexible [(Gly)4-Ser]3 linker. Following isolation of these constructs, each SEER-FP system was assayed for preferential refolding in the presence of an optimized DNA target, Zif268-0-PBSII, by monitoring fluorescence emission of the reassembled complex. We were unable to generate a functional SEER-Cerulean system due to difficulties encountered during expression and purification of NCerulean-Zif268. However, all other tested split-FP constructs functioned effectively in the SEER context (Figure 2). Of note, split-mCherry was capable of DNA templated reassembly, thus providing the first success of a DsRED derived FP in the SEER context. Each SEER-FP system was optimized with respect to protein concentration, and a DNA titration (Figure 2, right panels) established sensitivities of the systems, which were qualitatively equivalent, and each reassembled FP produced signal over background at 10 nM (1.0 pmol) target DNA.
Figure 2.
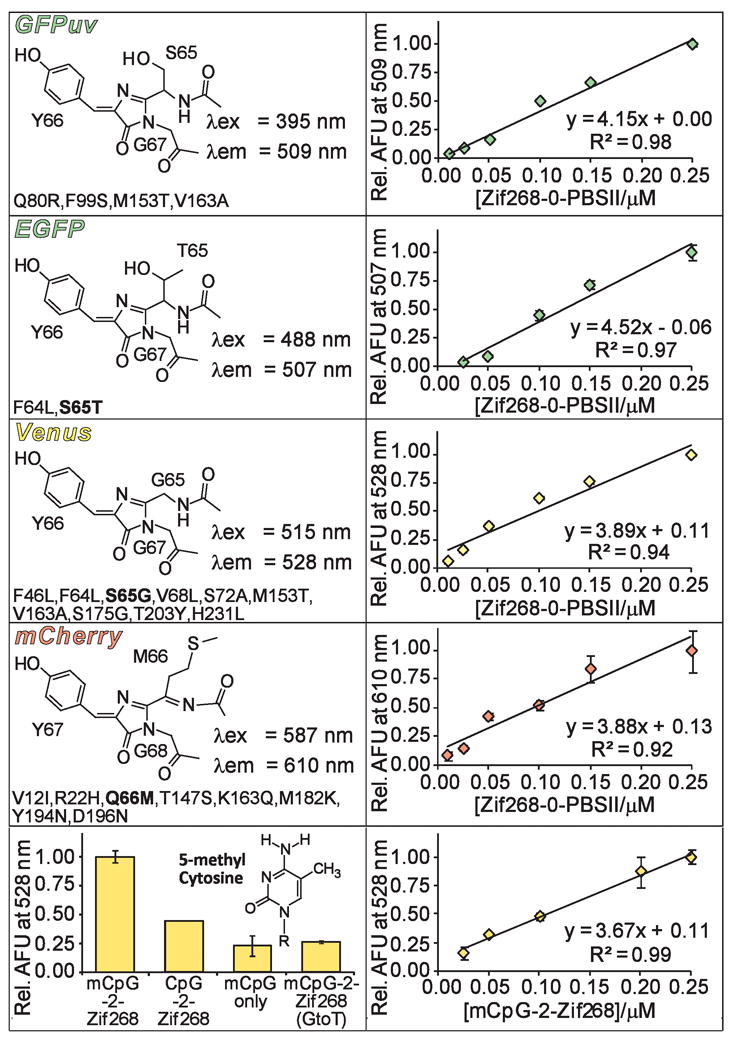
Properties of FPs utilized in SEER. The left panels display chromophore structures and wavelengths of maximum excitation (λex) and emission (λem) for each of the full length FP variants. Mutations (A. victoria numbering) are listed below the corresponding structures with chromophore mutations indicated in bold. mCherry includes additional GFP-type residues on both its N- and C-terminus.19 The right panels show a DNA titration for each SEER-FP system. The bottom panels show specificity data and a DNA titration for the mCpG-SEER system.
Due to the high fluorescence intensity of the SEER-Venus constructs, these proteins were selected to modify for detection of cytosine methylation, since this covalent DNA modification has emerged as a promising cancer biomarker.25 By simply replacing ZF PBSII with a methyl cytosine-guanine dinucleotide (mCpG) binding domain, MBD2, we were able to generate a system called mCpG-SEER-Venus for site-specific evaluation of the methylation status at individual CpG islands. We previously characterized two mCpG-SEER systems that conferred specificity for methylation at CpG sites, one of which employed an A. victoria FP variant.26, 27 To assess the specificity of the new system, we refolded NVenus-Zif268 and CVenus-MBD2 in the presence of target and off-target oligonucleotides (Figure 2, bottom panel). The mCpG-SEER-Venus system was shown to clearly distinguish between an mCpG site and its non-methylated equivalent with a 2.3-fold preference. This value seems slightly low since MBD2 has a 70-fold preference for binding methylated CpG islands (Kd = 2.7 nM) over the corresponding non-methylated site.28 An improvement in specificity for methylated sites may be achieved through further optimization of the mCpG-SEER-Venus system. Additional controls revealed a 4.3-fold reduction in fluorescence signal upon removal of the Zif268 binding site in the target (mCpG only) and a 3.8-fold reduction for a target with only a single guanine to thymine mutation in the Zif268 binding site (mCpG-2-Zif268 (G to T)). These results demonstrate the ability of the mCpG-SEER-Venus system to discriminate between cognate target sites and non-methylated, non-specific, or mutated DNA sequences. Finally, a DNA titration curve was generated for the mCpG-SEER-Venus system, resulting in detection of at least 25 nM DNA target (Figure 2, bottom panel).
The construction of several functional SEER-FP systems provides the potential for simultaneous detection of multiple DNA sequences, which we are currently pursuing with several new ZF DNA binding domains. An interesting alternative is the possibility of complementing different halves of the split-FPs to generate hybrid FPs with distinct spectral properties, which has previously been demonstrated for the detection of protein-protein interactions.29 We first attempted to visualize two different DNA sequences by complementing NVenus with both CVenus and CGFPuv (Figure 3A). The three fusion proteins were allowed to refold in the presence of one or two target oligonucleotides. Excitation and emission wavelengths were systematically scanned to identify major peaks. A fluorescence emission signal at 506 nm was attributed to the NVenus/CGFPuv hybrid and indicated the presence of the Zif268-0-PBSII target, while a 528 nm emission signal resulted from NVenus reassembling with CVenus in the presence of the Zif268-0-PE8B target (Figure 3B). When both targets were simultaneously present in solution, two distinct fluorescence emission maxima were observed by alternating the wavelength of excitation from 395 to 515 nm, corresponding to the formation of two distinct reassembled FPs. Only a minimal degree of off-target fluorescence was observed as emission at 506 nm in the absence of the Zif268-0-PBSII target (< 20%) and 528 nm emission in the absence of the Zif268-0-PE8B target (< 15%). Therefore, by using Zif268 to direct the constructs to a common target site, detection of two distinct downstream sequences was achieved. As a second example of mixed complementation, we reassembled NVenus and CCerulean (Figure 3C). We observed characteristic Venus fluorescence in the presence of the Zif268-0-PE8B target DNA and fluorescence attributable to the NVenus/CCerulean hybrid in the presence of Zif268-0-PBSII DNA (Figure 3D). A minimum of off-target fluorescence was observed using these constructs. These two examples of mixed complementation illustrate a straightforward method for the simultaneous detection of two distinct DNA sequences using a minimum of protein constructs. Thus far, we have generated a variety of SEER-FP constructs with distinct spectroscopic properties that are capable of sequence-specifically detecting dsDNA, reporting on chemical modifications to dsDNA, and simultaneously detecting two distinct DNA sequences.
Figure 3.

Mixed complementation of FPs. (A) A mixture of NVenus-Zif268, CGFPuv-PBSII, and CVenus-PE8B bind their respective targets. Wavelengths of maximum excitation (λex) and emission (λem) are indicated for each reassembled species. (B) NVenus-Zif268, CGFPuv-PBSII, and CVenus-PE8B were mixed in the presence of target DNA, and fluorescence was monitored at the indicated wavelengths. (C) SEER-Venus/Cerulean mixed complementation. The three constructs, NVenus-Zif268, CCerulean-PBSII, and CVenus-PE8B, bind their cognate sequences. Wavelengths of maximum excitation and emission are indicated next to the reassembled FPs. (D) NVenus-Zif268, CCerulean-PBSII, and CVenus-PE8B were mixed in the presence of target DNA, and fluorescence was monitored at the indicated wavelengths.
To demonstrate the specificity of our methodology, we have attempted the detection of cognate binding sites present in plasmid DNA using the SEER-Venus system. Specifically, we constructed a plasmid, pRSF Z0P10, which contains ten copies of the Zif268-0-PBSII target site, and attempted to visualize this plasmid in both a native (supercoiled) and linearized state, using the pRSF empty plasmid as background. We observed 3.1-fold signal over background for the linear pRSF Z0P10 plasmid, indicating that our system is not only capable of detecting discrete targets in solution, but also capable of detecting a target in the context of a nearly 23-fold excess of non-cognate DNA base pairs. A 2.1-fold signal over background was achieved when detecting Zif268-0-PBSII binding sites in supercoiled pRSF Z0P10. These results suggest a high level of specificity for a cognate target with little background binding to off-target DNA.
In conclusion, SEER offers a number of advantages over current approaches available for the direct detection of native dsDNA. These protein-based biosensors provide modularity, convenience of preparation, and most importantly, selectivity for targeting a user-defined nucleic acid sequence. We are currently focused on expanding the utility and broadening the applicability of our methodology by incorporating distinct signaling and detection domains. To this end, we have successfully incorporated four spectrally distinct split-FP variants, GFPuv, EGFP, Venus, and mCherry, each of which provide many opportunities for enhancing nucleic acid detection, including the ability to simultaneously detect two DNA sequences. Additionally, we determined that our SEER proteins are capable of detecting specific DNA sequences present in a plasmid DNA context, representing the high level of selectivity associated with the use of sequence-specific DNA binding proteins. These new advances in the SEER system open the door for expanding applications and further developments. For example, using appended ZFs to direct the system to a specific region of DNA, site-specific cytosine methylation has been achieved through reconstitution of a split methyltransferase.30 Additionally, real-time imaging of mitochondrial RNA localization was accomplished using an RNA targeted split-FP reassembly approach.31 Thus, we have shown that SEquence-Enabled Reassembly is an adaptable system, capable of performing a variety of functions, limited only by the availability of split-proteins and nucleic acid targeting domains and the imagination of the experimenter.
Supplementary Material
Acknowledgments
Funding was obtained from the National Institutes of Health (NIH): GM077403, CA122630. J.L.F. and C.I.S. were supported by a NIH Training Grant. We thank Jennifer Lippincott-Schwartz and Roberta A. Gottlieb for plasmids containing Cerulean and mCherry, respectively.
Footnotes
Supplementary data
Experimental details regarding cloning and protein purification, reassembly conditions, and fluorescence measurements can be found, in the online version, in the Supplementary material. Supplementary data associated with this article can be found, in the online version, at doi: xxxxx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Levsky JM, Singer RH. J Cell Sci. 2003;116:2833. doi: 10.1242/jcs.00633. [DOI] [PubMed] [Google Scholar]
- 2.Hoheisel JD. Nat Rev Genet. 2006;7:200. doi: 10.1038/nrg1809. [DOI] [PubMed] [Google Scholar]
- 3.Dervan PB. Bioorg Med Chem. 2001;9:2215. doi: 10.1016/s0968-0896(01)00262-0. [DOI] [PubMed] [Google Scholar]
- 4.Chan PP, Glazer PM. J Mol Med. 1997;75:267. doi: 10.1007/s001090050112. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh I, Stains CI, Ooi AT, Segal DJ. Mol BioSyst. 2006;11:551. doi: 10.1039/b611169f. [DOI] [PubMed] [Google Scholar]
- 6.Segal DJ, Barbas CF., III Curr Opin Biotechnol. 2001;12:632. doi: 10.1016/s0958-1669(01)00272-5. [DOI] [PubMed] [Google Scholar]
- 7.Blancafort P, Segal DJ, Barbas CF., III Mol Pharmacol. 2004;66:1361. doi: 10.1124/mol.104.002758. [DOI] [PubMed] [Google Scholar]
- 8.Johnsson N, varshavsky A. Proc Natl Acad Sci USA. 1994;94:8405. [Google Scholar]
- 9.Pelletier JN, Campbell-Valois FX, Michnick SW. Proc Natl Acad Sci USA. 1998;95:12141. doi: 10.1073/pnas.95.21.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh I, Hamilton AD, Regan L. J Am Chem Soc. 2000;122:5658. [Google Scholar]
- 11.Galarneau A, Primeau M, Trudeau LE, Michnick SW. Nat Biotechnol. 2002;20:619. doi: 10.1038/nbt0602-619. [DOI] [PubMed] [Google Scholar]
- 12.Paulmurugan R, Gambhir SS. Anal Chem (Wash) 2003;75:1584. doi: 10.1021/ac020731c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Science (Wash) 1994;263:802. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 14.Stains CI, Porter JR, Ooi AT, Segal DJ, Ghosh I. J Am Chem Soc. 2005;127:10782. doi: 10.1021/ja051969w. [DOI] [PubMed] [Google Scholar]
- 15.Ooi AT, Stains CI, Segal DJ, Ghosh I. Biochemistry (Wash) 2006;45:3620. doi: 10.1021/bi0517032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porter JR, Stains CI, Jester BW, Ghosh I. J Am Chem Soc. 2008;130:6488. doi: 10.1021/ja7114579. [DOI] [PubMed] [Google Scholar]
- 17.Tsien RY. Annu Rev Biochem. 1998;67:509. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 18.Mishin AS, Subach FV, Yampolsky IV, King W, Lukyanov KA, Verkhusha VV. Biochemistry. 2008;47:4666. doi: 10.1021/bi702130s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Nat Biotechnol. 2004;22:1567. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 20.Shaner NC, Steinbach PA, Tsien RY. Nat Methods. 2005;2:905. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 21.Crameri A, Whitehorn EA, Tate E, Stemmer WPC. Nat Biotechnol. 1996;14:315. doi: 10.1038/nbt0396-315. [DOI] [PubMed] [Google Scholar]
- 22.Rizzo MA, Springer GH, Granada B, Piston DW. Nat Biotechnol. 2004;22:445. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- 23.Yang TT, Cheng L, Kain SR. Nucleic Acids Res. 1996;24:4592. doi: 10.1093/nar/24.22.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. Nat Biotechnol. 2002;20:87. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 25.Esteller M, Corn PG, Baylin SB, Herman JG. Cancer Res. 2001;61:3225. [PubMed] [Google Scholar]
- 26.Stains CI, Furman JL, Segal DJ, Ghosh I. J Am Chem Soc. 2006;128:9761. doi: 10.1021/ja060681j. [DOI] [PubMed] [Google Scholar]
- 27.Porter JR, Stains CI, Segal DJ, Ghosh I. Anal Chem (Wash) 2007;79:6702. doi: 10.1021/ac071163+. [DOI] [PubMed] [Google Scholar]
- 28.Fraga MF, Ballestar E, Montoya G, Taysavang P, Wade PA, Esteller M. Nucleic Acids Res. 2003;31:1765. doi: 10.1093/nar/gkg249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu CD, Kerppola TK. Nat Biotechnol. 2003;21:539. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nomura WCFB., III J Am Chem Soc. 2007;129:8676. doi: 10.1021/ja0705588. [DOI] [PubMed] [Google Scholar]
- 31.Ozawa T, Natori Y, Sato M, Umezawa Y. Nat Methods. 2007;4:413. doi: 10.1038/nmeth1030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.