

Abstract
While studying T helper (Th) responses induced by cardiac transplantation, we observed that mice deficient in the Th1 transcription factor T-bet (T-bet-/-) mount both Th1 and Th17 responses, while wild type (WT) recipients mount only Th1 responses. Cells producing both IFNγ and IL-17 were readily detectable within the rejecting graft of T-bet-/- recipients, but were absent from the spleen indicating that the in vivo microenvironment influences Th function. In addition, disrupting CD40 – CD40 ligand (CD40L) costimulatory interactions was highly effective at prolonging allograft survival in WT mice, but ineffective in T-bet-/- recipients. Herein, we report that CD8+ Th17 mediate costimulation blockade resistant rejection in T-bet-/- allograft recipients. Depleting CD8+ cells or neutralizing IL-17 or the Th17-inducing cytokine IL-6 ablated the Th17 response and reversed costimulation blockade resistant graft rejection. Neutralizing IL-4 in IFNγ-/- allograft recipients did not induce Th17, suggesting that T-bet, rather than IL-4 and IFNγ (known inhibitors of Th17), plays a critical role in negatively regulating Th17 in the transplant setting.
Keywords: costimulation, cytokines, T cells, transplantation
Introduction
Multiple effector mechanisms of allograft rejection exist and the roles of distinct Th subsets have not been completely defined (reviewed in (1-3)). The well established Th1-Th2 paradigm (4) has been extensively investigated in the transplant setting, yet the roles of Th1 and Th2 cytokines in allograft rejection vs. acceptance remain controversial (1). In wild type (WT)3 mice, cardiac allograft rejection is characterized by a dominant Th1 response and mononuclear cell infiltration of the transplant (5-7). These IFNγ producing Th1 are predominantly CD8+ and require CD4+ help (7-9) and the CD40 – CD40L costimulatory pathway (7, 10) for their generation. The transcription factor T-bet is a positive regulator of the Th1 response and is expressed by CD4+ and CD8+ T cells, NK cells, dendritic cells, and B cells (reviewed in (11)). T-bet induces and supports IFNγ production (reviewed in (11)) by both remodeling the IFNγ gene (12) and inducing expression of the IL-12Rβ2 chain of the IL-12 receptor (13). While T-bet is critical for CD4+ T cell commitment to a Th1 lineage, CD8+ T cells may express the T-bet homologue eomesodermin (14) as an alternative Th1 transcription factor resulting in IFNγ production. Though IFNγ has been historically associated with allograft rejection, other studies suggest that this cytokine may be required for graft acceptance (7, 15). Hence, this study was initiated to explore the requirement for T-bet in the generation of graft-reactive Th1 and its role in allograft rejection.
IL-4 producing Th2 were initially believed to play a protective role in the transplant setting due to their ability to antagonize Th1 responses (reviewed in (1)). TCR and IL-4 receptor signaling upregulate the Th2 transcription factor GATA3, which drives remodeling of the Th2 cytokine cluster and Th2 cytokine gene expression (16). In addition, GATA3 inhibits Th1 differentiation by suppressing STAT4 and IL-12Rβ2 expression (17). Despite this Th1 antagonizing activity, Th2 responses have been associated with an alternate form of rejection which is characterized by eosinophil and neutrophil infiltration of the graft (5, 18). Hence, the role of Th2 in transplant rejection vs. acceptance remains controversial.
The role of Th17 in the transplant setting has not been explored in depth and the few reports have been associative in nature (reviewed in (19)). However, IL-17 producing Th17 have been implicated in a number of chronic inflammatory diseases (20, 21) including autoimmune myocarditis (22), allergic asthma (19), autoimmune arthritis (19), and mycobacterial infection (23). Th17 induced pathologies are characterized by granulocytic infiltration of the tissues (21). The transcription factor RORγT and the cytokines TGFβ and IL-6 induce IL-17 secretion, while IL-23 perpetuates the Th17 response (21). Negative regulators of Th17 include IFNγ (20, 21), IL-4 (20, 21), IL-2 (24), and interestingly T-bet (25).
In many transplant models disrupting CD40 – CD40L costimulatory interactions is a highly effective means of preventing graft-reactive T cell priming and promoting allograft acceptance (reviewed in (26)). However, this form of costimulatory blockade is less effective under certain conditions (reviewed in (27, 28)). Costimulatory blockade resistant rejection has been attributed to redundancies in costimulatory molecules (29), a high frequency of graft-reactive T cells (30), heterologous memory cells (28), IL-2 (31), and the absence of IFNγ (7, 15). Herein, we describe a novel form of costimulatory blockade resistant rejection which is mediated by CD8+ Th17 in T-bet-/- allograft recipients.
Materials and Methods
Culture medium
RPMI supplemented with 2% FCS, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 1.6 mM L-glutamine, 10 mM HEPES buffer (all from Invitrogen, Grand Island, NY), 0.27 mM L-asparagine, 1.4 mM L-arginine HCl, 14 μM folic acid and 50 μM 2-mercaptoethanol (all from Sigma Chemicals, St. Louis, MO).
Mice
Female C57BL/6 WT and IFNγ-/- (H-2b) mice, and BALB/c (H-2d) mice were purchased from The Jackson Laboratories (Bar Harbor, ME). Breeder pairs of T-bet-/- mice (32) on the C57BL/6 background were provided by Dr. Kevin T. McDonagh (University of Michigan) with permission from Dr. Laurie H. Glimcher (Harvard School of Public Health). Colonies of T-bet-/- mice were established, and all mice were housed under specific pathogen-fee conditions in the Unit for Laboratory Animal Medicine at the University of Michigan. Mice used were between 6 and 12 weeks of age. These experiments were approved by the University Committee on Use and Care of Animals (UCUCA) at the University of Michigan.
Heterotopic cardiac transplantation
C57BL/6 WT, T-bet-/-, or IFNγ-/- mice were transplanted with intact BALB/c cardiac allografts, as described (33). In this model, the donor heart is anastomosed to the great vessels of the abdomen, perfused with the recipient mouse’s blood, and resumes contraction. Transplant function was monitored by abdominal palpation.
In vivo antibody treatment
Hybridomas secreting anti-CD4 (clone GK1.5), anti-CD8 (clone 2.43), anti-IL-4 (clone 11.B11), and anti-IL-6 (clone MPR-20F30) were obtained from American Type Culture Collection (Manassas, VA). The hybridoma secreting anti-CD40L (clone MR1) was provided by Dr. Randy Noelle (Dartmouth, Lebanon, NH). Anti-CD4, anti-CD8, anti-IL-4, anti-IL-6, and anti-CD40L mAb were purified and resuspended in PBS by BioExpress (West Lebanon, NH). Neutralizing anti-IL-17 mAb (clone 50104) was obtained from R&D Systems (Minneapolis, MN). Mice received 1 mg of anti-CD40L i.p. days 0, 1 and 2 (7, 10). To neutralize IL-4 or IL-6, recipients received 1 mg i.p. of neutralizing mAb days 0, 1, 3, and 5. To neutralize IL-17, recipients received 0.2 mg i.p. anti-IL-17 mAb days 1, 4, 6 and 8 (34). To deplete T cell subsets, recipients received 1 mg i.p. anti-CD4 or anti-CD8 mAb on days -1, 0 and 7. Where indicated, cardiac allograft recipients were given an additional 1 mg of anti-CD8 mAb weekly to prevent CD8+ T cell repopulation. All doses are relative to day of transplant. Depletion was verified by flow cytometry at the time of organ harvest.
Histology
Allografts were recovered at the times indicated post-transplantation, fixed in formalin, and embedded in paraffin. Sections were stained with hematoxylin-eosin (H & E) to assess myocyte viability (presence of cross striation and myocyte nuclei), and the nature and intensity of graft infiltrating cells.
Recovery of graft-infiltrating cells (GIC)
Groups of 3 transplanted hearts were removed, pooled, minced, and digested with 1 mg/ml collagenase A (Roche, Indianapolis, IN) for 30 min at 37°C. Tissue debris were allowed to settle at 1 × g, and the suspension containing GIC was harvested by pipette. RBC were lysed by hypotonic shock, GIC were passed through a 30-μm pore size nylon mesh, and viable leukocytes were enumerated by Trypan blue exclusion. For differential enumeration, GIC were placed on slides with a cytocentrifuge and stained with Wright stain.
ELISPOT assay for cytokine-producing cells
ELISPOT assays were performed as previously described (35). Capture and detection antibodies specific for IFNγ (RA-6A2, XMG1.2), IL-4 (BVD4-1D11, BVD6-24G2) and IL-17 (TC11-18H10, TC11-BH4.1) were purchased from Pharmingen (San Diego, CA). PVDF-backed microtiter plates (Millipore, Bedford, MA) were coated with unlabeled mAb and blocked with 1% BSA in PBS. Irradiated (1,000 rad) donor splenocytes (4×105) and 1×106 recipient splenocytes were added to each well. After washing, biotinylated detection mAb were added to the plates. After washing, a 1:1,000 dilution of anti-biotin alkaline phosphatase (AP) conjugate (Vector Laboratories, Burlingame, CA) was added to IFNγ and IL-17 plates, and a 1:2,000 dilution of horseradish peroxidase-conjugated streptavidin (SA-HRP; Dako, Carpinteria, CA) was added to IL-4 plates. Plates were washed and spots visualized by addition of nitroblue tetrazolium (NBT; Biorad, Hercules, CA)/3 Bromo 4 Chloro Inolyl Phosphate (BCIP; Sigma) to IFNγ and IL-17 plates, or 3-amino-9-ethylcarbazole (AEC; Pierce, Rockford, IL) to IL-4 plates. Color development continued until spots were visible and stopped adding H2O. Plates were dried and spots quantified with an Immunospot Series 1 ELISPOT analyzer (Cellular Technology Ltd., Cleveland, OH).
Competitive IL-4 ELISA assay
The competitive IL-4 ELISA assay was performed as previously described (7) to demonstrate adequate levels of neutralizing anti-IL4 mAb in anti-IL-4 treated mice. Briefly, rIL-4 samples (2.5 ng/ml and 5 ng/ml) were incubated with PBS, dilutions of experimental sera, or purified anti-IL-4 (clone 11.B11, 1, 0.1, or 0.01 μg/ml) for 30 minutes at room temperature. Samples were then assessed for serologically detectable IL-4 by ELISA according to manufacture’s suggestion (Pharmingen). Absorbance was determined at 405 nm using an EL 800 microplate reader (Bio-Tek Instruments, Winooski, VT).
In vitro depletion of T cell subsets
CD4+ or CD8+ T cells were depleted using Dynal Beads (Invitrogen, Carlsbad, CA) as per manufacturer’s directions. Single cell suspensions of splenocytes were incubated with anti-CD4 or anti-CD8 coated beads (5 beads to 1 target cell) for 30 min. Bead-bound cells were removed magnetically and unbound cells were confirmed to be either CD4+ or CD8+ T cell depleted by flow cytometry (approx. 1% contamination; FACSCalibur, Becton-Dickinson, Franklin Lakes, NJ). Cells were then suspended at 5×106/ml in RPMI for use in ELISPOT assays.
Intracellular cytokine staining
Splenocytes and GIC from T-bet-/- allograft recipients were enriched for CD8+ cells by depleting splenocytes of B220+ and CD4+ cells and depleting GIC of Gr-1+ cells with Dynal Beads. Resulting splenocyte and GIC populations were approximately 85% and 50% CD8+, respectively. Enriched populations were stimulated with 5 ng/ml Phorbol 12-myristate 13-acetate (PMA) and 500 ng/ml ionomyocin (Sigma-Aldrich) and cultured with brefeldin A (BD Biosciences). Cells were then washed, permeabilized, and intracellular cytokine staining was performed using the BD Cytofix/Cytoperm Fixation/Permeabilization Kit (with BD GolgiPlug protein transport inhibitor containing brefeldin A) as per manufacture’s instructions (BD Biosciences). Cells were dual stained with PE-conjugated anti-IL-17 and FITC-conjugated anti-IFNγ. Cell analysis was performed on a Becton Dickinson FACSCalibur.
RNA isolation and RT-PCR
Cardiac allografts were homogenized in 1 ml TRIzol® (Invitrogen Life Technologies, Carlsbad, CA) and RNA was isolated as per manufacturer’s protocol. Five μg of total RNA were reverse transcribed using 10× PCR buffer (Roche), 10 mM dNTPs, Oligo (dT), M-MLV-RT (all from Invitrogen), and RNAsin (Promega). Products were then cleaned with 1:1 phenol/chloroform/isoamyl (25:24:1) and re-precipitated with 7.5 M NH4OAC in pure EtOH overnight at -80°C.
Real-time PCR was performed on cDNA using a Rotor-Gene 3000 ™ (Corbett Life Science, San Francisco, CA). Primer binding to DNA was detected by SYBR Green I™ dye (Roche, Indianapolis, IN). Relative expression of the gene of interest was expressed as the comparative concentration of the gene product compared to CycA product as calculated by accompanying Rotor-Gene software. Significance was determined with an unpaired t-test with Welch’s correction.
Primer sequences:
IL-17 sense: 5’ GGACTCTCCACCGCAATGA
anti-sense: 5’ GACCAGGATCTCTTGCTGGA;
T-bet sense: 5’ GCAGAGATCACTCAGCTGAAAA
anti-sense: 5’ GAGGGGACACTCGTATCAACA;
RORγT sense: 5’ TGCGACTGGAGGACCTTCTAC
anti-sense: 5’ CTCCCACATTGACTTCCTCTGG;
CycA sense: 5’ AGGGTGGTGACTTTACACGC
anti-sense: 5’ ATCCAGCCATTCAGTCTTGG.
Statistical analysis
Data were analyzed with GraphPad Prism 4.0c software using unpaired t-tests with Welch’s correction or log rank tests as appropriate. P values of ≤ 0.05 were considered statistically significant.
Results
Unconventional rejection in T-bet-/- allograft recipients
WT and T-bet-/- mice were transplanted with cardiac allografts to study the impact of T-bet on cytokine production and allograft rejection. Allografts were acutely rejected by day 10 post-transplant in both groups of recipients (Fig. 1A). However, the histopathology and composition of the graft infiltrating cells (GIC) were strikingly different (Fig. 1B & C). In WT recipients, rejection was characterized by a diffuse mononuclear cell infiltrate (Fig. 1B) that was composed primarily of lymphocytes and macrophages, with few granulocytes (Fig. 1C). In contrast, rejection in T-bet-/- recipients was associated with massive infiltration of the graft by neutrophils. Since granulocytic infiltration of the graft has been reported to be associated with costimulation blockade resistant rejection (7), we treated WT and T-bet-/- allograft recipients with the anti-CD40L mAb MR1 to disrupt the CD40 – CD40L costimulatory pathway. As previously reported (7, 10, 26, 36), disrupting CD40 – CD40L interactions was highly effective in preventing rejection (Fig. 1A) and graft infiltration (Fig. 1B) in WT recipients. However, anti-CD40L treatment was completely ineffective at preventing acute rejection and granulocytic infiltration of the graft in T-bet-/- mice.
FIGURE 1.
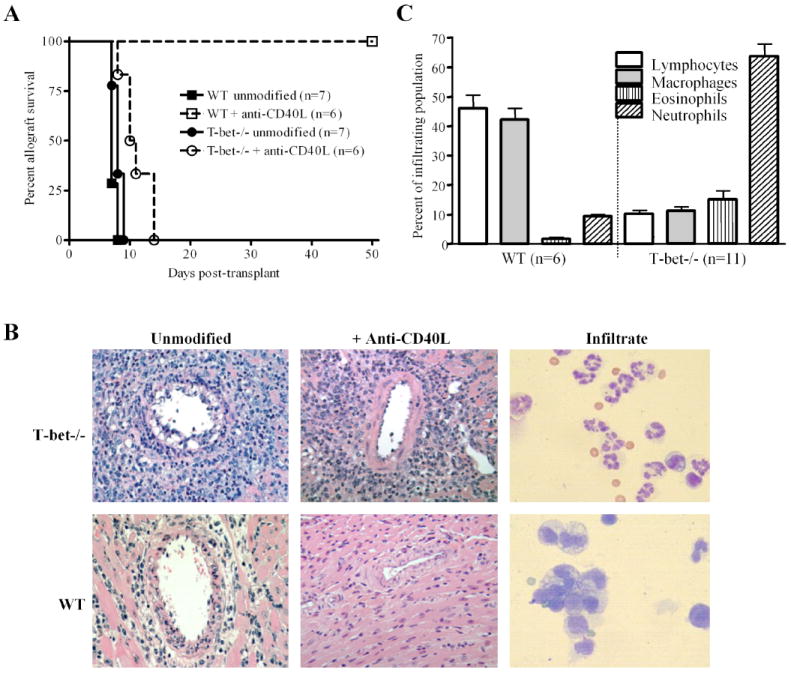
T-bet-/- allograft recipients mount unconventional costimulation blockade resistant rejection responses. A, C57BL/6 WT (squares) and T-bet-/- (circles) recipients of BALB/c cardiac allografts were either treated with anti-CD40L mAb (open symbols) or left untreated (closed symbols) and graft function was monitored. Numbers in parentheses represent the number of recipients in each group. B, Allograft histology in T-bet-/- and WT recipients ± anti-CD40L therapy (400X). Allografts were harvested on day 7 post-transplantation and sections were stained with H & E. Harvested GIC were Wright stained (right panel, 1000X). Note the aggressive neutrophil rich infiltrate which is not prevented by anti-CD40L mAb treatment of T-bet-/- recipients, and the diffuse mononuclear cell infiltrate which is prevented by anti-CD40L treatment of WT recipients. Tissue sections and GIC preparations are representative of at least 6 separate experiments. C, Differential cell counts of Wright stained GIC verified a dominance of lymphocytes (open bars) and macrophages (shaded bars) in WT recipients compared to a dominance of neutrophils (hatched bars) in T-bet-/- recipients. Bars represent averages + S.E.M. Numbers in parenthesis represent the number of differential counts.
Donor-reactive Th1, Th2, and Th17 responses in WT and T-bet-/- allograft recipients
Mononuclear cell infiltration of the graft has been associated with a dominant Th1 response in this transplant model (5-8). We and others have reported that granulocytic infiltration of allografts may be associated with the induction of Th2 responses (5, 7, 18). However, Th17 responses have also been implicated in pathologies that are characterized by neutrophil infiltration of tissues (21). Hence, we employed ELISPOT assays (35) to quantify in vivo primed, donor-reactive Th1 (IFNγ), Th2 (IL-4), and Th17 (IL-17) responses by splenocytes and GIC of WT and T-bet-/- mice (Fig. 2). Allograft recipients were either unmodified or were treated with anti-CD40L mAb. As expected, high numbers of donor-reactive Th1 (Fig. 2A, left panel) were present in the spleens and grafts of unmodified WT recipients. Interestingly, Th1 responses were also observed in unmodified T-bet-/- recipients, likely due to the presence of the alternate Th1 transcription factor eomesodermin in CD8+ cells (14). Indeed, depleting CD8+ cells, but not CD4+ cells, prior to addition to the ELISPOT cultures eliminated the Th1 response in T-bet-/- mice (Fig. 4A). Anti-CD40L mAb treatment of either WT or T-bet-/- allograft recipients eliminated the Th1 response (Fig. 2A, right panel).
FIGURE 2.
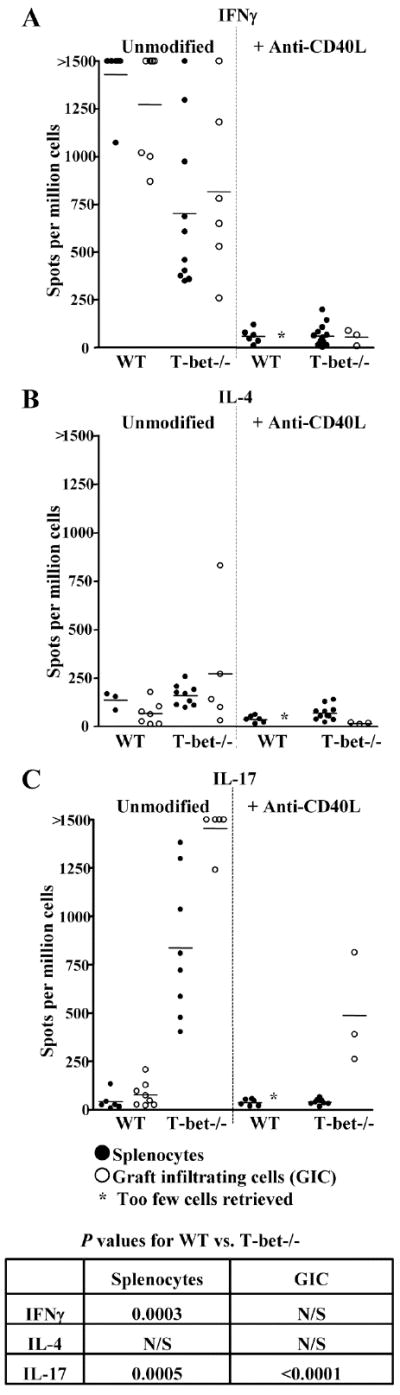
T-bet-/-, but not WT allograft recipients mount donor-reactive Th17 responses. WT and T-bet-/- allograft recipients were left either untreated or treated with anti-CD40L mAb. At the time of rejection, splenocytes (closed symbols) and GIC (open symbols) were harvested and processed for ELISPOT assays to quantify primed, donor-reactive IFNγ (A), IL-4 (B), or IL-17 (C) producing cells. Cells from WT recipients which were treated with anti-CD40L mAb and did not reject their allografts were harvested on day 7 post-transplant. Data points for splenocytes represent responses for individual allograft recipients. GIC from 3 individual mice were pooled to ensure enough cells for analysis. An insufficient number of GIC were obtained from WT recipients treated with anti-CD40L for ELISPOT analysis to be performed. Horizontal bars represent the mean responses for each experimental group. Insert table depicts p values for unmodified WT vs. unmodified T-bet-/- recipients. N/S = not significant.
FIGURE 4.
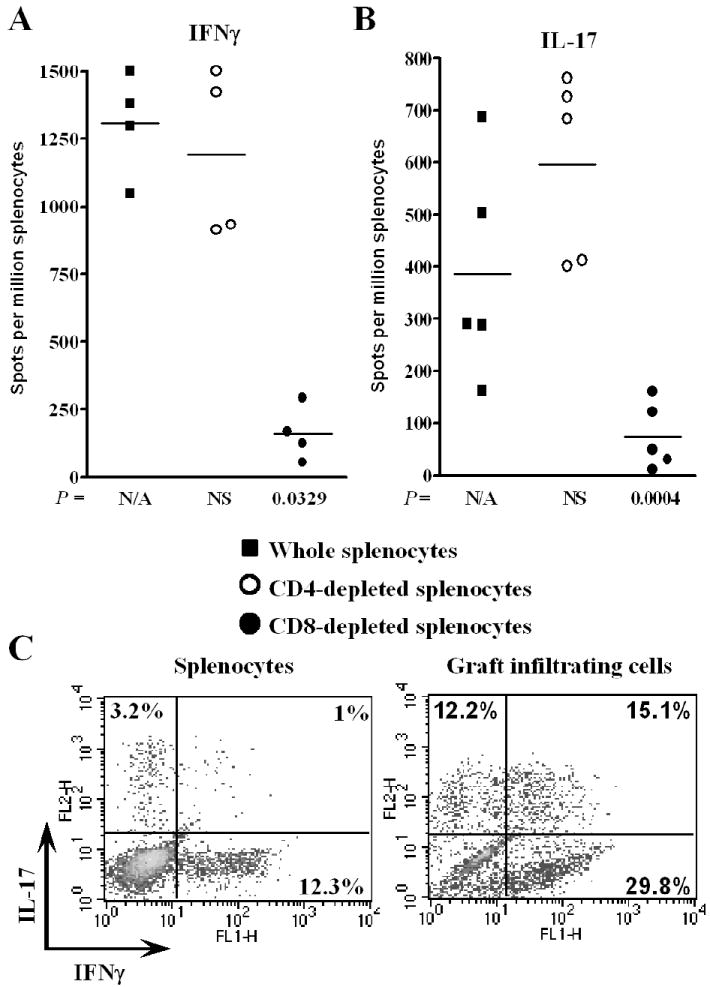
CD8+ T cells produce IFNγ and IL-17 in T-bet-/- allograft recipients. Splenocytes were harvested from T-bet-/- allograft recipients on day 7 post-transplant. Splenocytes were either left untreated (closed squares) or were depleted of CD4+ (open circles) or CD8+ (closed circles) cells prior to addition to ELISPOT assays to quantify primed, donor-reactive IFNγ (A) or IL-17 (B) producing cells. Responses of individual allograft recipients are depicted and bars represent the mean response of experimental groups. C, Splenocytes and GIC from T-bet-/- allograft recipients were enriched for CD8+ cells by depleting splenocytes of B220+ and CD4+ cells and depleting GIC of Gr-1+ cells. Enriched populations were stained for intracellular IFNγ and IL-17. Representative flow cytometric analyses of splenocytes (n = 7) and GIC (n = 3) are depicted. Note that GIC, but not splenocytes, contain an appreciable population of cells expressing both IFNγ and IL-17.
Donor-reactive Th2 responses (Fig. 2B) were unremarkable in both WT and T-bet-/- recipients, suggesting that the aggressive neutrophil infiltration of grafts in T-bet-/- recipients (Fig. 1B & C) was not due to the induction of Th2. However, the Th17 response (Fig. 2C) was markedly different in WT and T-bet-/- mice. While the Th17 response was negligible in unmodified WT recipients, strong Th17 responses were observed in both the spleens and allografts of unmodified T-bet-/- mice. Of particular note, anti-CD40L treatment of T-bet-/- recipients did not eliminate the Th17 response within the allograft. Hence, costimulation blockade resistant allograft rejection in T-bet-/- mice was associated with the presence of donor-reactive Th17 in the transplant.
IFNγ-/- allograft recipients do not mount Th17 responses
We have reported that costimulation blockade resistant rejection also occurs in IFNγ-/- allograft recipients and is associated with donor-reactive Th2 responses and granulocytic infiltration of the transplant. Since IFNγ is inhibitory to Th17 (20, 21), we asked whether the absence of IFNγ would promote donor-reactive Th17 development (Fig. 3A). While donor-reactive Th2 were readily detectable by ELISPOT, Th17 were rare in IFNγ-/- mice. Since IL-4 has also been reported to be inhibitory to Th17 (20, 21), we treated IFNγ-/- recipients with neutralizing anti-IL-4 mAb using a dosing schedule with proven in vivo bioactivity (7). However, Th17 were also rare in this setting (Fig. 3B). To verify that the sera of these mice contained adequate levels of neutralizing anti-IL-4 mAb, we employed a competitive IL-4 ELISA technique (7) (Fig. 3C). This analysis revealed that a 1:10,000 dilution of sera from anti-IL-4 treated mice contained at least enough anti-IL-4 mAb to neutralize 5 ng/ml of recombinant IL-4. Hence, eliminating IFNγ and IL-4, both known inhibitors of Th17, did not promote the emergence of Th17 in this transplant system. Since IFNγ-/- mice express T-bet (Fig. 5D), these observations emphasize the importance of T-bet as an inhibitor of Th17 in transplantation.
FIGURE 3.
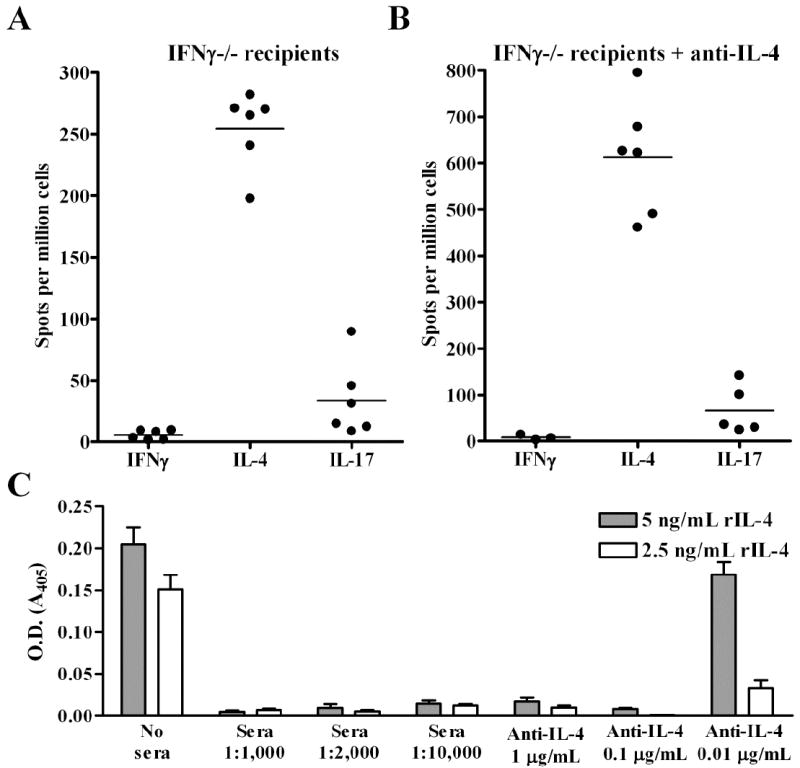
Eliminating IFNγ and IL-4 does not induce donor-reactive Th17. IFNγ-/- allograft recipients were either left untreated (A) or treated with neutralizing anti-IL-4 mAb (B). At the time of rejection (day 7 post-transplant), splenocytes were processed for ELISPOT analysis to quantify primed donor-reactive IFNγ, IL-4, and IL-17 producing cells. C, To verify that adequate levels of neutralizing anti-IL-4 mAb were present in the circulation of anti-IL-4 treated mice, dilutions of sera were assessed for their ability to neutralize 2.5 (open bars) or 5 (shaded bars) ng/ml rIL-4 in a competitive ELISA. For reference, the IL-4 neutralizing capacity of diluted sera was compared to the IL-4 neutralizing capacity of 1, 0.1, and 0.01 μg of purified anti-IL-4 mAb. The mean + S.E.M. IL-4 neutralizing capacity of 6 anti-IL-4 treated allograft recipients is depicted.
FIGURE 5.
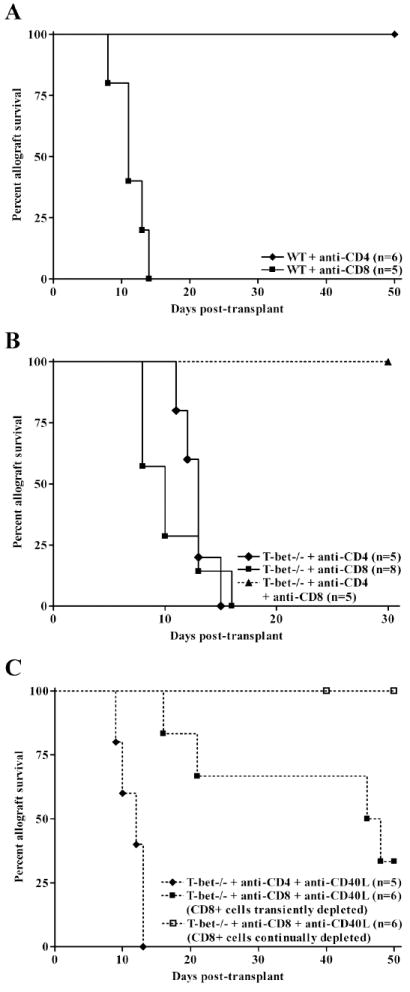
CD8+ cells mediate costimulatory blockade resistant rejection in T-bet-/- allograft recipients. WT (A) or T-bet-/- (B) allograft recipients were depleted of CD4+ (closed diamonds) or CD8+ (closed squares) cells by i.p. injections with anti-CD4 or anti-CD8 mAb. In addition, T-bet-/- recipients were depleted of both CD4+ and CD8+ cells (closed triangles). In Panel C, T-bet-/- recipients were treated with anti-CD40L mAb and were either initially depleted of CD4+ (closed diamonds) or CD8+ cells as above (transient depletion, closed squares), or were given weekly injections of anti-CD8 mAb (continued depletion, open squares) to assess the contribution of CD8+ cells to costimulation blockade resistant rejection.
CD8+ cells produce IFNγ and IL-17 in T-bet-/- allograft recipients
To identify the T cell subset(s) which produce IFNγ and IL-17 in T-bet-/- allograft recipients, splenocytes were harvested on day 7 post-transplant and were left intact or were depleted of CD4+ or CD8+ T cells with immunomagnetic beads prior to addition to ELISPOT assays. Depletion of CD4+ cells did not reduce the number of IFNγ (Fig. 4A) or IL-17 (Fig. 4B) producing cells, indicating that CD4+ cells were not the principle source of these cytokines. In contrast, depletion of CD8+ cells significantly reduced the magnitude of both Th1 and Th17 responses.
To determine whether individual CD8+ cells produced both IFNγ and IL-17, splenocytes and GIC from T-bet-/- allograft recipients were enriched for CD8+ cells and processed for dual intracellular cytokine staining (Fig. 4C). While CD8+ splenocytes produced either IFNγ or IL-17, an appreciable population of GIC produced both cytokines.
CD8+ cells mediate costimulation blockade resistant rejection and are the principle source of IL-17 in vivo
To identify the T cell subset responsible for costimulation blockade resistant rejection, we depleted cardiac allograft recipients of CD4+ or CD8+ cells in vivo by peri-operative injections of anti-CD4 or anti-CD8 mAb (Fig. 5). For comparison, Fig. 5A depicts T cell subset requirements for costimulation blockade sensitive rejection in WT mice. As previously reported (5, 7, 8), acute rejection in WT mice is dependent upon the presence of CD4+, but not CD8+ T cells.
In contrast, T-bet-/- mice rejected allografts regardless of whether CD4+ or CD8+ cells were depleted (Fig. 5B), indicating that effector CD8+ cells emerge independent of CD4+ cell help. When both T cell subsets were depleted, allografts continued to function for at least 30 days post-transplant. To test whether CD8+ cells were responsible for costimulation blockade resistant rejection, T-bet-/- allograft recipients were depleted of CD8+ cells and treated with anti-CD40L mAb (Fig. 5C). While anti-CD40L mAb significantly prolonged graft survival in T-bet-/- recipients that were initially depleted of CD8+ cells, allografts were rejected as CD8+ cells repopulated the periphery (closed squares). However, if CD8+ cells were continually depleted by weekly injections of anti-CD8 mAb (open squares), anti-CD40L therapy was effective at preventing rejection. Anti-CD40L therapy had no impact on survival when used in conjunction with CD4+ T cell depletion (closed diamonds). These data indicate that CD8+ cells are responsible for costimulation blockade resistant rejection in T-bet-/- mice.
Since ELISPOT revealed that anti-CD40L treatment did not eliminate the Th17 response within the grafts of T-bet-/- mice (Fig. 2C), we used quantitative real-time PCR to assess the level of IL-17 expression within the grafts of mice under various conditions (Fig. 6A). As expected, IL-17 was not expressed in the grafts of WT recipients. In contrast, IL-17 expression was readily detected in the grafts of unmodified, anti-CD40L treated, and CD4+ cell depleted T-bet-/- mice. However, intragraft expression of IL-17 was significantly reduced when T-bet-/- mice were depleted of CD8+ cells. Of note, intragraft IL-17 expression was negligible in unmodified or anti-IL-4 treated IFNγ-/- mice.
FIGURE 6.
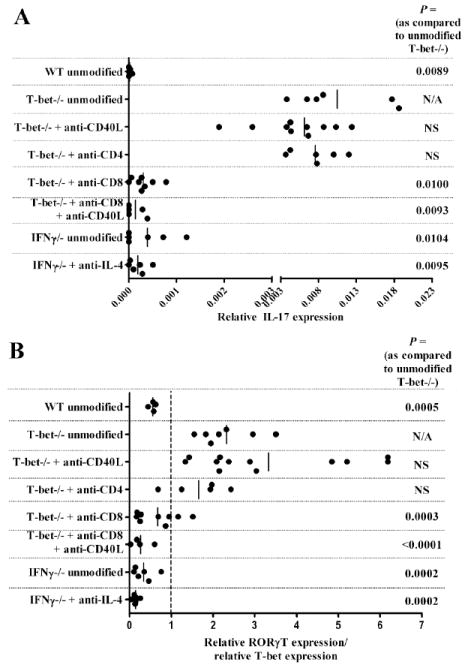
CD8+ cells express IL-17 within the allografts of T-bet-/- recipients. A, Intragraft expression of IL-17 was assessed by real-time RT-PCR. From top to bottom, RNA was harvested from allografts transplanted into the following mice: unmodified WT, unmodified T-bet-/-, anti-CD40L treated T-bet-/-, T-bet-/- depleted of CD4+ cells, T-bet-/- depleted of CD8+ cells, T-bet-/- continually depleted of CD8+ cells and treated with anti-CD40L, unmodified IFNγ-/-, or IFNγ-/- treated with anti-IL-4. Data points reflect IL-17 expression levels in individual allografts and bars depict the mean for each group. B, Intragraft expression of RORγT relative to T-bet was determined. RNA samples from the allografts in (A) were assessed for RORγT and T-bet expression by real-time RT-PCR. To normalize responses in individual recipients, data points represent the ratio of RORγT:T-bet in individual grafts. Bars depict the mean RORγT:T-bet ratios in each group.
We also assessed the intragraft expression of the Th17 transcription factor RORγT (21) relative to the expression of T-bet in these mice (Fig. 6B). In this analysis, a value of 1 indicates that RORγT and T-bet are equivalently expressed relative to the housekeeping gene cyclophylin A. It should be noted that T-bet-/- mice have a nonsense transcription of the T-bet gene, thereby allowing RT-PCR amplification of the nonsense T-bet cDNA (32, 37). This analysis revealed that the intragraft RORγT:T-bet ratio mirrored the expression of IL-17 in that T-bet-/- recipients expressed higher levels of RORγT unless they were depleted of CD8+ cells. Collectively, the data presented in Fig. 5 and 6 indicate a strong correlation between the expression of IL-17 by CD8+ cells and costimulation blockade resistant rejection in T-bet-/- allograft recipients.
In vivo neutralization of IL-17 or IL-6 reverses costimulation blockade resistant rejection
To confirm a role for IL-17 in costimulation blockade resistant rejection, T-bet-/- allograft recipients were treated with anti-CD40L mAb and either control rat IgG or neutralizing anti-IL-17 mAb. Anti-IL-17 mAb was given i.p. at 0.2 mg every other day, similar to a dosing regimen which is effective at inhibiting IL-17 mediated experimental autoimmune encephalomyelitis (EAE) (34). Allografts continued to function normally until the termination of the experiment (day 10 post-transplant) in anti-IL-17 treated mice, while grafts were undergoing acute rejection in the rat IgG treated controls. As shown in Fig. 7A, neutralizing IL-17 prevented allograft infiltration by neutrophils and preserved myocardial architecture of the graft in T-bet-/- recipients that were treated with anti-CD40L mAb. Fig. 7B revealed that neutralizing IL-17 reduced intragraft IL-17 expression relative to treatment with control rat IgG, reflecting the absence graft infiltration. Since IL-6 is critical in the for the development of the Th17 lineage (21), we treated T-bet-/- allograft recipients with anti-CD40L and neutralizing anti-IL-6 mAb. Allografts continued to function normally until the termination of the experiment on day 10 (n = 3) or day 14 (n = 6) post-transplant. As was the case with anti-IL-17 treatment, neutralizing IL-6 prevented allograft infiltration, intragraft IL-17 expression, and IL-17 associated graft pathology (Fig. 7A & B). Indeed, continued neutralization of IL-6 in anti-CD40L treated T-bet-/- recipients prevented graft infiltration, inhibited Th17 responses, and prevented rejection for at least 50 days post-transplant (n = 5, data not shown). Thus, neutralizing IL-17, or the Th17 inducing cytokine IL-6, reversed costimulation blockade resistant allograft rejection in T-bet-/- mice.
FIGURE 7.

Neutralizing IL-17 or IL-6 reverses costimulation blockade resistant rejection. T-bet-/- allograft recipients were treated with anti-CD40L mAb and either neutralizing anti-IL-17 mAb (n = 5), anti-IL-6 (n = 9), or control rat IgG (n = 7). At day 10 post-transplant, allografts were processed for H & E staining and histologic analysis (A) and RNA was isolated to assess intragraft IL-17 expression by real-time RT-PCR (B). An additional 6 anti-IL-6 treated recipients were harvested on day 14 post-transplant. At the termination of the experiment, all allografts placed in anti-IL-17 or anti-IL-6 treated recipients continued to function normally while 3 of the control rat IgG allografts were rejected and the remaining 4 control allografts exhibited deteriorating function.
Discussion
Both Th1 and Th2 responses have been implicated in acute allograft rejection, albeit with distinct pathologies (1, 3). Th1 mediated rejection is generally associated with mononuclear cell infiltration of the graft (5-7) (Fig. 1B & C) and CD8+ T cells are the principle source of IFNγ in unmodified rejection. Th2 mediated rejection is associated with graft infiltration by eosinophils (5, 7, 18) and emerges under conditions where IFNγ is limited. The contributions of Th17 to allograft rejection have not been rigorously investigated. While several associative studies indicate that Th17 may contribute to acute rejection (reviewed in (19)), elements that regulate graft-reactive Th17 development have not been defined.
While studying the requirement for the Th1 transcription factor T-bet in graft-reactive Th1 responses, we observed that rejection in T-bet-/- recipients was characterized by heavy infiltration of the graft by neutrophils (Fig. 1B & C). Since neutrophil rich infiltrates are characteristic of Th17 induced pathologies (21) and T-bet has been reported to be a negative regulator of Th17 responses (25), we assessed Th1, Th2, and Th17 responses in WT and T-bet-/- allograft recipients (Fig. 2). While the Th2 response was not remarkable in either group, strong Th17 responses were observed in T-bet-/- recipients but were not induced in WT recipients (Fig. 2C). As expected, WT recipients mounted strong Th1 responses (Fig. 2A). T-bet-/- recipients also mounted donor-reactive Th1 responses, albeit to a lesser degree than WT mice. This is not surprising, since CD8+ cells were the principle source of IFNγ in T-bet-/- recipients (Fig. 4A) and these cells express the alternate Th1 transcription factor eomesodermin (14). Indeed, real-time RT-PCR revealed that eomesodermin was expressed within the allografts of both WT and T-bet-/- recipients (data not shown). Interestingly, CD8+ cells were also the source of IL-17 in this system (Fig. 4B & 6A). Regarding CD4+ cells, Th1 and Th17 are believed to represent distinct lineages (20). To determine whether the same CD8+ cells produced both IFNγ and IL-17, or if each cytokine was produced by distinct populations of CD8+ cells, we employed dual intracellular cytokine staining (Fig. 4C). An appreciable number of GIC, but not splenocytes, produced both IFNγ and IL-17. Similar observations have been reported by Korn et al. (38) in an EAE model where IFNγ and IL-17 expressing cells are detectable in the inflamed CNS, but not in the draining lymph nodes. Hence, Th1 and Th17 do not necessarily represent distinct Th cell lineages. Importantly, the plasticity of these Th subsets is in part dependent on the in vivo microenvironment.
It is not clear whether CD8+ cells that produce IFNγ and/or IL-17 are comparable to their CD4+ counterparts. We have reported that CD8+ cells are “hard wired” to produce IFNγ, in that they do not require IL-12 to acquire a Th1 phenotype (9). Indeed, alloantigen-reactive CD4+ and CD8+ cells differ in several additional aspects, including their susceptibility to the suppressive activities of TGFβ and their relative contribution to costimulation blockade resistant rejection (reviewed in (39)).
The absence of a Th17 response in WT allograft recipients could be attributed to the overwhelming Th1 response in these mice, since IFNγ inhibits IL-17 production (20). Indeed, in experimental autoimmune myocarditis (EAM), IFNγ producing CD8+ heart infiltrating cells inhibit Th17 responses and protect against severe EAM (40). Further, T-bet-/- CD8+ heart infiltrating cells lose the capacity to release IFNγ within the heart in EAM. Hence, it is possible that the relatively reduced Th1 response in T-bet-/- recipients (compared to WT, Fig. 2A) may result in insufficient levels of IFNγ to inhibit Th17 induction. To this end, we assessed Th17 responses in IFNγ-/- allograft recipients (Fig. 3). However, Th17 were not induced in IFNγ-/- recipients, though IL-4 producing Th2 were detectable. Since IL-4 is also antagonistic to Th17 (20), we treated IFNγ-/- allograft recipients with neutralizing anti-IL-4 mAb (Fig. 3B). However, neutralizing IL-4 failed to induce Th17 in IFNγ-/- recipients, suggesting that T-bet, rather than IFNγ or IL-4, negatively regulates Th17 in the transplant setting. It should be noted that Komiyama et al. (41) reported that Th17 were detectable in IFNγ-/- mice with EAE. This apparent difference from our findings may be due to the relative dominance of CD8+ (cardiac allograft rejection) vs. CD4+ (EAE) cells in these distinct experimental models.
The mechanism(s) by which T-bet represses graft-reactive CD8+ Th17 responses in vivo are not yet known. In vitro, T-bet has been shown to interfere with IL-23 stimulated CD4+ Th17 development by diverting these cells into the Th1 lineage (25). IL-23 plays an important role in amplifying and/or stabilizing the Th17 phenotype (reviewed in (21, 42)). In addition, T-bet represses IL-21 transcription by inhibiting NFATc2 binding to the IL-21 promoter in CD4+ cells (43). IL-21, which is produced primarily by Th17, cooperates with TGFβ in the absence of IL-6 to promote Th17 development and therefore may serve as an autocrine amplification factor for Th17 (38). However, it is not known whether CD4+ and CD8+ Th17 have the same cytokine requirements for their development, amplification, and maintenance. The vast majority of information regarding Th17 biology has been generated in CD4+ cells (reviewed in (21, 42, 44)), while reports documenting CD8+ Th17 are limited (i.e. (45-48)).
To our knowledge, a role for Th17 in costimulation blockade resistant rejection has not been reported. It is unlikely that Th17 mediated costimulation blockade resistant rejection is due to heterologous memory cells (28, 49), in that the mice used in these studies were maintained under pathogen free conditions and were not subjected to overt antigenic challenge prior to transplantation. Indeed, primed alloantigen-reactive Th1, Th2, and Th17 are not detectable in non-transplanted T-bet-/- mice (data not shown). Further, it is unlikely that resistance to costimulatory blockade is due to the absence of IFNγ (7, 15) since graft-reactive Th1 were readily detectable in T-bet-/- allograft recipients (Fig. 2A). It should be noted that Th17 may be poorly susceptible to regulation by regulatory T cells (50), and regulatory T cells are believed to contribute to allograft acceptance following costimulatory blockade (26).
In summary, these data reveal a novel relationship between T-bet, CD8+ Th17, and costimulatory blockade resistant allograft rejection. These findings have implications for tolerance induction in the transplant setting, and provide further insight into the complexity of Th17 biology and Th17 associated pathologies.
Footnotes
This work was supported by R01 AI061469 (DKB) and R01 HL070613 (DKB) from the National Institutes of Health.
Abbreviations used in this paper: EAE, experimental autoimmune encephalomyelitis; EAM, experimental autoimmune myocarditis; GIC, graft infiltrating cells; IFNγ-/-, interferon gamma deficient; T-bet-/-, T-bet deficient; WT, wild type.
Disclosures The authors declare no competing financial interests.
References
- 1.Piccotti JR, Chan SY, VanBuskirk AM, Eichwald EJ, Bishop DK. Are Th2 helper T lymphocytes beneficial, deleterious, or irrelevant in promoting allograft survival? Transplantation. 1997;63:619–624. doi: 10.1097/00007890-199703150-00001. [DOI] [PubMed] [Google Scholar]
- 2.Rocha PN, Plumb TJ, Crowley SD, Coffman TM. Effector mechanisms in transplant rejection. Immunol Rev. 2003;196:51–64. doi: 10.1046/j.1600-065x.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 3.Le Moine A, Goldman M. Non-classical pathways of cell-mediated allograft rejection: new challenges for tolerance induction? Am J Transplant. 2003;3:101–106. doi: 10.1034/j.1600-6143.2002.00026.x. [DOI] [PubMed] [Google Scholar]
- 4.Coffman RL. Origins of the T(H)1-T(H)2 model: a personal perspective. Nat Immunol. 2006;7:539–541. doi: 10.1038/ni0606-539. [DOI] [PubMed] [Google Scholar]
- 5.Chan SY, DeBruyne LA, Goodman RE, Eichwald EJ, Bishop DK. In vivo depletion of CD8+ T cells results in Th2 cytokine production and alternate mechanisms of allograft rejection. Transplantation. 1995;59:1155–1161. [PubMed] [Google Scholar]
- 6.Piccotti JR, Chan SY, Goodman RE, Magram J, Eichwald EJ, Bishop DK. IL-12 antagonism induces T helper 2 responses, yet exacerbates cardiac allograft rejection. Evidence against a dominant protective role for T helper 2 cytokines in alloimmunity. J Immunol. 1996;157:1951–1957. [PubMed] [Google Scholar]
- 7.Bishop DK, Chan Wood S, Eichwald EJ, Orosz CG. Immunobiology of allograft rejection in the absence of IFN-gamma: CD8+ effector cells develop independently of CD4+ cells and CD40-CD40 ligand interactions. J Immunol. 2001;166:3248–3255. doi: 10.4049/jimmunol.166.5.3248. [DOI] [PubMed] [Google Scholar]
- 8.Bishop DK, Shelby J, Eichwald EJ. Mobilization of T lymphocytes following cardiac transplantation. Evidence that CD4-positive cells are required for cytotoxic T lymphocyte activation, inflammatory endothelial development, graft infiltration, and acute allograft rejection. Transplantation. 1992;53:849–857. [PubMed] [Google Scholar]
- 9.Piccotti JR, Li K, Chan SY, Ferrante J, Magram J, Eichwald EJ, Bishop DK. Alloantigen-reactive Th1 development in IL-12-deficient mice. J Immunol. 1998;160:1132–1138. [PubMed] [Google Scholar]
- 10.Nathan MJ, Yin D, Eichwald EJ, Bishop DK. The immunobiology of inductive anti-CD40L therapy in transplantation: allograft acceptance is not dependent upon the deletion of graft-reactive T cells. Am J Transplant. 2002;2:323–332. doi: 10.1034/j.1600-6143.2002.20406.x. [DOI] [PubMed] [Google Scholar]
- 11.Peng SL. The T-box transcription factor T-bet in immunity and autoimmunity. Cell Mol Immunol. 2006;3:87–95. [PubMed] [Google Scholar]
- 12.Grogan JL, Mohrs M, Harmon B, Lacy DA, Sedat JW, Locksley RM. Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity. 2001;14:205–215. doi: 10.1016/s1074-7613(01)00103-0. [DOI] [PubMed] [Google Scholar]
- 13.Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, Kung AL, Cereb N, Yao TP, Yang SY, Reiner SL. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 2001;292:1907–1910. doi: 10.1126/science.1059835. [DOI] [PubMed] [Google Scholar]
- 14.Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, Shen H, Cereb N, Yang SY, Lindsten T, Rossant J, Hunter CA, Reiner SL. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–1043. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 15.Konieczny BT, Dai Z, Elwood ET, Saleem S, Linsley PS, Baddoura FK, Larsen CP, Pearson TC, Lakkis FG. IFN-gamma is critical for long-term allograft survival induced by blocking the CD28 and CD40 ligand T cell costimulation pathways. J Immunol. 1998;160:2059–2064. [PubMed] [Google Scholar]
- 16.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 17.Ouyang W, Ranganath SH, Weindel K, Bhattacharya D, Murphy TL, Sha WC, Murphy KM. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 1998;9:745–755. doi: 10.1016/s1074-7613(00)80671-8. [DOI] [PubMed] [Google Scholar]
- 18.Martinez OM, Ascher NL, Ferrell L, Villanueva J, Lake J, Roberts JP, Krams SM. Evidence for a nonclassical pathway of graft rejection involving interleukin 5 and eosinophils. Transplantation. 1993;55:909–918. doi: 10.1097/00007890-199304000-00041. [DOI] [PubMed] [Google Scholar]
- 19.Afzali B, Lombardi G, Lechler RI, Lord GM. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol. 2007;148:32–46. doi: 10.1111/j.1365-2249.2007.03356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 21.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 22.Sonderegger I, Rohn TA, Kurrer MO, Iezzi G, Zou Y, Kastelein RA, Bachmann MF, Kopf M. Neutralization of IL-17 by active vaccination inhibits IL-23-dependent autoimmune myocarditis. Eur J Immunol. 2006;36:2849–2856. doi: 10.1002/eji.200636484. [DOI] [PubMed] [Google Scholar]
- 23.Cruz A, Khader SA, Torrado E, Fraga A, Pearl JE, Pedrosa J, Cooper AM, Castro AG. Cutting edge: IFN-gamma regulates the induction and expansion of IL-17-producing CD4 T cells during mycobacterial infection. J Immunol. 2006;177:1416–1420. doi: 10.4049/jimmunol.177.3.1416. [DOI] [PubMed] [Google Scholar]
- 24.Lohr J, Knoechel B, Wang JJ, Villarino AV, Abbas AK. Role of IL-17 and regulatory T lymphocytes in a systemic autoimmune disease. J Exp Med. 2006;203:2785–2791. doi: 10.1084/jem.20061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathur AN, Chang HC, Zisoulis DG, Kapur R, Belladonna ML, Kansas GS, Kaplan MH. T-bet is a critical determinant in the instability of the IL-17-secreting T-helper phenotype. Blood. 2006;108:1595–1601. doi: 10.1182/blood-2006-04-015016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol. 2004;22:307–328. doi: 10.1146/annurev.immunol.22.012703.104533. [DOI] [PubMed] [Google Scholar]
- 27.Adams AB, Larsen CP, Pearson TC, Newell KA. The role of TNF receptor and TNF superfamily molecules in organ transplantation. Am J Transplant. 2002;2:12–18. doi: 10.1034/j.1600-6143.2002.020104.x. [DOI] [PubMed] [Google Scholar]
- 28.Adams AB, Pearson TC, Larsen CP. Heterologous immunity: an overlooked barrier to tolerance. Immunol Rev. 2003;196:147–160. doi: 10.1046/j.1600-065x.2003.00082.x. [DOI] [PubMed] [Google Scholar]
- 29.Clarkson MR, Sayegh MH. T-cell costimulatory pathways in allograft rejection and tolerance. Transplantation. 2005;80:555–563. doi: 10.1097/01.tp.0000168432.60022.99. [DOI] [PubMed] [Google Scholar]
- 30.Ford ML, Koehn BH, Wagener ME, Jiang W, Gangappa S, Pearson TC, Larsen CP. Antigen-specific precursor frequency impacts T cell proliferation, differentiation, and requirement for costimulation. J Exp Med. 2007;204:299–309. doi: 10.1084/jem.20062319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones TR, Ha J, Williams MA, Adams AB, Durham MM, Rees PA, Cowan SR, Pearson TC, Larsen CP. The role of the IL-2 pathway in costimulation blockade-resistant rejection of allografts. J Immunol. 2002;168:1123–1130. doi: 10.4049/jimmunol.168.3.1123. [DOI] [PubMed] [Google Scholar]
- 32.Finotto S, Neurath MF, Glickman JN, Qin S, Lehr HA, Green FH, Ackerman K, Haley K, Galle PR, Szabo SJ, Drazen JM, De Sanctis GT, Glimcher LH. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 2002;295:336–338. doi: 10.1126/science.1065544. [DOI] [PubMed] [Google Scholar]
- 33.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- 34.Hofstetter HH, Ibrahim SM, Koczan D, Kruse N, Weishaupt A, Toyka KV, Gold R. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol. 2005;237:123–130. doi: 10.1016/j.cellimm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 35.Matesic D, Lehmann PV, Heeger PS. High-resolution characterization of cytokine-producing alloreactivity in naive and allograft-primed mice. Transplantation. 1998;65:906–914. doi: 10.1097/00007890-199804150-00008. [DOI] [PubMed] [Google Scholar]
- 36.Kirk AD, Blair PJ, Tadaki DK, Xu H, Harlan DM. The role of CD154 in organ transplant rejection and acceptance. Philos Trans R Soc Lond B Biol Sci. 2001;356:691–702. doi: 10.1098/rstb.2001.0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 38.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Csencsits KL, Bishop DK. Contrasting alloreactive CD4+ and CD8+ T cells: there’s more to it than MHC restriction. Am J Transplant. 2003;3:107–115. doi: 10.1034/j.1600-6143.2003.00036.x. [DOI] [PubMed] [Google Scholar]
- 40.Rangachari M, Mauermann N, Marty RR, Dirnhofer S, Kurrer MO, Komnenovic V, Penninger JM, Eriksson U. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med. 2006;203:2009–2019. doi: 10.1084/jem.20052222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 42.Tato CM, O’Shea JJ. Immunology: what does it mean to be just 17? Nature. 2006;441:166–168. doi: 10.1038/441166a. [DOI] [PubMed] [Google Scholar]
- 43.Mehta DS, Wurster AL, Weinmann AS, Grusby MJ. NFATc2 and T-bet contribute to T-helper-cell-subset-specific regulation of IL-21 expression. Proc Natl Acad Sci U S A. 2005;102:2016–2021. doi: 10.1073/pnas.0409512102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006;27:17–23. doi: 10.1016/j.it.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 45.Happel KI, Zheng M, Young E, Quinton LJ, Lockhart E, Ramsay AJ, Shellito JE, Schurr JR, Bagby GJ, Nelson S, Kolls JK. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J Immunol. 2003;170:4432–4436. doi: 10.4049/jimmunol.170.9.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He D, Wu L, Kim HK, Li H, Elmets CA, Xu H. CD8+ IL-17-producing T cells are important in effector functions for the elicitation of contact hypersensitivity responses. J Immunol. 2006;177:6852–6858. doi: 10.4049/jimmunol.177.10.6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kryczek I, Wei S, Zou L, Altuwaijri S, Szeliga W, Kolls J, Chang A, Zou W. Cutting Edge: Th17 and Regulatory T Cell Dynamics and the Regulation by IL-2 in the Tumor Microenvironment. J Immunol. 2007;178:6730–6733. doi: 10.4049/jimmunol.178.11.6730. [DOI] [PubMed] [Google Scholar]
- 48.Liu SJ, Tsai JP, Shen CR, Sher YP, Hsieh CL, Yeh YC, Chou AH, Chang SR, Hsiao KN, Yu FW, Chen HW. Induction of a distinct CD8 Tnc17 subset by transforming growth factor-beta and interleukin-6. J Leukoc Biol. 2007;82:354–360. doi: 10.1189/jlb.0207111. [DOI] [PubMed] [Google Scholar]
- 49.Adams AB, Williams MA, Jones TR, Shirasugi N, Durham MM, Kaech SM, Wherry EJ, Onami T, Lanier JG, Kokko KE, Pearson TC, Ahmed R, Larsen CP. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111:1887–1895. doi: 10.1172/JCI17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, Romagnani S. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]