

Abstract
Early postnatal blockade of NMDA receptors by phencyclidine (PCP) causes cortical apoptosis in animals. This is associated with the development of schizophrenia-like behaviors in rats later in life. Recent studies show that the mechanism involves a loss of neurotrophic support from the phosphoinositol-3 kinase/Akt pathway, which is normally maintained by synaptic NMDA receptor activation. Here we report that activation of dopamine D1 receptors (D1R) with dihydrexidine (DHX) prevents PCP-induced neurotoxicity in cortical neurons by enhancing the efficacy of NMDAergic synapses. DHX increases serine phosphorylation of the NR1 subunit through protein kinase A (PKA) activation and tyrosine phosphorylation of the NR2B subunit via Src kinase. DHX enhances recruitment of NR1 and NR2B, but not NR2A, into synapses. DHX also facilitated the synaptic response in cortical slices and this was blocked by an NR2B antagonist. DHX pretreatment of rat pups prior to PCP on postnatal days 7, 9 and 11 inhibited PCP-induced caspase-3 activation on PN11 and deficits in pre-pulse inhibition of acoustic startle measured on PN 26–28. In summary, these data demonstrate that PCP-induced deficits in NMDA receptor function, neurotoxicity and subsequent behavioral deficits may be prevented by D1R activation in the cortex and further, it is suggested that D1R activation may be beneficial in treating schizophrenia.
Keywords: phencyclidine, NMDA receptors, NR1, NR2B, D1 receptor, phosphoinositol-3 kinase, Src kinase, schizophrenia
Introduction
The cortex, particularly the prefrontal cortex (PFC), is important in cognition and working memory and deficits in these important functions are central features of schizophrenia (Elvevag & Goldberg 2000). Blockade of N-methyl-D-aspartate receptors (NMDAR) by open channel blockers such as phencyclidine (PCP), MK801 and ketamine, leads to a reduction of NMDAR function and substantially altered behaviors that mimic several aspects of schizophrenia (Olney et al. 1999). This is the core of the hypoglutamatergic hypothesis of schizophrenia (Olney & Farber 1995). Administration of NMDAR channel blockers to primates and rodents early in postnatal life produces neurodegeneration in several brain regions relevant to schizophrenia, including the cortex, striatum, hippocampus, and thalamus (Ikonomidou et al. 1999, Slikker et al. 2007). Previous studies from this lab and several others have shown that administration of PCP to rats on post-natal (PN) days 7, 9, 11 causes behavioral deficits that resemble certain features of schizophrenia in adult rats (Wang et al. 2001, du Bois & Huang 2007, Broberg et al. 2008). These studies include those that have shown that antipsychotic drugs block, or significantly dampen these behaviors in adolescent or adult rodents (Duncan et al. 2006, Kargieman et al. 2007, Anastasio & Johnson 2008), thereby further supporting the link between neurotoxicity during an early postnatal period and schizophrenia-like symptoms later in life.
Knowledge of the mechanisms of NMDAR antagonist-induced neuronal damage could lead to novel approaches for the treatment of schizophrenia. Cepeda et al (1993) first reported that dopamine, through activation of the D1 receptor (D1R), potentiates NMDA receptor-mediated synaptic responses in the striatum. This finding has been extended to the PFC and hippocampus (Yang 2000, Flores-Hernandez et al. 2002). Seamans et al (2001) showed that D1R agonists caused a slight reduction in the size of the non-NMDA component of excitatory postsynaptic currents (EPSCs) in layer V PFC neurons, while significantly increasing, through a postsynaptic mechanism, the size of the NMDA component of EPSCs. Gonzalez-Islas and Hablitz (2003) also reported that bath application of dopamine in layer II-III pyramidal neurons in the rat PFC significantly enhanced EPSC amplitudes via a mechanism in which both NMDA and AMPA receptors contributed. This effect resulted from D1, but not D2 receptor activation. Furthermore, it has been suggested that D1R- mediated potentiation of NMDAR in PFC may be attributable to a postsynaptic signaling cascade predominantly involving PKA and Ca2+ (Gonzalez-Islas & Hablitz 2003). We recently reported that enhancing synaptic efficacy by increasing glutamate release with bicuculline, a GABA antagonist, or increasing intracellular Ca2+ with an L-type calcium channel agonist protects against PCP-induced neurotoxicity in neuronal culture (Lei et al. 2008). Stimulation of dopamine D1R in the presence of bicuculline has been reported to increase the amplitude of EPSCs in layer IIIII cortical pyramidal neurons evoked by weak intra-cortical stimulus (Bandyopadhyay et al. 2005). Therefore, these experiments were designed to determine whether activation of D1 receptors could prevent PCP-induced neurotoxicity, and if so, to determine the intracellular signaling mechanism responsible for this action.
Materials and Methods
Chemicals and antibodies
PCP was acquired from the National Institute on Drug Abuse (Rockville, MD, USA). PP2 (3-(4-chlorophenyl) 1 – (1,1-dimethylethyl) – 1 H-pyrazolo [3,4-d] pyrimidin-4-amine), lavendustin A (5 –[[(2,5-dihydroxyphenyl) methyl][(2-hydroxyphenyl) methyl] amino] -2- hydroxybenzoic acid), SCH23390, dihydrexidine ((±) –trans-10,11- dihydroxy −5,6,6a,7,8,12b-hexahydrobenzo[a] phenanthridine hydrochloride), and bicuculline methobromide, DL-2-amino-5-phosphonopentanoic acid (AP5), 6-Cyano-7-nitroquinoxaline-2,3-dione disodium (CNQX), and KT5720 were purchased from Tocris Cookson Inc.(Ellisville, MO, USA). SKF38393, phosphatase inhibitor cocktail 1 and 2, and 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO). Bisindolylmaleimide I, H-89 (N-[2-((p-bromocinnamyl) amino) ethyl]-5-isoquinolinesulfonamide) and PKI14–22 were purchased from EMD Biosciences Inc. (San Diego, CA, USA). Cell Death Detection ELISA kit was purchased from Roche Applied Science (Indianapolis, IN, USA). Neurobasal™ medium and B27 supplement were purchased from Invitrogen (Carlsbad, CA, USA). All antibodies used were commercially available. NR1 antibody (clone 54.1, mouse IgG) was purchased from BD Biosciences (San Jose, CA 95131). NR2A and NR2B antibodies (mouse IgG) were purchased from Invitrogen (Grand Island, NY 14072). Antibodies against PSD95, pNR1Ser897, pPKAThr197, pNR2BTyr1472, pAktSer473, Akt, pGSK-3βSer9 and GSK-3β were purchased from Cell Signaling Technology (Beverly, MA 01915).
Animals
Timed, day 14 pregnant female Sprague–Dawley rats were obtained from Charles River Laboratories (Wilmington, MA). The dams were housed individually with a regular 12 h light–dark cycle (lights on 0700, off at 1900) with food and water available ad libitum. For in vitro experiments, cultures were prepared from embryonic day 18 rat brains. For all other experiments, following parturition, male and female pups from 3 dams were combined and randomly cross-fostered to one of the lactating dams. Each litter consisted of ten to twelve pups with approximately equal numbers of each gender. All experiments were conducted in accordance with the NIH and the University of Texas Medical Branch at Galveston Institutional Animal Care and Use Committee.
Neuronal culture and cell viability assays
Neuronal cultures were prepared following the procedure published by Lei et al (2008). Briefly, cortices from E18 SD rats were dissociated in cold Hanks solution and cells were plated at 10 × 105 cells/ml and grown on polylysine (5 mg/ml)-coated multiwell plates in Neuorbasal™ medium containing 0.5 mM L-glutamine, supplemented with 10% B27 at 37°C and 5% CO2. Experiments were conducted on DIV14 -17. Neuronal apoptosis was assessed by measuring DNA-associated histone complexes using the Cell Death Detection ELISA® kit according to the manufacturer's instructions (Roche Diagnostics Corporation, Indianapolis, IN). Cell viability was determined by measuring mitochondrial metabolism of MTT (Lei et al. 2008).
Membrane protein preparation and immunoprecipitation
Cultured neurons were mechanically dissociated from the dish after two washes with ice-cold PBS. Following washing with ice-cold PBS two times, the cells were kept on ice for 1hr and sonicated in ice-cold buffer containing 320 mM sucrose, 10 mM Tris–HCl pH 7.4, 1 mM NaHCO3 pH 7.4, 1 mM MgCl2 supplemented with 1 mM sodium orthovanadate and 1% protease inhibitor cocktail, and subsequently spun at 5,000 g for 15 min. The supernatant was collected and centrifuged at 40,000 g (Optima™ MZX-E ultracentrifuge, Beckman) for 60 min. The pellets were dissolved in a lysis buffer (10 mM Tris–HCl pH 9, 150 mM NaCl, 0.5% Triton X-100, 1% sodium deoxycholate, 0.5% SDS, 2 mM EDTA, 1 mM sodium orthovanadate and 1% protease inhibitor cocktail) for subsequent experiments.
For immunoprecipitation, 300 µg membrane protein in 300 µl was incubated with antibodies against PSD95 (5 µl) or rabbit IgG at 4°C and gently shaken overnight. Following precipitation with protein G–Sepharose beads, the immunoprecipitates were washed 4 times with ice-cold PBS, and then resuspended in 2x Laemmli sample buffer for Western blot analysis.
ELISA assay for cell surface expression
This assay was done essentially following a published protocol. PFC cultured neurons on DIV14 were treated with 10 µM DHX for 24h and then fixed in 4% paraformaldehyde for 10 min in the absence (non-permeant conditions) or the presence (permeant conditions) of 1% Triton X-100. Cells were incubated with a monoclonal antibody against the Flag epitope (BD Bioscience, 1 µg/ml to detect the Flag epitope inserted into the N terminus of NR1 subunit). After incubation with the corresponding HRP-conjugated secondary antibodies, the HRP substrate OPD was added to produce a color reaction that was stopped with 3N HCl. The degree of cell surface expression of NR1 was presented as the ratio of the colorimetric reading under non-permeabilized conditions to those under permeabilized conditions and then normalized to their respective control groups. The analyses were done using at least six separate dishes in each group.
Whole-cell patch-clamp recording and synaptic stimulation
Brain slices were obtained from PN6-8 rats. Rats were decapitated and the brains were quickly dissected out and immersed in cold (∼4°C) artificial cerebrospinal fluid (ACSF) bubbled continuously with 95% O2/5% CO2 to maintain proper pH (7.3–7.4). The composition of the ACSF solution was as follows (in mM): 117 NaCl, 4.7 KCl, 1.2 NaH2PO4, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, and 11 glucose. Coronal brain slices (300 µm) were prepared using a Vibroslice (Camden Instruments, London, UK). After incubation in ACSF at room temperature for at least 1 h, a single brain slice was transferred to the recording chamber and submerged in ACSF (31 ± 1°C), which was also used to superfuse the slice at ∼2 ml/min. One or two brain slices per animal were used and activity from one neuron was recorded in each slice and a fresh slice was used for each new experimental protocol. Numbers in the manuscript refer to the number of neurons tested for each parameter.
Whole-cell recordings were obtained from neurons in layer V of the PFC. Patch electrodes (4–6 M Ohm tip resistance) were made from borosilicate glass capillaries (1.5 mm and 1.12 mm, outer and inner diameter, respectively; Drummond, Broomall, PA) using a Flaming-Brown micropipette puller (P-80/PC, Sutter Instrument Co., Novato, CA). The internal solution of the recording electrodes contained (in mM): 122 K-gluconate, 5 NaCl, 0.3 CaCl2, 1 EGTA, 10 HEPES, 5 Na2-ATP, 0.4 Na2-GTP; pH was adjusted to 7.2–7.3 with KOH and the osmolarity to 280 mOsm/kg with sucrose. After tight (>1 G Ohm) seals were formed and the whole-cell configuration was obtained, neurons were included in the sample if the resting membrane potential was more negative than −50 mV and action potentials overshooting 0 mV were evoked by direct depolarizing current injections. Voltage and current signals were low-pass filtered at 1 KHz with a dual 4-pole Bessel filter (Waner Instrument Corp., Hamden, CT), digitized at 5 KHz (Digidata 1322A interface, Axon instrument, Molecular Devices, Sunnyvale, CA), and stored on a computer (Dell Pentium 4). Data were also continually recorded on an ink chart recorder (Gould 3400, Could Instr., Valley View, OH). Current- and voltage-clamp (d-SEVC) recordings were made using an Axoclamp-2B amplifier (Axon Instr.) with a switching frequency of 5–6 kHz (30% duty cycle), gain of 3–8 nA/mV, and time constant of 20 ms. Phase shift and anti-alias filter were optimized. The headstage voltage was monitored continuously on a digital oscilloscope (Gould 400, Could Instr.) to ensure precise performance of the amplifier. If series resistance (monitored with pClamp 8 software, Axon Instr.) changed more than 10%, the neuron was discarded to minimize any potentially confounding influences of series and space clamp changes. Voltage- and current data were analyzed with pClamp 8 software (Axon Instruments). Neurons were voltage-clamped at −60 or +40 mV for isolating the NMDA component of EPSCs. Using a concentric bipolar stimulating electrodes (SNE-100, Kopf Instr.; 22 k Ohm), monosynaptic excitatory postsynaptic currents (EPSCs) were evoked in layer V neurons by electrical stimulation (using a Grass S88 stimulator) of a layer II-III input. Electrical stimuli (150 micro-s square-wave pulses) were delivered at low frequencies (<0.25 Hz). Input-output functions were obtained by increasing the stimulus intensity in 100 µA steps. For evaluation of a drug effect on synaptically evoked responses, the stimulus intensity was adjusted to 75–80% of the intensity required for orthodromic spike generation.
Measurement of pre-pulse inhibition (PPI) of acoustic startle
Male and female rat pups were treated on PN7, 9, and 11 with 10 mg/kg PCP or saline (s.c). Saline, DHX (3 mg/kg, s.c.) or SCH23390 (1 mg/kg, s.c.) were given 30 min prior to PCP or saline either alone or in combination. The treated animals were assessed for PPI of acoustic startle on PN26-28 according to published procedures with minor modifications (Anastasio & Johnson 2008, Wang et al. 2001). Briefly, rat pups were tested in a startle chambers (SR-Lab, San Diego Instruments, San Diego, CA) with a background noise level of 65 dB. Following a 10 min acclimation, rats were exposed to three randomly administered stimuli: no stimulus, a 73 dB 20 ms pre-pulse 100 ms prior to a 120 dB pulse, or a 120 dB 40 ms pulse alone with a variable inter-trial interval (5–20 s) for a total of 63 trials (21 no stimulus, 21 pulse alone, and 21 pre-pulse+pulse). % PPI of acoustic startle was calculated as the [pulse–(pre-pulse+pulse)] /pulse×100.
Results
The effect of dopamine D1R activation on PCP-induced neurotoxicity in PFC cultured neurons
To test the effect of D1R activation on PCP- induced neurotoxicity, we examined cell viability by measuring mitochondrial metabolism of MTT. Nucleosomal DNA fragmentation was assessed by measuring histone-associated DNA fragments using the Cell Death Detection ELISA kit. We first tested the effect of DHX, dopamine D1R full agonist. Pretreatment with DHX alone 30 min prior to a 48 hr treatment with PCP had no significant effect on PCP-induced neurotoxicity (Fig 1A and Fig 1B). As D1R activation has been shown to potentiate NMDA receptor function in the PFC through postsynaptic mechanisms (Seamans et al. 2001, Gonzalez-Islas & Hablitz 2003), we proposed that any effect of DHX may require activation of NMDAR by glutamate. Moreover, bath application of DHX can suppress presynaptic glutamate release (Yang & Chen 2005). Thus, a sub-threshold concentration (2.5 µM) of bicuculline (Bic), a GABAA receptor antagonist known to facilitate synaptic glutamate release in cultured neurons (Hardingham et al. 2002, Ivanov et al. 2006), was added following the stimulation of D1R with DHX. The results showed that 2.5 µM Bic alone did not block PCP- induced neurotoxicity, but pretreatment with 10 µM DHX 30 min prior to Bic addition significantly attenuated PCP-induced neurotoxicity (Fig. 1A and 1B). This protection was abolished by SCH23390, a selective D1R antagonist, in a concentration-dependent manner (Fig.1C), but not by haloperidol, a typical D2 antagonist (data not shown). We also examined the effect of SKF38390, a partial D1R agonist, anticipating that SKF38390 would partially block PCP- induced toxicity. The result confirmed that SKF38390 could only partially reverse PCP-induced neurotoxicity (Fig.1D).
Figure 1. D1 receptor-mediated neuroprotection by dihydrexidine (DHX) requires activation by GABA receptor blockade with bicuculline.
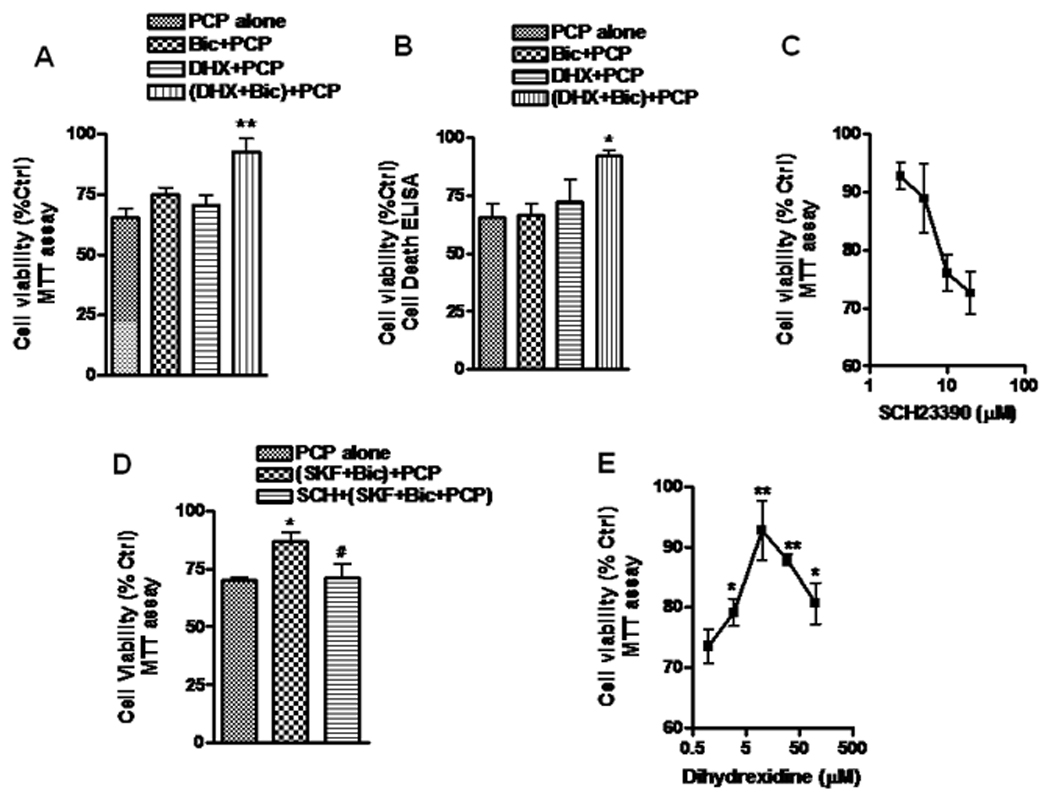
Neuronal cells prepared from the prefrontal cortex (PFC) were cultured for 14 days (DIV14) in Neurobasal ™ medium supplemented with B27. The cultured cells were first treated with 10 µM DHX for 30 min, then incubated with 2.5 µM bicuculline (Bic, a GABAA antagonist) for 3h. After the treatment, the cells were exposed to 1 µM PCP for 48h and assayed for cell viability by measuring MTT metabolism (A) or for 24h and assessed for DNA fragmentation using the Cell Death Detection ELISA kit (B). C. Cultured neurons were first exposed to a series of concentrations of the D1R antagonist, SCH23390, for 30 min, followed by treatment with DHX and PCP as described in panel A. D. Cells were pretreated with 10 µM SCH23390 or vehicle as a control for 1h, then stimulated with 10 µM SKF38390 (a partial D1 agonist) for 30 min followed by 2.5 µM Bic for 3h. Following this treatment, the cells were exposed to 1 µM PCP for 48 h and assessed for viability. E. Cultured neurons were incubated with different concentrations of DHX 30 min before treatment of cells with Bic and PCP as described in Fig.1A. DHX alone control experiments showed no significant effect on cell viability as assessed by measurement of MTT 48 hrs later (data not shown). Values shown are mean ± SEM (* P<0.05, ** P<0.01 vs PCP treated group, # P < 0.05, SKF protection vs SCH inhibition, one-way ANOVA with Newman-Keuls post-hoc analysis, N = 3–4/group).
It has been reported that there is an inverted “U” dose-response profile for D1Rmediated PFC -based cognitive task performance and NMDA synaptic responses (Williams & Goldman-Rakic 1995, Yang & Chen 2005). These reports showed that a low-level of D1R stimulation increases the synaptic response, while a stronger activation of D1R resulted in a lesser response. To determine whether the protection of DHX against PCP-induced toxicity showed a similar biphasic or inverted “U” profile, different concentrations of DHX were applied to the cultured neurons, and cell viability examined by measuring mitochondrial metabolism of MTT. These data (Fig.1E) demonstrated that the protection afforded by DHX increased in a concentration-dependent manner up to 10 µM, but higher concentrations were less effective. DHX alone in these experiments did not cause significant toxicity in cultured neurons (data not shown). Thus, this neuroprotective effect of DHX has a dose-response similar to that seen in performance of cognitive tasks.
Roles of PKA in dopamine D1R - mediated neuroprotection
It has been proposed that PKA plays a major role in D1R-mediated potentiation of NMDAR in the striatum, hippocampus and PFC (Greengard 2001). However, Chen et al (2004) reported that potentiation of NMDAR-mediated currents in neurons prepared from prefrontal cortex by selective activation of D1R was independent of PKA. That study demonstrated that application of calmodulin or PKC inhibitors, but not PKA inhibitors, could block the increase in NMDAR-mediated currents induced by D1R stimulation. Therefore, we examined the role of these kinases in D1R mediated neuroprotection. These results showed that the protection against PCP-induced neurotoxicity by D1R stimulation (along with Bic-induced depolarization) could be prevented by either 0.5 µM PKI14–22 or 10 µM H89, both of which are membrane-permeable PKA inhibitors (Fig. 2A). However, pretreatment with 1 µM bisindolylmaleimide I (BIS), a selective PKC inhibitor, failed to prevent D1R-mediated protection. The protective effect was also intact in the presence of 2 µM KN93, a selective, but not specific, CaMKII inhibitor. KN93, BIS, PKI and H89 at these concentrations were not significantly toxic to the cells (data not shown).
Figure 2. The role of PKA on D1R-mediated protection and attenuation of PCP-induced reduction of NR1 phosphorylation.
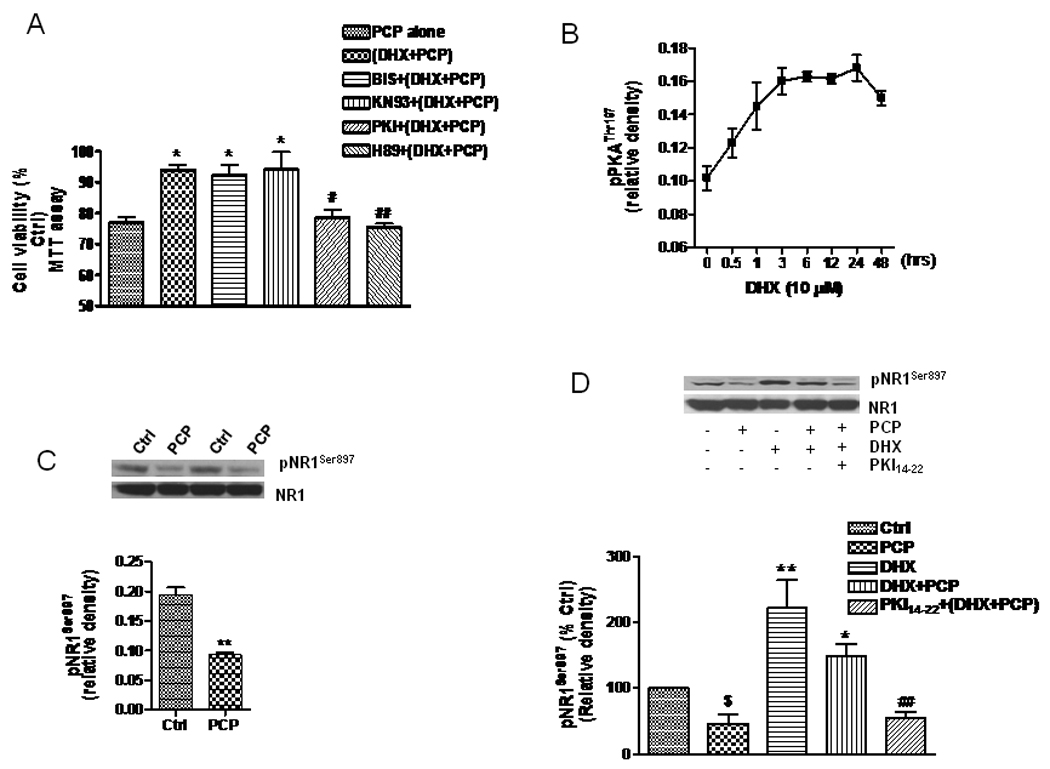
A. PCP-treated neuronal cells (DIV14) were treated for three hrs with either DHX (10 µM) alone or 2.5 µM with Bic, or they were pretreated with BIS (1 µM), KN93 (2 µM), H89 (10 µM) or PKI14–22 (0.5 µM) for 1h, followed by DHX (10 µM) plus Bic (2.5 µM) for another 3h. The treated cells were then exposed to PCP (1 µM) for 48h. Cell viability was determined by assessing MTT metabolism. B. Cultured neurons were treated with DHX (10 µM) for different times and lysed in ice-cold RIPA buffer containing inhibitors of proteinases, kinases and phosphatases. The cell lysate (15 µg) was used for WB to determine pPKAThr197. The time course data, normalized to β-actin, were quantified from three independent WB experiments. C. Cultured neurons were treated on DIV 14 with 1 µM PCP for 24h, then lysed for WB. The upper panel shows 2 examples of reduction of pNR1Ser897 after PCP treatment, while the lower panel is the quantification of 3 different WB. D. The cells were treated with PCP (1 µM), DHX (10 µM), or combination of PCP and DHX in the presence or absence of the PKA inhibitor, PKI14–42 (1 µM). Phosphorylation of NR1 (pNR1S897) was determined by WB (upper panel). Data are summarized from 3 independent experiments (lower panel). The values shown are mean ± SEM (*, # and $ P<0.05, ** and ## P< 0.01, * and ** compared with PCP group; # and ##, compared with DHX+PCP group; $ vs control group, one way ANOVA with Newman-Keuls posthoc analysis, N= 3–4/group).
The PKA-holoenzyme is made up of two regulatory subunits and two catalytic subunits. The catalytic subunit of PKA has two main phosphorylation sites, Thr197 and Ser338, and full activity requires phosphorylation. We observed that PCP could decrease Thr197 phosphorylation of PKA in cultured neurons (data not shown). Therefore, we determined the effect of D1R stimulation on the phosphorylation of Thr197 (pPKAThr197). As shown in Fig. 2B, the phosphorylation of Thr197 of the PKA catalytic subunit was elevated when the neurons were treated with 10 µM DHX. An increased phosphorylation of pPKAThr197 could be observed as early as 30 min after DHX treatment, with the peak effect at about 3 h. Interestingly, the elevated phosphorylation of PKA was very stable, maintaining a high level for at least 24 hr (Fig. 2B).
NR1Ser897 has been suggested to be a substrate for PKA. Tingley et al (1997) reported that D1R-induced PKA activation could directly upregulate NMDA receptor currents via increased phosphorylation of the NR1 subunit. Moreover, a reduced phosphorylation of pNR1Ser897 has been observed in brains from patients with schizophrenia (Emamian et al. 2004) and mice treated with PCP (Mouri et al. 2007). Thus, we determined whether PCP caused any alteration of pNR1Ser897 in cultured neurons. As shown in Fig. 2C, pNR1Ser897, but not total NR1, was reduced by about half when the cells were exposed to 1 µM PCP for 24h. Following a 24 h treatment with PCP, the IC50 value for PCP-induced reduction of pNR1Ser897 was approximately 0.5 µM (data not shown). We then determined whether activation of D1R could increase phosphorylation of Ser897 on the NR1 subunit via the PKA-mediated pathway. The data shown in Fig. 2D demonstrates that DHX can increase pNR1Ser897 as well as reverse the PCP-induced reduction of pNR1Ser897. This effect of DHX on pNR1Ser897 was abolished by the selective PKA inhibitor PKI14–22, but not by the PKC inhibitors, BIS or KN93 (data not shown).
Tyrosine phosphorylation contributes to the protection afforded by D1R stimulation
Phosphorylation of tyrosine residues in NMDAR has been reported to play roles in regulation of the NMDAR in the brain. Among 25 tyrosine residues in the C-terminal tail of the NR2B subunit, 7 residues can be phosphorylated by FYN in vitro. Of these 7 residues, Tyr-1252, Tyr-1336 and Tyr-1472 were phosphorylated in human embryonic kidney fibroblasts when co-expressed with FYN, and Tyr-1472 was the predominant phosphorylation site. Phosphorylation of NR2BTyr1472 is well known to provide stabilization of NR2B on the cell surface (Prybylowski et al. 2005, Snyder et al. 2005, Braithwaite et al. 2006). Moreover, phosphorylation of NR2BTyr1472 may be involved in long-term potentiation in the hippocampal CA1 region (Nakazawa et al. 2001). Further, several groups have recently reported that activation of D1R increased phosphorylation of pNR2BTyr 1472 in striatal neurons (Dunah & Standaert 2001, Dunah et al. 2004, Hallett et al. 2006)and Gao and Wolf (2008) reported similar findings in the cortex. Therefore, we examined whether activation of D1R in the PFC affected either the phosphorylation of pNR2BTyr 1472 or its role in PCP-induced neurotoxicity. PFC neurons at DIV14 were treated with 10 µM DHX in the presence of 1 µM lavendustin A, a pan tyrosine kinase inhibitor or 1 µM PP2, a selective Src kinase inhibitor (Fig. 3A). This experiment showed that activation of D1R by DHX increases pNR2BTyr 1472. This effect was blocked by LvA, PP2, or SCH23390. As phosphorylation of Tyr1472 on NR2B increases NMDA receptor currents (Salter & Kalia 2004), we proposed that tyrosine phosphorylation by D1R activation would also play a role in D1R-mediated protection against PCP-induced neurotoxicity. Thus, we examined the role of tyrosine phosphorylation of NMDAR in the protection afforded by D1R activation. Fig. 3B and Fig. 3C demonstrated that the protection afforded by DHX against PCP-induced neurotoxicity was diminished in the presence of either LvA or PP2, suggesting that inhibition of tyrosine kinase can partially block the protection demonstrated by D1R activation.
Figure 3. The effect of Src kinase inhibition on D1R-mediated phosphorylation of NR2B and protection against PCP-induced neurotoxicity.
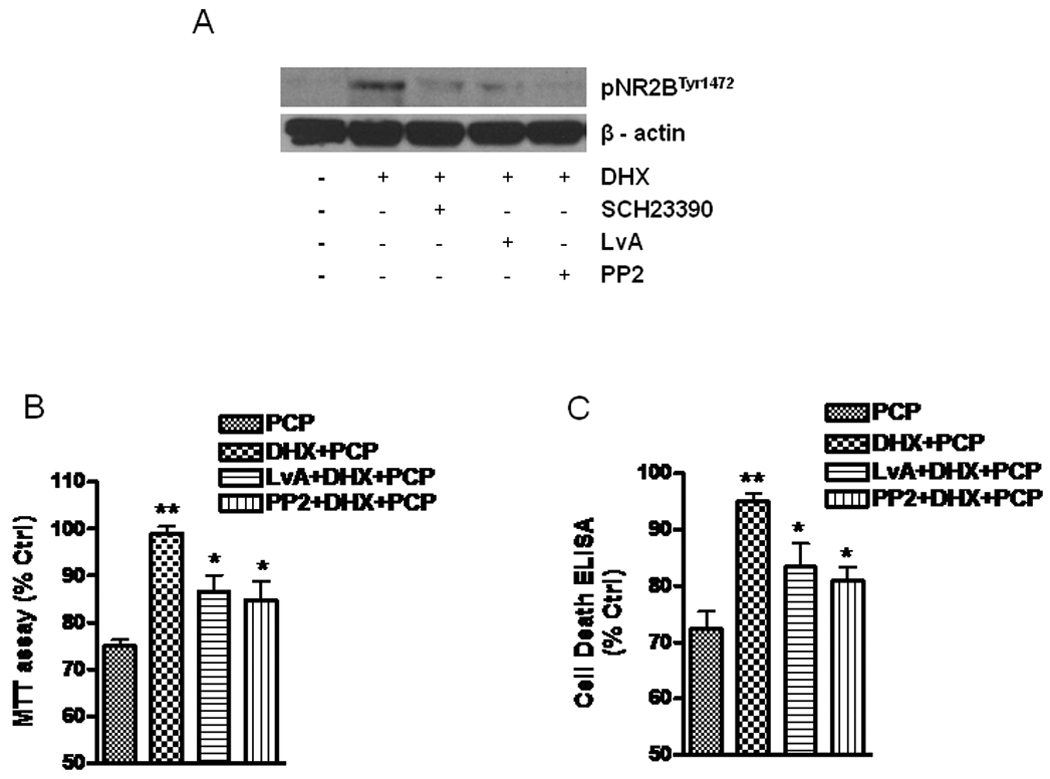
A. D1R-mediated increase of NR2B (pNR2BTyr1472) is inhibited by the Src kinase inhibitors, lavendustin A (LvA) or PP2, at 1 µM each. Cultured neurons were treated for 24 h with DHX (10 µM) or a combination of DHX and either SCH23390 (1 µM), or the Src kinase inhibitors as indicated. The treated cells were then lysed with RIPA buffer and equal amounts of protein (20 µg) were loaded into each lane of 7.5% SDS-PAGE. The blots were probed with a specific antibody against pNR2BTyr1472. B and C. Inhibition of tyrosine kinase partially blocks the protection afforded by D1R activation. Cells were pretreated with 1 µM LvA or 1 µM PP2 for 30 min, followed by exposure to PCP or a combination of PCP and DHX (plus 2.5µM bicuculline) for 48h (B) or 24h (C) prior to assessment of viability. Values shown are mean ± SEM (** P<0.01 vs PCP treated group; *P<0.05 vs cells treated with DHX+PCP, one-way ANOVA with Newman-Keuls post-hoc analysis, N = 3/group).
D1R activation increases synaptic strength and promotes NMDA receptor surface expression and synaptic insertion
Our previous report demonstrated that increasing synaptic strength could prevent PCP-induced neurotoxicity (Lei et al. 2008). Moreover, it has been shown that enhancing NMDA receptor function by application of glycine, an NMDA receptor co-agonist, could significantly improve negative symptoms and cognitive impairment in schizophrenic subjects (Heresco-Levy et al. 1999, Javitt 2001, Shim et al. 2008). Further, activation of D1R in cultured striatal neurons by tyrosine phosphorylation has been reported to lead to increased clustering of NR1 and NR2B subunits in dendrites with postsynaptic density 95 (PSD95), a scaffolding protein (Hallett et al. 2006, Dunah et al. 2004). Recently, Gao and Wolf (2008) reported that activation of D1R by SKF81297 increased NR1 and NR2B, but not NR2A surface expression in rat PFC neuronal culture. Consistent with this report, our results shown in Fig. 4A and Fig. 4B further demonstrate that DHX increases the association of PSD95 with NR1 and NR2B, but not NR2A subunits in postsynaptic sites. This association was blocked by SCH23390, suggesting that it is dependent on D1R activation. Additionally, inhibition of PKA by PKI14–22 or Src tyrosine kinase by PP2 also decreased the formation of both NR1 and NR2B complexes with PSD95 (Fig. 4B).
Figure 4. D1R activation increases the association of NR1 and NR2B subunits with PSD95 and increases NMDAR cell surface expression.
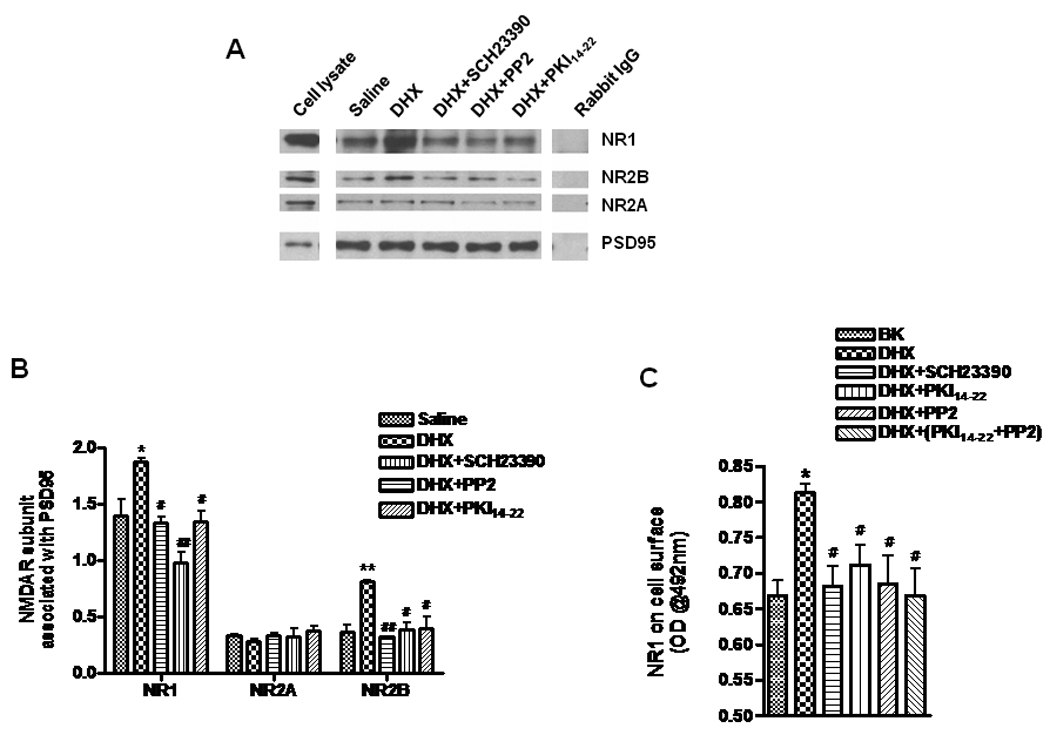
A. The cell lysate lane was loaded with solubilized membrane protein (20 µg) prepared from cells used for immunoprecipitation. Membrane protein (200 µg) from cells treated as indicated were incubated with 1 µg antibody against PSD95 or rabbit IgG overnight at 4 °C. The blots were probed sequentially, after stripping, with antibodies against NR1, NR2A, NR2B, and PSD95 on the same membrane. B. Quantification of results from 3 experiments, as indicated in panel A, show the association of PSD95 with NR1, NR2A and NR2B (left, middle and right group of columns, respectively). C. Activation of D1R also enhances NR1 subunit cell surface expression. Cells were treated with 10 µM of DHX and a combination of either 10 µM SCH23390, 1 µM PKI14–22 or 1µM PP2 or both 1 µM PKI14–22 and 1 µM PP2. After treatment, the cells were labeled with a specific antibody against NR1 (clone 54.1) and the receptor cell surface ELISA assay was performed. Values shown are mean ± SEM (* and # P < 0.05, ** and ## P < 0.01, * or ** vs saline treated, # and ## vs DHX treated, one-way ANOVA and Newman-Keuls posthoc analysis, N = 3/group).
It has been reported that activation of PKC and PKA regulate NMDA receptor trafficking by phosphorylating NR1 and promoting export from the endoplasmic reticulum (ER) as well as delivery of NMDAR to the plasma membrane (Scott et al. 2001). As the NR1 subunit is the obligatory subunit of functional NMDA receptors, we examined the expression of the NR1 subunit on the cell surface using the Cell Surface ELISA assay. Stimulation of D1R by DHX for 24 hrs significantly increased the amount of NR1 subunit protein on the cell surface. This was blocked by either PKI14–22 or PP2, but the combination of PKI14–22 and PP2 had no additive effect, implying that either PKA or tyrosine kinase mediated phosphorylation is sufficient for receptor export from the ER (Fig 4C).
To further examine the effects of DHX stimulation on synaptic strength, we measured the NMDA receptor – mediated synaptic response in PFC slices following DHX stimulation. As shown in figure 5A, DHX increases excitatory postsynaptic currents (EPSCs) in PFC neurons through activation of SCH23390-sensitive D1R. This effect was blocked by KT5720, a selective PKA inhibitor (Fig. 5B). Next, the relative role of NR2A and NR2B receptor subunits in the D1R-mediated increase in the synaptic response measured by recording the EPSCs following pretreatment of PFC slices with either PEAQX or Ro25-6981, selective inhibitors of NR2A and 2B, respectively. The NR2B antagonist, Ro25-6981 (1µM), essentially completely blocked the enhanced response due to D1R stimulation (Fig. 5C). On the other hand, pretreatment with the NR2A-selective drug, PEAQX, was without effect on the enhanced synaptic response following DHX pretreatment (Fig 5D). The relevance of this selective D1R-mediated enhancement of the NR2B component of the synaptic response by DHX is shown in Fig. 5E. Here, the protection against PCP neurotoxicity by D1R activation was abolished in the presence of the NR2B-selective antagonist, Ro25-6981, but the NR2A-selective antagonist PEAQX had no effect.
Figure 5. Facilitation of NMDA receptor-mediated synaptic transmission by dihydrexidine (DHX) is occluded by blocking NR2B, but not by an NR2A antagonist.
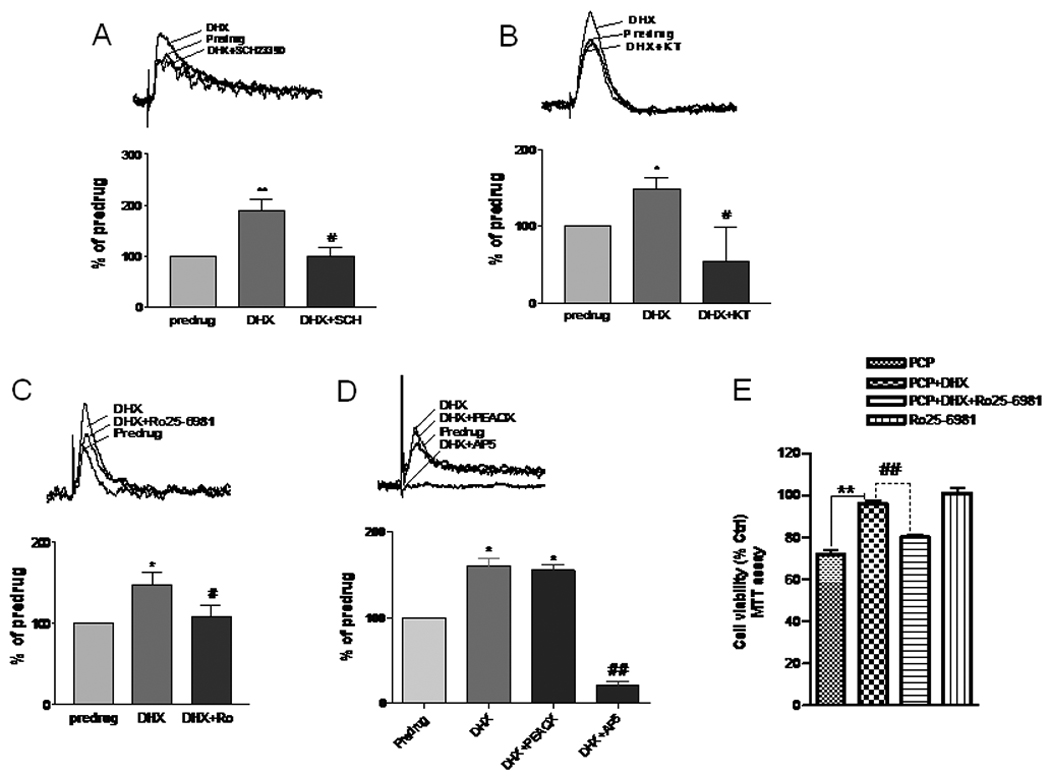
Whole-cell patch clamp recordings of layer V prefrontal cortical (PFC) neurons were made in brain slices from postnatal day 6–8 rats. The NMDA component of synaptic EPSCs (average of 8–10 traces) was recorded in the PFC neurons of brain slices dissected from postnatal day 6–8 rat pups (N=10) at +40 mV holding potential in the presence of CNQX (20 µM) to block AMPA-receptor dependent current. A. A D1 receptor agonist DHX (1 µM) facilitated NMDA receptor-mediated EPSCs (N=10). A selective D1R antagonist, SCH23390 (10 µM), inhibited the DHX-induced NMDA receptor-mediated synaptic component increase (N=2/group). B. A selective membrane-permeable PKA inhibitor (KT5720, 1µM) reduced the NMDA receptor mediated EPSCs and occluded the effect of DHX (N=2/group). C. A selective NR2B subunit antagonist, Ro25–6981 (1 µM), robustly diminished the potentiation of the NMDA receptor mediated EPSC by DHX (N=3/group). D. PEAQX (1 µM), a relatively selective NR2A subunit antagonist, failed to reduce the NMDA receptor mediated EPSCs or occlude the effect of DHX (N=3/group), but the EPSCs were completely inhibited by subsequent application of 30 µM AP5, an NMDA receptor antagonist. E. Cells were treated with DHX (10 µM) followed by PCP (1 µM) for 48h in the presence or absence of Ro 25–6981(1 µM), then assayed for cell viability. RO25–6981 significantly reduced the neuroprotective effect of DHX (N=3/group). *P < 0.05, ** P < 0.01 vs. pre-drug or PCP groups, #P < 0.05, ## P < 0.01 vs. DHX treatment (ANOVA followed by Newman-Keuls post-hoc analysis).
D1R activation increases signaling downstream of the NMDA receptor
Akt (PKB) is a predominant downstream molecule regulated by synaptic NMDA receptors. Activation of synaptic NMDA receptors facilitates Akt activity though phosphorylation of Ser473 on Akt (pAktSer473) with the consequence of activating transcription and translation of prosurvival genes such as CREB and BDNF, thereby promoting neuronal survival (Soriano et al. 2006). Since we previously demonstrated that PCP caused dephosphorylation and inactivation of the PI-3K/Akt survival pathway (Lei et al. 2008), we anticipated that the protection afforded by D1R activation may result from increasing Akt activity by phosphorylation of Ser473. Fig. 6A shows that DHX increases pAktSer473, but not total Akt. Glycogen synthase kinase 3β (GSK3β) is a prominent substrate for Akt, which phosphorylates GSK3β on Ser9 (pGSK3βSer9), resulting in inhibition of GSK3β and promoting neuronal survival. Fig. 6B shows that PCP decreased pGSK3βSer9, but DHX, by activation of D1R, reversed this reduction. It was further demonstrated (Fig. 6C and Fig. 6D) that inhibition of either PKA by PKI14–22 or tyrosine kinase by PP2 abolished the increased phosphorylation of pAktSer473 and pGSK3βSer9 caused by D1R activation. This suggests that phosphorylation of both the NR1 subunit by PKA and the NR2B subunit by tyrosine phosphorylation contribute to the protection against PCP-induced neurotoxicity by D1R activation.
Figure 6. D1R activation enhances Akt and GSK-3β phosphorylation.
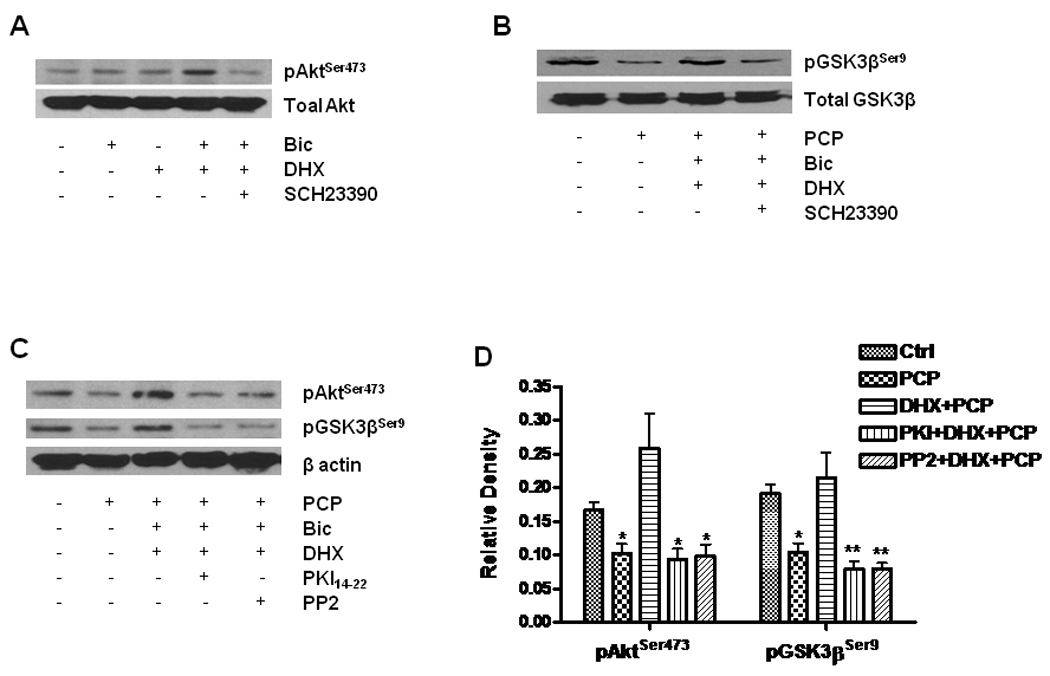
Cultured neurons at DIV14 were treated with 10 µM DHX in the presence or absence of 1 µM SCH23390 for 6h. The treated cells were lysed with RIPA buffer. Equal amounts of protein (20 µg) were loaded in each lane for WB. A. Effect of DHX on phosphorylation of Akt. Total Akt and phosphorylated Akt were determined by WB with specific antibodies against pAktSer473 and Akt in the same membrane after stripping. B. DHX reverses the reduction of pGSK-3β induced by PCP. WB shows alterations of pGSK3β and total GSK3β by probing with specific antibodies against pGSK-3βSer9 and total GSK-3β. C. The role of PKA and tyrosine kinase activation in D1R-mediated phosphorylation of Akt and GSK-3β. The cells were pretreated with 1 µM PKI14–22 or 1 µM PP2 for 30 min and then exposed to 1 µM PCP in the presence or absence of 10 µM DHX for 24h. D. Quantification of β-actin-normalized data from 3 different experiments done as shown in panel C. Values shown are mean ± SE (* p< 0.05, ** p< 0.01, one-way ANOVA, Newman-Keuls post-hoc analysis, N = 3/group).
In vivo effects of D1R activation on PCP-induced neurotoxicity and deficits in prepulse inhibition of acoustic startle
Caspase-3 activation has been reported to be a cell death marker in vivo and in vitro for neurotoxicity caused by PCP and other noncompetitive NMDA receptor antagonists (Monti & Contestabile 2000, Adams et al. 2004, Wang & Johnson 2007). Therefore, we examined the effect of DHX on caspase-3 activation in the cortex of rat pups treated with DHX (0.3 – 30 mg/kg) 30 min prior to PCP (10 mg/kg, s.c.) on postnatal day (PN) 7, 9 and11. DHX inhibited PCP-induced caspase-3 activity in a dosedependent manner (data not shown) and at 3 mg/kg DHX essentially completely inhibited PCP-induced caspase-3 activation in rat pups. This effect was specific for D1R, as it was completely inhibited by a 30 min pretreatment with 3 mg/kg SCH23390, a specific D1R antagonist. This laboratory has previously proposed that PCP-induced neurotoxicity in rat pups gives rise to abnormal behaviors later in life that may model certain aspects of schizophrenia (Anastasio & Johnson 2008, Wang et al. 2001). Therefore, we determined whether DHX had any effect on abnormal behavior induced by PCP. While deficits in pre-pulse inhibition (PPI) of acoustic startle are routinely observed in schizophrenia, this deficit is not considered to be a specific hallmark of schizophrenia, as it is also observed in other disease states (Braff et al. 2001). Importantly, PPI deficits are significantly more responsive to treatment with atypical antipsychotics compared to typical drugs such as haloperidol. Since deficits in PPI are observed in this PCP model (Anastasio & Johnson 2008), PPI was used here to test whether our observations in vitro with DHX could be extended to an in vivo paradigm. No effect on startle amplitude was measured in any of the treatment groups (data not shown). However, as shown in Fig.7C, PCP treatment (10 mg/kg) on PN7, 9 and 11 results in an approximate 50% deficit in pre-pulse inhibition (PPI) of the acoustic startle reflex measured on PN26-28 (F4, 29 = 33.03). Pretreatment with 3 mg/kg DHX 30 min prior to PCP on PN7, 9 and 11 protects against deficits in PPI caused by PCP.
Figure 7. Dihydrexidine inhibits caspase-3 activation in vivo and reverses deficits in prepulse inhibition of acoustic startle.
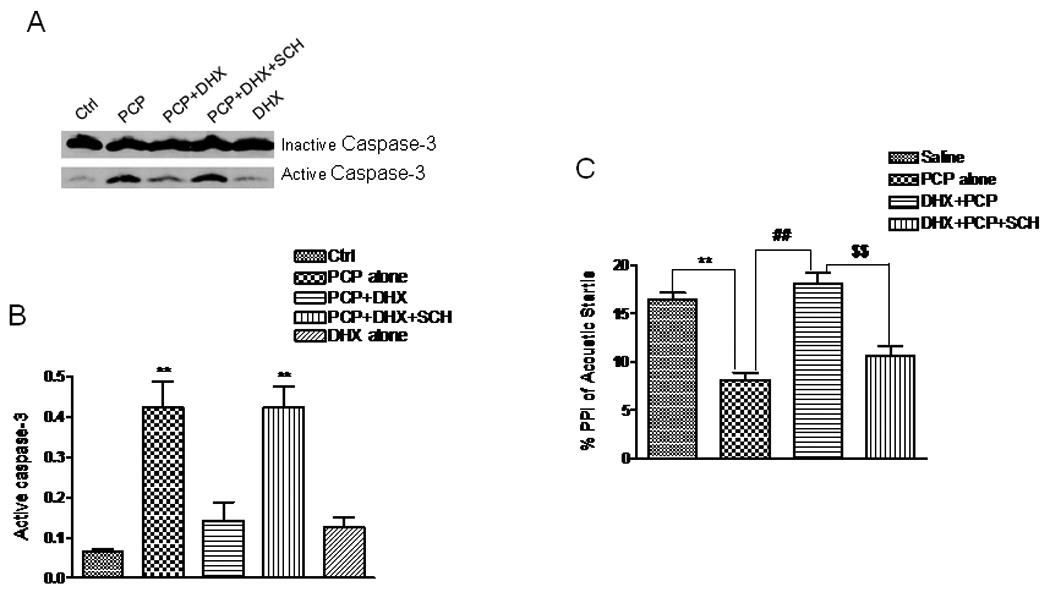
Rat pups on PN7, 9, 11 were treated with PCP (10 mg/kg, i.p.) or saline. DHX (3 mg/kg, s.c.) or SCH23390 (1 mg/kg, s.c.) was administered 30 min prior to PCP or saline either alone or in combination. Pups were sacrificed by decapitation 8 hr following the treatment. Tissue from the frontal cortex was dissected and homogenized in ice-cold RIPA buffer containing proteinase inhibitors. A. Representative western blots showing procaspase-3 (∼32 kDa) and the active caspase-3 fragment (17 kDa) from the control and treated rat pups. B. Activated caspase-3 normalized as its ratio to procaspase-3 in the four treatment groups. The quantified WB data are from 4 separate experiments. C. The effect of DHX (with or without pretreatment with SCH23390) on PCP-induced (PN7, 9 and 11) deficits in prepulse inhibition (PPI) of acoustic startle. Following the indicated drug treatment on PN7, 9, 11, PPI of acoustic startle was measured on PN26–28. Values shown are mean ± SEM (** P< 0.01 vs saline or control, ## P< 0.01 vs PCP treatment, $$ P< 0.01 vs DHX+PCP group, one-way ANOVA with Newman-Keuls post hoc test, N = 4 for (B), N= 5–7/group for (C).
Discussion
This study demonstrated that activation of D1 receptors in the cortex protects against PCP-induced neurotoxicity in developing rat pups by enhancing synaptic strength through synergistic activation of at least two signaling pathways. Stimulation of D1R by DHX increases phosphorylation of the NR1 subunit at Ser897 via PKA activation as well as phosphorylation of the NR2B subunit at Tyr1472 through the Src kinase family pathway. It was also demonstrated that D1R activation facilitates the recruitment of the NMDA NR1 and NR2B receptor subunits into cortical synapses, which also play a role in the protection by D1R activation by enhancing synaptic efficacy. The enhanced recruitment of NR1 and NR2B subunits into synaptic sites is associated with an elevated expression of NMDA receptors on the cell surface, as well as an increased synaptic response to electrical stimulation. Inhibition of either PKA with PKI14–22 or Src kinase with PP2 reduces the interaction between PSD95 and the NR1 and NR2B subunits, implying that following D1R activation, both pathways contribute to NMDA receptor trafficking and externalization. Further, pAktSer473 and pGSK3βSer9, which are both regulated by synaptic NMDAR, were significantly increased by D1R activation. Activation of the PI-3K/Akt/GSK3β pathway is known to promote neuronal cell survival. Finally, in vivo stimulation of D1R by dihydrexidine inhibits PCP-induced caspase-3 activation in the PFC and blunts the blockade of pre-pulse inhibition of acoustic startle caused by PCP treatment. Thus, these data demonstrate a novel mechanism for protection against PCP-induced neurotoxicity through activation of D1R, and provide further evidence that neurotoxicity induced by PCP may be linked to abnormal behaviors such as impaired prepulse inhibition of acoustic startle later in life. These data also suggest that this model may be useful in the discovery of novel therapeutic approaches to schizophrenia.
Stimulation of D1R in the striatum increases cytosolic cAMP levels and PKA activity, leading to phosphorylation-activation of dopamine receptor-activated phosphoprotein 32kDa (Greengard 2001, Neve et al. 2004). Further, D1R-mediated modulation of intrinsic excitability and glutamatergic signaling has been observed (Snyder et al. 1998, Flores-Hernandez et al. 2002), though the mechanism of D1R regulation of NMDA receptor function in the PFC is uncertain. Chen et al.(2004) reported that D1R-mediated enhancement of NMDAR function in acutely isolated PFC pyramidal neurons was independent of PKA or protein phosphatase 1 (PP1), but that inhibition of protein kinase C (PKC) or calmodulin activity significantly diminished the effect of D1R activation of NMDA receptor mediated current. In opposition to this, the current study demonstrated that neither PKC nor CaMKII inhibition affected the ability of D1R activation to protect against PCP-induced neurotoxicity. Further, the experiments reported here show that blockade of PKA activity with selective inhibitors reversed the protective effect of D1R activation, as well as the phosphorylation of PKA at Thr197. Moreover, stimulation of D1R diminished the reduction of pNR1Ser897 induced by PCP, which is consistent with the result that SKF81297, a D1R agonist, increased pNR1Ser897 phosphorylation in PCP-treated mice (Mouri et al. 2007). All of these results support a dominant role for PKA, but not PKC, in the action of D1 receptor activation in this context. The reason for the discrepancy between these results and those of Chen et al (2004) is unclear, but the differential location of the stimulated NMDA receptors may be an explanation. That is, Chen et al. (2004) stimulated NMDA receptors in cultured neurons by bath application of 500 µM NMDA, a concentration that likely activates both synaptic and extrasynaptic NMDAR, while the current experiments used a low concentration of bicuculline to diminish GABAergic inhibition, which is known to facilitate synaptic glutamate release and likely stimulates only synaptic NMDAR (Hardingham et al. 2002, Ivanov et al. 2006). Furthermore, the proposal that PKA activation contributes to D1R-mediated increases in synaptic NMDAR in PFC has been demonstrated by others (Gonzalez-Islas & Hablitz 2003, Tseng & O'Donnell 2004), though Gao and Wolf (2008) observed no effect of presumptive activation of PKA with SpcAMPS on NR1 surface expression.
Phosphorylation is a major mechanism for regulation of NMDAR function. D1R activation of accumbens medium spiny neurons increases NR1 phosphorylation in a PKA-dependent manner (Snyder et al. 1998). Tingley et al. (1997) demonstrated that PKA can phosphorylate NR1Ser897. This study demonstrated that phosphorylation of NR1Ser897 was elevated by DHX in PFC neurons. Inhibition of PKA by PKI14–22 or H89 decreased phosphorylation of NR1Ser897 and prevented the protective effects of DHX against PCP-induced neurotoxicity, suggesting that phosphorylation of NR1Ser897 improves NMDAR function even in the face of blockade by PCP. Reduction of pNR1Ser897 in PFC cultures by PCP is similar to that observed in schizophrenia. Emamian et al. (2004) reported a significant decrease of pNR1Ser897 in brains from patients with schizophrenia, and an increase in pNR1Ser897 in those treated with antipsychotics. In striatal tissue from DARPP-32-depleted mice, basal tyrosine and serine phosphorylation of striatal NMDA receptor subunits NR1, NR2A, and NR2B was normal, and dopamine D1 receptor activation has been demonstrated to redistribute NMDA receptors from vesicular compartments to synaptosomal membranes by a mechanism that requires Fyn kinase (Dunah et al. 2004). In addition, Hallett et al (2006) reported that treatment of striatal neurons with a D1R agonist increased phosphorylation of pNR2BTyr1472. Tyr1472 is a substrate for Fyn tyrosine kinase, and is the predominant site for phosphorylation of NR2B (Suzuki & Okumura-Noji 1995). While the present study was in progress, it was reported that D1 receptor stimulation resulted in tyrosine kinase-dependent phosphorylation of pNR2BTyr1472 (Gao & Wolf 2008). The present study confirms that D1R activation of significantly increased pNR2BTyr1472 in PFC neurons. Additionally, we demonstrated that blocking tyrosine kinase with lavendustin A or PP2 inhibited the protective effect of DHX against PCP-induced neurotoxicity. This further supports the hypothesis that tyrosine phosphorylation of the NR2B subunit of NMDAR also plays a role in the D1R-mediated neuroprotection against PCP neurotoxicity.
In addition to increasing the association of NR1 and NR2B subunits with PSD95, D1R activation may facilitate synaptic function by increasing NMDA receptor trafficking through the tyrosine phosphorylation-dependent pathway or by enhancing AMPA receptor synaptic insertion (Sun et al. 2005, Hallett et al. 2006). The latter interaction is modulated by activation of both serine/threonine kinase (PKA) and protein tyrosine kinases, particularly Fyn kinase, as inhibition of either prevented the effect of DHX on the interaction of NR2B and NR1 with PSD95, and NMDA receptor externalization. Hallett et al (2006) demonstrated that stimulation of striatal D1R leads to rapid trafficking of NR2B and NR1 subunits in dendrites as well as co-clustering of these subunits with PSD95 in a tyrosine kinase dependent manner. However, it is less clear why D1R activation has no effect on NR2A trafficking to the PSD. The NR2A subunit has been well documented to be located predominantly in synapses (in adults), while NR2B is preferentially located extrasynaptically (Cull-Candy & Leszkiewicz 2004, Groc et al. 2007). Thus, D1R activation may increase NR2B trafficking to synaptic sites through lateral diffusion from extrasynaptic sites. In contrast, the extrasynaptic pool of NR2A receptors may be more limited and thus, this mechanism of compensation would be less effective. A similar lack of effect of D1R stimulation on NR2A trafficking into the synapse has been recently reported (Gao & Wolf 2008). It should also be noted that the mechanism described above for D1 receptor-mediated neuroprotection against NMDA receptor blockade is quite distinct from the mechanism described for D1-receptor-mediated protection against NMDA-induced excitotoxicity (Lee et al. 2002). That mechanism involved a decrease in the association between NR1-1a and NR2A subunits with the D1 receptor via activation of the PI-3 kinase pathway. Therefore, it appears that D1 receptor activation plays an important role in tuning the function of NMDA receptors by bidirectional modulation of the association of either NR2A or 2B subunits with NR1.
Although deficient sensorimotor gating, i.e. the inability to filter competing stimuli correctly, is not specific for schizophrenia (Braff et al. 2001), decreased prepulse inhibition (PPI) of acoustic startle is characteristic of schizophrenia, and this deficit may underlie sensory flooding and cognitive fragmentation (McGhie & Chapman 1961). Unlike many other symptoms of schizophrenia, this particular deficit can be closely modeled in rodents by PPI of the acoustic startle response. It has been suggested that the regulation of PPI, as well as the “negative symptoms” of schizophrenia, is due to a functional deficit in dopamine neurotransmission in the PFC (Carlsson et al. 2001, de Jong & van den Buuse 2006) as well as an alteration in glutamatergic projections from the PFC to the nucleus accumbens (Bortolato et al. 2005). The role of dopamine receptor involvement in the regulation of PPI is complex and may depend on the stage of development (Curzon & Decker 1998, Geyer et al. 2001). In this study DHX prevented PCP-induced PPI deficits and this effect was blocked by the D1R antagonist, SCH23390, thereby suggesting that the D1R is also crucial to this regulation. Interestingly, these results contrast with the observation that the partial D1R agonist, SKF38393, potentiated MK801-induced disruption of PPI in adult rats (Bortolato et al. 2005). The reason for this apparent discrepancy is not known, but it may be related to the different stages of brain development or the different levels of D1 receptor occupancy in these two studies. Nevertheless, the current study shows that activation of D1R by a full agonist at this early stage of development is able to protect against the neurotoxic effects of PCP and to ameliorate the detrimental behavioral effects of PCP on PPI.
Previous studies from this laboratory have shown that PCP-induced neurotoxicity in vitro could be prevented by either inhibition of caspase-3 activation (Wang & Johnson 2007), facilitation of presynaptic glutamate release or stimulation of L-type Ca2+ channels (Lei et al. 2008). In this study, another mechanism for blockade of PCP-induced neurotoxicity was demonstrated. Stimulation of D1R provides neuroprotection associated with enhanced synaptic strength. DHX stimulation of D1R also promotes phosphorylation of NR1 and NR2B subunits and increases recruitment of NR1 and NR2B subunits into the synapse. This, in turn, is postulated to enhance synaptic NMDA receptor downstream signaling, particularly through activation of the PI3-K-Akt pathway, which could then enhance neuronal survival (Lei et al. 2008). Alternatively, the increased recruitment of NR1 and NR2B observed here could also be linked to other protective mechanisms such as that recently proposed by Papadia et al. (2008), who provided evidence that NMDA receptor stimulation can also increase antioxidant defenses by making changes to the thioredoxin-peroxiredoxin system. That study indicated that synaptic activity enhanced thioredoxin activity, facilitated the reduction of over-oxidized peroxiredoxins and promoted resistance to oxidative stress. Thus, multiple mechanisms could be involved in the ultimate neuroprotective effects of increased synaptic activity. Taken together, the present results demonstrate that D1R agonists are able to both prevent the neurotoxic effects of perinatal PCP administration as well as the development of subsequent behavioral deficits. To the extent that PCP-induced neurotoxicity and behavioral deficits model schizophrenia, these data suggest that further understanding of the mechanisms responsible for mitigating the effects of PCP-induced neurotoxicity may be useful in the discovery of novel strategies for the treatment of this disease.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH grants RO1 DA-02073 and F31 DA-022831.
Abbreviations
- (PCP)
phencyclidine
- (NMDAR)
N-Methyl-D-Aspartate Receptor
- (DHX)
dihydrexidine
- (PFC)
prefrontal cortex
- (D1R)
dopamine D1 receptors
- (PKA)
protein kinase A
- (NR1, NR2A, NR2B)
NMDAR subunits
- (EPSCs)
excitatory postsynaptic currents
- (PN)
postnatal
- (PP2)
3-(4-chlorophenyl) 1 — (1,1-dimethylethyl) — 1 H-pyrazolo [3,4-d] pyrimidin-4-amine
- (SCH23390)
dihydrexidine ((±) —trans-10,11- dihydroxy -5,6,6a,7,8,12b-hexahydrobenzo[a] phenanthridine hydrochloride)
- (AP5)
DL-2-amino-5-phosphonopentanoic acid
- (CNQX)
6-Cyano-7-nitroquinoxaline-2,3-dione disodium
- (MTT)
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide
- (PSD)
postsynaptic density
- (ACSF)
artificial cerebrospinal fluid
- (BDNF)
brain derived neuronal growth factor
- (GSK)
glycogen synthase kinase
- (BIS)
bisindolylmaleimide I
- (DIV)
days in vitro
REFERENCES
- Adams SM, de Rivero Vaccari JC, Corriveau RA. Pronounced cell death in the absence of NMDA receptors in the developing somatosensory thalamus. J Neurosci. 2004;24:9441–9450. doi: 10.1523/JNEUROSCI.3290-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasio NC, Johnson KM. Atypical anti-schizophrenic drugs prevent changes in cortical N-methyl-D-aspartate receptors and behavior following sub-chronic phencyclidine administration in developing rat pups. Pharmacol Biochem Behav. 2008;90:569–577. doi: 10.1016/j.pbb.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Gonzalez-Islas C, Hablitz JJ. Dopamine enhances spatiotemporal spread of activity in rat prefrontal cortex. J Neurophysiol. 2005;93:864–872. doi: 10.1152/jn.00922.2004. [DOI] [PubMed] [Google Scholar]
- Bortolato M, Aru GN, Fa M, et al. Activation of D1, but not D2 receptors potentiates dizocilpine-mediated disruption of prepulse inhibition of the startle. Neuropsychopharmacology. 2005;30:561–574. doi: 10.1038/sj.npp.1300547. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Braithwaite SP, Adkisson M, Leung J, Nava A, Masterson B, Urfer R, Oksenberg D, Nikolich K. Regulation of NMDA receptor trafficking and function by striatal-enriched tyrosine phosphatase (STEP) Eur J Neurosci. 2006;23:2847–2856. doi: 10.1111/j.1460-9568.2006.04837.x. [DOI] [PubMed] [Google Scholar]
- Broberg BV, Dias R, Glenthoj BY, Olsen CK. Evaluation of a neurodevelopmental model of schizophrenia--early postnatal PCP treatment in attentional set-shifting. Behav Brain Res. 2008;190:160–163. doi: 10.1016/j.bbr.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML. Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol. 2001;41:237–260. doi: 10.1146/annurev.pharmtox.41.1.237. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci U S A. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Greengard P, Yan Z. Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101:2596–2600. doi: 10.1073/pnas.0308618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Curzon P, Decker MW. Effects of phencyclidine (PCP) and (+)MK-801 on sensorimotor gating in CD-1 mice. Prog Neuropsychopharmacol Biol Psychiatry. 1998;22:129–146. doi: 10.1016/s0278-5846(97)00184-x. [DOI] [PubMed] [Google Scholar]
- de Jong IE, van den Buuse M. SCH 23390 in the prefrontal cortex enhances the effect of apomorphine on prepulse inhibition of rats. Neuropharmacology. 2006;51:438–446. doi: 10.1016/j.neuropharm.2006.04.002. [DOI] [PubMed] [Google Scholar]
- du Bois TM, Huang XF. Early brain development disruption from NMDA receptor hypofunction: relevance to schizophrenia. Brain Res Rev. 2007;53:260–270. doi: 10.1016/j.brainresrev.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Sirianni AC, Fienberg AA, Bastia E, Schwarzschild MA, Standaert DG. Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol Pharmacol. 2004;65:121–129. doi: 10.1124/mol.65.1.121. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Standaert DG. Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J Neurosci. 2001;21:5546–5558. doi: 10.1523/JNEUROSCI.21-15-05546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan GE, Moy SS, Lieberman JA, Koller BH. Typical and atypical antipsychotic drug effects on locomotor hyperactivity and deficits in sensorimotor gating in a genetic model of NMDA receptor hypofunction. Pharmacol Biochem Behav. 2006;85:481–491. doi: 10.1016/j.pbb.2006.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvevag B, Goldberg TE. Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol. 2000;14:1–21. [PubMed] [Google Scholar]
- Emamian ES, Karayiorgou M, Gogos JA. Decreased phosphorylation of NMDA receptor type 1 at serine 897 in brains of patients with Schizophrenia. J Neurosci. 2004;24:1561–1564. doi: 10.1523/JNEUROSCI.4650-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Hernandez J, Cepeda C, Hernandez-Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P, Levine MS. Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: role of D1 receptors and DARPP-32. J Neurophysiol. 2002;88:3010–3020. doi: 10.1152/jn.00361.2002. [DOI] [PubMed] [Google Scholar]
- Gao C, Wolf ME. Dopamine receptors regulate NMDA receptor surface expression in prefrontal cortex neurons. J Neurochem. 2008;106:2489–2501. doi: 10.1111/j.1471-4159.2008.05597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 2001;156:117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Hablitz JJ. Dopamine enhances EPSCs in layer II-III pyramidal neurons in rat prefrontal cortex. J Neurosci. 2003;23:867–875. doi: 10.1523/JNEUROSCI.23-03-00867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P. The neurobiology of dopamine signaling. Biosci Rep. 2001;21:247–269. doi: 10.1023/a:1013205230142. [DOI] [PubMed] [Google Scholar]
- Groc L, Choquet D, Stephenson FA, Verrier D, Manzoni OJ, Chavis P. NMDA receptor surface trafficking and synaptic subunit composition are developmentally regulated by the extracellular matrix protein Reelin. J Neurosci. 2007;27:10165–10175. doi: 10.1523/JNEUROSCI.1772-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, Spoelgen R, Hyman BT, Standaert DG, Dunah AW. Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking. J Neurosci. 2006;26:4690–4700. doi: 10.1523/JNEUROSCI.0792-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Ermilov M, Giltsinsky B, Lichtenstein M, Blander D. Treatment-resistant schizophrenia and staff rejection. Schizophr Bull. 1999;25:457–465. doi: 10.1093/oxfordjournals.schbul.a033393. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC. Management of negative symptoms of schizophrenia. Curr Psychiatry Rep. 2001;3:413–417. doi: 10.1007/s11920-996-0036-9. [DOI] [PubMed] [Google Scholar]
- Kargieman L, Santana N, Mengod G, Celada P, Artigas F. Antipsychotic drugs reverse the disruption in prefrontal cortex function produced by NMDA receptor blockade with phencyclidine. Proc Natl Acad Sci U S A. 2007;104:14843–14848. doi: 10.1073/pnas.0704848104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FJ, Xue S, Pei L, et al. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell. 2002;111:219–230. doi: 10.1016/s0092-8674(02)00962-5. [DOI] [PubMed] [Google Scholar]
- Lei G, Xia Y, Johnson KM. The role of Akt-GSK-3beta signaling and synaptic strength in phencyclidine-induced neurodegeneration. Neuropsychopharmacology. 2008;33:1343–1353. doi: 10.1038/sj.npp.1301511. [DOI] [PubMed] [Google Scholar]
- McGhie A, Chapman J. Disorders of attention and perception in early schizophrenia. Br J Med Psychol. 1961;34:103–116. doi: 10.1111/j.2044-8341.1961.tb00936.x. [DOI] [PubMed] [Google Scholar]
- Monti B, Contestabile A. Blockade of the NMDA receptor increases developmental apoptotic elimination of granule neurons and activates caspases in the rat cerebellum. Eur J Neurosci. 2000;12:3117–3123. doi: 10.1046/j.1460-9568.2000.00189.x. [DOI] [PubMed] [Google Scholar]
- Mouri A, Noda Y, Noda A, Nakamura T, Tokura T, Yura Y, Nitta A, Furukawa H, Nabeshima T. Involvement of a dysfunctional dopamine-D1/N-methyl-d-aspartate-NR1 and Ca2+/calmodulin-dependent protein kinase II pathway in the impairment of latent learning in a model of schizophrenia induced by phencyclidine. Mol Pharmacol. 2007;71:1598–1609. doi: 10.1124/mol.106.032961. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, Yamamoto T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2001;276:693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Olney JW, Farber NB. NMDA antagonists as neurotherapeutic drugs, psychotogens, neurotoxins, and research tools for studying schizophrenia. Neuropsychopharmacology. 1995;13:335–345. doi: 10.1016/0893-133X(95)00079-S. [DOI] [PubMed] [Google Scholar]
- Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Leveille F, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Swanson GT, Zhang C, Ehlers MD. An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci. 2001;21:3063–3072. doi: 10.1523/JNEUROSCI.21-09-03063.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci U S A. 2001;98:301–306. doi: 10.1073/pnas.011518798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim SS, Hammonds MD, Kee BS. Potentiation of the NMDA receptor in the treatment of schizophrenia: focused on the glycine site. Eur Arch Psychiatry Clin Neurosci. 2008;258:16–27. doi: 10.1007/s00406-007-0757-8. [DOI] [PubMed] [Google Scholar]
- Slikker W, Jr, Zou X, Hotchkiss CE, et al. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol Sci. 2007;98:145–158. doi: 10.1093/toxsci/kfm084. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine-and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297–10303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci. 2006;26:4509–4518. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Okumura-Noji K. NMDA receptor subunits epsilon 1 (NR2A) and epsilon 2 (NR2B) are substrates for Fyn in the postsynaptic density fraction isolated from the rat brain. Biochem Biophys Res Commun. 1995;216:582–588. doi: 10.1006/bbrc.1995.2662. [DOI] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–5166. doi: 10.1074/jbc.272.8.5157. [DOI] [PubMed] [Google Scholar]
- Tseng KY, O'Donnell P. Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci. 2004;24:5131–5139. doi: 10.1523/JNEUROSCI.1021-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, McInnis J, Ross-Sanchez M, Shinnick-Gallagher P, Wiley JL, Johnson KM. Long-term behavioral and neurodegenerative effects of perinatal phencyclidine administration: implications for schizophrenia. Neuroscience. 2001;107:535–550. doi: 10.1016/s0306-4522(01)00384-0. [DOI] [PubMed] [Google Scholar]
- Wang CZ, Johnson KM. The role of caspase-3 activation in phencyclidine-induced neuronal death in postnatal rats. Neuropsychopharmacology. 2007;32:1178–1194. doi: 10.1038/sj.npp.1301202. [DOI] [PubMed] [Google Scholar]
- Williams GV, Goldman-Rakic PS. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature. 1995;376:572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- Yang CR, Chen L. Targeting prefrontal cortical dopamine D1 and N-methyl-D-aspartate receptor interactions in schizophrenia treatment. Neuroscientist. 2005;11:452–470. doi: 10.1177/1073858405279692. [DOI] [PubMed] [Google Scholar]
- Yang SN. Sustained enhancement of AMPA receptor-and NMDA receptor-mediated currents induced by dopamine D1/D5 receptor activation in the hippocampus: an essential role of postsynaptic Ca2+ Hippocampus. 2000;10:57–63. doi: 10.1002/(SICI)1098-1063(2000)10:1<57::AID-HIPO6>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.