

Abstract
Disrupting the CD40-CD40L costimulation pathway promotes allograft acceptance in many settings. Herein, we demonstrate that stimulating OX40 overrides cardiac allograft acceptance induced by disrupting CD40-CD40L interactions. This effect of OX40 stimulation was dependent upon CD4+ T cells, which in turn provided help for CD8+ T cells and B cells. Allograft rejection was associated with donor-reactive Th1 and Th2 responses and an unconventional granulocytic infiltrate and thrombosis of the arteries. Interestingly, OX40 stimulation induced a donor-reactive IgG class switch in the absence of CD40-CD40L interactions, and the timing of OX40 stimulation relative to transplantation affected the isotype of donor-reactive antibody produced. Inductive OX40 stimulation induced acute graft rejection, which correlated with both IgG1 and IgG2a deposition within the graft. Once graft acceptance was established following CD40-CD40L blockade, delayed OX40 stimulation did not induce acute allograft rejection despite priming of graft-reactive Th1 and Th2. Rather, chronic rejection was induced, which was characterized by IgG1 but not IgG2a deposition within the graft. These studies reveal both redundancy and key differences in function among costimulatory molecules that manifest in distinct pathologies of allograft rejection. These findings may help guide development of therapeutics aimed at promoting graft acceptance in transplant recipients.
Keywords: costimulation, T cells, Th1/Th2 cells, transplantation
Introduction
Costimulation is central to T cell activation, survival, and effector differentiation (reviewed in (1-3)). Unlike CD28, CD40L and OX40 are expressed almost exclusively by activated CD4+ T cells and to a lesser degree by activated CD8+ T cells (reviewed in (2, 3)). These molecules interact with CD40 and OX40 ligand (OX40L3) on the APC, respectively (reviewed in (1)). Stimulation through OX40 induces expansion of T cells activated by T cell receptor engagement (4, 5). Whereas CD40-CD40L interactions support a Th1 response by inducing APC to produce IL-12 (reviewed in (6, 7)), OX40 signaling promotes Th2 responses (8, 9) or may paradoxically further drive the development of a Th1 response (5, 10, 11).
Disrupting CD40-CD40L interactions results in prolonged allograft survival (reviewed in (2, 12)),(13-17). However, there are differences in the requirement for CD40-CD40L interactions among different strains of mice and graft protection resulting from CD40-CD40L inhibition is in large part dictated by the model in which it is studied. For example, whereas C57BL/6 mice that are deficient in CD40 (CD40-/-) fail to reject cardiac allografts from CD40-/- BALB/c donors, CD40-/- BALB/c recipients acutely reject CD40-/- C57BL/6 allografts (18). In C3H mice, blocking both CD28 and CD40L interactions results in long-term skin allograft survival (19, 20), whereas C57BL/6 mice deficient in both CD28 and CD40L acutely reject skin grafts (20, 21). However, blocking OX40-OX40L interactions in CD28 and CD40L deficient C57BL/6 recipients promotes long-term skin graft survival (21), and both CD4+ and CD8+ T cell mediated rejection is sensitive to this blockade (22).
Similarly, differences in costimulatory pathway usage and cytokine profiles have been observed in the model of Leishmania infection (8, 23). In response to infection, BALB/c mice mount a dominant Th2 response resulting in the “non-healer” phenotype and succumb to progressive lesions (8, 23). This Th2 response is driven by OX40-OX40L interactions (8). Conversely, C57BL/6 mice infected with Leishmania mount a dominant Th1 response, resulting in the “healer” phenotype and clearance of the pathogen (23). Indeed, when the BALB/c recipient response is skewed towards a Th1 response by neutralizing IL-4 (24), the infection is cleared and the mice recover. Additionally, if the immune response of C57BL/6 mice is skewed away from a Th1 response by genetic deficiency in IFNγ (25), T-bet (26), or CD40L (27), or by over expression of OX40L (28) the infection is not cleared and the mice respond similarly to their unmodified BALB/c counterparts. Interestingly, BALB/c mice infected with Leishmania are able to clear the disease (8) or significantly delay disease pathology (28) following OX40-OX40L blockade. Thus, preferential usage of the OX40-OX40L costimulatory pathway may influence Th1/Th2 balance and impact on the effectiveness of the immune response, but the exact relevance of this notion in transplant models is unknown.
The current study demonstrates that cardiac allograft acceptance resulting from disrupting CD40-CD40L interactions in C57BL/6 mice is prevented by OX40 stimulation. Acute rejection driven by OX40 stimulation was associated with donor-reactive T cell priming and the generation of a donor-reactive IgG antibody response, with IgG2a detectable within the graft. Once allograft acceptance was established, OX40 stimulation failed to induce acute rejection but promoted the progression of chronic rejection and deposition of IgG1 within the graft. These data suggest that despite the importance of CD40-CD40L engagement in the C57BL/6 recipient response to allografts, perturbation through an alternative costimulatory molecule pathway may supersede the necessity for these interactions in the progression of both acute and chronic rejection.
Materials and Methods
Mice
Female C57BL/6 (H-2b) mice, CD40L-/- C57BL/6 mice, and BALB/c (H-2d) mice were purchased from The Jackson Laboratories (Bar Harbor, ME). Breeder pairs of CD40-/-C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Breeder pairs of CD40-/- BALB/c mice were provided by Dr. Randy Noelle (Dartmouth College, Lebanon, NH). Colonies of CD40-/- mice were established, and all mice were housed under specific pathogen-free conditions maintained by the Unit for Laboratory Animal Medicine at University of Michigan. Mice used were between 6 and 12 weeks of age. These experiments were approved by the University Committee on Use and Care of Animals at the University of Michigan.
Culture medium
RPMI 1640 (for ELISPOT) or DMEM (for cell culture) was supplemented with 2% FCS, 1 mM sodium pyruvate, 100 U/mL penicillin, 100 μg/mL streptomycin, 1.6 mM L-glutamine, 10 mM HEPES buffer (all from Invitrogen, Grand Island, NY), 0.27 mM L-asparagine, 1.4 mM L-arginine HCl, 14 μM folic acid and 50 μM 2-mercaptoethanol (all from Sigma Chemicals, St. Louis, MO).
Flow cytometry for OX40
One million splenocytes per mL were cultured for times indicated with 1 μg/mL Concavalin A (Sigma). After separating viable cells by Ficol-Hypaque gradient, splenocytes were triple labeled with FITC-conjugated anti-OX40 mAb (OX86, Cedar Lane), PE-conjugated anti-CD4 mAb (GK1.5, Pharmingen), and Cy5-conjugated anti-CD8 mAb (2.43, Pharmingen). Cell analyses were performed on a Becton Dickinson FACSCalibur (San Jose, CA) using forward versus side scatter to gate on lymphocytes.
Heterotopic cardiac transplantation
C57BL/6 WT, CD40L-/-, or CD40-/- mice were transplanted with intact WT or CD40-/-BALB/c cardiac allografts, as described (29). In this model, the donor heart is anastomosed to the great vessels of the abdomen, perfused with the recipient mouse’s blood, and resumes contraction. Transplant function was monitored by abdominal palpation.
In vivo treatment with mAb
The hybridomas secreting anti-CD4 (clone GK1.5) and anti-CD8 (clone 2.43) were obtained from American Type Culture Collection (Manassas, VA). The hybridoma secreting anti-CD40L (clone MR1) was provided by Dr. Randy Noelle (Dartmouth, Lebanon, NH). Anti-CD4, anti-CD8, anti-CD40L, and anti-OX40 (clone OX86 (30)) mAb were purified and resuspended in PBS by Bio Express (West Lebanon, NH). To deplete CD4+ or CD8+ T cells, allograft recipients were injected i.p. with 1 mg of anti-CD4 or anti-CD8 mAb days -1, 0 and 7 relative to transplant (31-33). For inductive anti-CD40L therapy, mice were injected i.p. with 1 mg of anti-CD40L on days 0, 1 and 2 post transplant (14, 32). To induce OX40 stimulation, recipients were injected with 0.25 mg i.p. of anti-OX40 mAb on days 0, 1, 3 and 7 post transplant, a regimen similar to what has been published regarding islet transplant (34). To induce delayed OX40 stimulation, recipients were injected with 0.25 mg i.p. of anti-OX40 mAb on days 30, 31, 33 and 37 relative to transplant.
Histology
Allografts were recovered at the times indicated post transplantation, fixed in formalin, and embedded in paraffin. Sections were stained with hematoxylin and eosin (H&E) to assess myocyte viability (presence of cross striation and nuclei) and the nature and intensity of graft-infiltrating cells (GIC). As described (35), Masson trichrome stain was used to identify collagen deposition.
Recovery of GIC
Three transplanted hearts were removed, pooled, minced and digested with 1 mg/mL collagenase A (Roche, Indianapolis, IN) for 30 min at 37°C. Tissue debris were allowed to settle at 1 × g and the suspension containing GIC was harvested by pipette. RBC were lysed by hypotonic shock, GIC were passed through a 30 μm pore size nylon mesh, and viable leukocytes were enumerated by Trypan blue exclusion. For differential enumeration, GIC were placed on slides with a cytocentrifuge and stained with Wright stain.
ELISPOT assay for in vivo primed, donor-reactive cytokine-producing cells
ELISPOT assays were performed as described (36). Capture and detection mAb specific for IFNγ (RA-6A2, XMG1.2), IL-4 (BVD4-1D11, BVD6-24G2) and IL-17 (TC11-18H10, TC11-BH4.1) were purchased from Pharmingen (San Diego, CA). PVDF-backed microtiter plates (Millipore, Bedford, MA) were coated with unlabeled mAb and blocked with 1% BSA in PBS. Irradiated (1,000 rad) donor splenocytes (4×105) and 106 recipient splenocytes were added to each well. After washing, biotinylated detection mAb were added to the plates. After washing, a 1:1,000 dilution of anti-biotin alkaline phosphatase (AP) conjugate (Vector Laboratories, Burlingame, CA) was added to IFNγ and IL-17 plates and a 1:2,000 dilution of horseradish peroxidase-conjugated streptavidin (SA-HRP; Dako, Carpinteria, CA) was added to IL-4 plates. Plates were washed and spots visualized by addition of nitroblue tetrazolium (NBT; BioRad, Hercules, CA)/3 Bromo 4 Chloro Indolyl Phosphate (BCIP; Sigma) to IFNγ and IL-17 plates or 3-amino-9-ethylcarbazole (AEC; Pierce, Rockford, IL) to IL-4 plates. Color development continued until spots were visible and stopped by adding H2O. Plates were dried and spots quantified using an Immunospot Series 1 ELISPOT analyzer (Cellular Technology Ltd., Cleveland, OH).
In vitro T cell subset depletion
CD4+ or CD8+ cells were depleted using Dynal Beads (Invitrogen, Carlsbad, CA) as per manufacturer’s protocol. Single cell suspensions of splenocytes were incubated with anti-CD4 or anti-CD8 coated beads for 30 min. Bead-bound cells were removed magnetically. Unbound cells were confirmed to be depleted by flow cytometry (approx. 1% contamination; FACSCalibur, Becton-Dickinson, Franklin Lakes, NJ).
Morphometric analysis of intragraft collagen deposition
Quantitative analyses of areas of intragraft collagen deposition were performed as previously described (37). Briefly, images of ten areas of each graft’s myocardium were captured and analyzed using the application program IP Lab Spectrum-R4 (BD Biosciences, Rockville MD). The color wavelengths of image copies were transformed into digital readings. Using the original image for comparison, the color spectrum of the copied image was adjusted until areas positive for collagen turned green, while unaffected surrounding myocytes remained black. Percent collagen was calculated by dividing the total pixel area of the field by the pixel area within areas of collagen deposition.
Immunohistochemistry
To detect IgG deposition within the graft, frozen sections of grafts were fixed in cold acetone and incubated with 1:150 dilution of goat anti-mouse IgG-HRP (Southern Biotech, Birmingham, AL) or either IgG1-AP or IgG2a-AP followed by incubation with 1:600 streptavidin-HRP (Vector Laboratories, Burlingame, CA). Incubation with AEC revealed antibody binding.
Alloantibody assay
As described (31, 38), 106 P815 (H-2d) cells (ATCC) were incubated with 1:50 dilutions of sera collected from CD40-/- C57BL/6 recipients at specified time points after transplantation. Cells were then washed and incubated with FITC-conjugated, affinity purified rabbit anti-mouse IgM or IgG antibody (The Binding Site, San Diego, CA, USA). The samples were analyzed on a FACSCalibur cytometer (BD-Pharmingen) equipped with Cell Quest software using forward versus side scatter for appropriate gating. Data are reported as the mean channel fluorescence.
ELISA for total serum IgG levels
Total concentrations of IgG present in serum samples were determined by ELISA as previously described (39, 40). Briefly, a mixture of unlabeled anti-IgG isotype-specific Abs (IgG1, IgG2b, IgG2c, IgG3; Southern Biotech) were diluted to 10 μg/mL in a carbonate buffer and used to coat Immulon 2, 96 well flat bottom ELISA plates (Costar, Corning Inc., Corning, NY) overnight. After washing, wells were blocked by the addition of 100 μL PBS + 10% FCS + 0.4% azide per well. Following washing, 100 μL per well of samples diluted in PBS + 10% FCS + 0.4% azide were added. After washing, a mixture of AP-tagged anti-IgG isotype-specific antibodies (Southern Biotech) were diluted 1:1000 in Tris buffer and added at 100 μL per well. After washing, plates were developed by the addition of 100 μL per well of substrate (Sigma Fast pNPP and Tris buffer tablets, diluted in distilled water; Sigma-Aldrich) and read at 405 nm by an EL 800 microtiter plate reader (Bio-Tek Instruments, Winooski, VT). All incubations were performed at room temperature. Data are reported as the mean absorbance + S.E.M. of 3 to 7 individual mice per experimental group.
Statistical analysis
Data were analyzed with GraphPad Prism 4.0c software using Student’s unpaired t-tests with Welch’s correction or log rank tests as appropriate. P values of ≤ 0.05 were considered statistically significant.
Results
OX40 is upregulated following CD4+ T cell activation
This study assessed the effects of stimulation via an alternative costimulatory molecule on allograft acceptance induced by CD40-CD40L blockade. We focused on the OX40-OX40L pathway as BALB/c mice have been reported to preferentially utilize the OX40-OX40L pathway relative to C57BL/6 mice (28) and CD40 is not required for the rejection response in BALB/c allograft recipients (18). To verify differential expression of OX40 by BALB/c and C57BL/6 mice, splenocytes from both WT and CD40-/-BALB/c and C57BL/6 mice were stimulated for 0, 24, 48 and 72 hours with Concavalin A (ConA) and analyzed for OX40 expression by flow cytometry (Fig. 1). Prior to stimulation, OX40 expression was negligible. After stimulation, OX40 was upregulated on CD4+ T cells (Fig. 1) but was not detected on CD8+ T cells (data not shown). BALB/c CD4+ T cells expressed OX40 to a greater extent than their C57BL/6 counterparts both in terms of surface density (Fig. 1A) and percentage of CD4+ T cells expressing OX40 (Fig. 1B). Further, BALB/c CD4+ cells maintained OX40 expression for a longer period of time as compared to their C57BL/6 counterparts (Fig. 1B).
Figure 1. Comparison of OX40 surface expression between C57BL/6 and BALB/c cells after stimulation.

Splenocytes from either WT or CD40-/- C57BL/6 and BALB/c mice were stimulated with ConA for 0h, 24h, 48h, or 72h before analysis of OX40 surface expression by flow cytometry. Data are represented as the mean channel fluorescence of CD4+ T cells stained for OX40, reflecting OX40 surface density (A), or the percentage of CD4+ splenocytes that are OX40+ (B). All data are representative of at least three separate experiments.
Agonistic anti-OX40 mAb stimulates CD4+ T cells and induces a distinct pathology of allograft rejection
In WT allograft recipients, transient depletion of CD4+ T cells allows for long-term graft survival (32, 33, 35, 38). These CD4+ T cells repopulate the periphery 3-4 weeks post-depletion, yet these repopulating cells are hyporesponsive (41). In contrast, depletion of CD8+ T cells allows only for a modest increase in graft survival time (42). Since only CD4+ T cells expressed OX40 (Fig. 1) following ConA stimulation, we hypothesized that stimulatory anti-OX40 mAb would directly activate CD4+, but not CD8+ T cells. To test this hypothesis WT allograft recipients were inductively depleted of either CD4+ or CD8+ T cells and treated with agonistic anti-OX40 mAb or control rat IgG. In recipients inductively depleted of CD8+ T cells, OX40 stimulation significantly accelerated allograft rejection when compared to rat IgG treated controls (Fig. 2A, p = 0.0079). Additionally, ELISPOT analysis revealed a significant increase in both donor-reactive Th1 (IFNγ) and Th2 (IL-4) cells following OX40 stimulation (Fig. 2B). In contrast, OX40 stimulation failed to induce acute graft rejection following inductive depletion of CD4+ T cells (Fig. 2A). Further, ELISPOT analysis revealed that donor-reactive Th1 and Th2 responses were not induced in recipients that were inductively depleted of CD4+ cells and given anti-OX40 (Fig. 2B). Thus, in the absence of CD4+ cells OX40 stimulation had no measurable effect on the CD8+ T cell response.
Figure 2. Stimulatory anti-OX40 mAb acts on CD4+ T cells.

(A) WT C57BL/6 allograft recipients were transiently depleted of CD4+ or CD8+ cells (1 mg i.p. anti-CD4 mAb or anti-CD8 mAb days -1, 0, and 7 relative to transplant) concurrent with agonistic anti-OX40 mAb (closed symbols, solid line) or control rat IgG treatment (open symbols, dashed line) (0.25 mg i.p. days 0, 1, 3 and 7 relative to transplant). (B) Recipients treated as described in Panel A were sacrificed either at time of rejection or 30 days post transplant. Donor-reactive Th1 (IFNγ) and Th2 (IL-4) splenocyte responses were quantified by ELISPOT. Individual data points represent the number of primed donor-reactive Th1 or Th2 cells in individual allograft recipients. Bars are indicative of the average number of donor-reactive splenocytes per experimental group. (C) WT recipients inductively depleted of CD4+ T cells were treated with stimulatory anti-OX40 mAb (closed circles, solid line) or control rat IgG (open circles, dashed line) 30 days post transplantation (0.25 mg i.p. days 30, 31, 33 and 40 relative to transplant) and monitored for function. (D) Recipients treated as described in Panel C were sacrificed either at the time of allograft rejection or 30 days after the initiation of treatment. Th1 (IFNγ) and Th2 (IL-4) splenocyte responses were quantified by ELISPOT. Individual data points represent the donor-reactive Th1 and Th2 responses of individual allograft recipients. Bars are indicative of the average number of donor-reactive Th1 or Th2 cells per experimental group.
We next asked whether OX40 stimulation would stimulate rejection once CD4+ cells returned following transient depletion. Following initial depletion, CD4+ T cells begin to repopulate the periphery between 3-4 weeks post transplantation (35, 38). Indeed, when recipients in this study were sacrificed 39-60 days post transplantation, 7-15% of the splenocytes were CD4+ (data not shown). Recipients received delayed anti-OX40 mAb 30 days post transplant as CD4+ T cells returned. All recipients treated with anti-OX40 mAb rejected their allografts by day 44 post transplant, or 14 days after receiving anti-OX40 mAb (Fig. 2C, p = 0.0101). In contrast, recipients given delayed control rat IgG maintained their grafts, verifying that donor-reactive CD4+ T cells were hyporesponsive (Fig. 2C). These observations indicate that the rejection mediated by OX40 stimulation is dependent upon CD4+ T cells.
ELISPOT analysis revealed primed, donor-reactive Th1 and Th2 responses in recipient splenocytes following delayed treatment with anti-OX40 (Fig. 2D). While unmodified recipients produce elevated levels of donor-reactive IFNγ, levels of donor-reactive IL-4 are comparatively rare (31). Hence, once CD4+ T cells repopulated the periphery OX40 stimulation reversed their state of hyporesponsiveness and induced both a Th1 and an atypical Th2 response.
OX40 stimulation overrides CD40-CD40L blockade induced allograft acceptance
Since BALB/c CD40-/- mice, but not C57BL/6 CD40-/- mice, reject cardiac allografts (18), and BALB/c mice employ the OX40-OX40L pathway to a greater extent than C57BL/6 mice (8, 28), we asked whether OX40 stimulation would override graft acceptance induced by CD40-CD40L blockade in C57BL/6 recipients. To this end CD40-CD40L interactions were disrupted in C57BL/6 allograft recipients by treating WT mice with anti-CD40L mAb, or using CD40L-/- or CD40-/- recipients. Allograft recipients were treated inductively with agonistic anti-OX40 mAb or rat IgG as a control. Regardless of the method of CD40-CD40L disruption, OX40 stimulation induced acute allograft rejection in all recipients by day 14 post transplant (Fig. 3). In contrast, grafts from rat IgG treated control recipients continued to function normally until the termination of the experiment at day 30. For reference, otherwise unmodified WT mice were treated with either anti-OX40 or control rat IgG. Following OX40 stimulation, WT recipients rejected their allografts at a significantly accelerated tempo as compared to the rat IgG treated controls (p = 0.0009, Fig. 3).
Figure 3. OX40 stimulation overrides induction of allograft acceptance induced by inhibition of CD40-CD40L interactions.
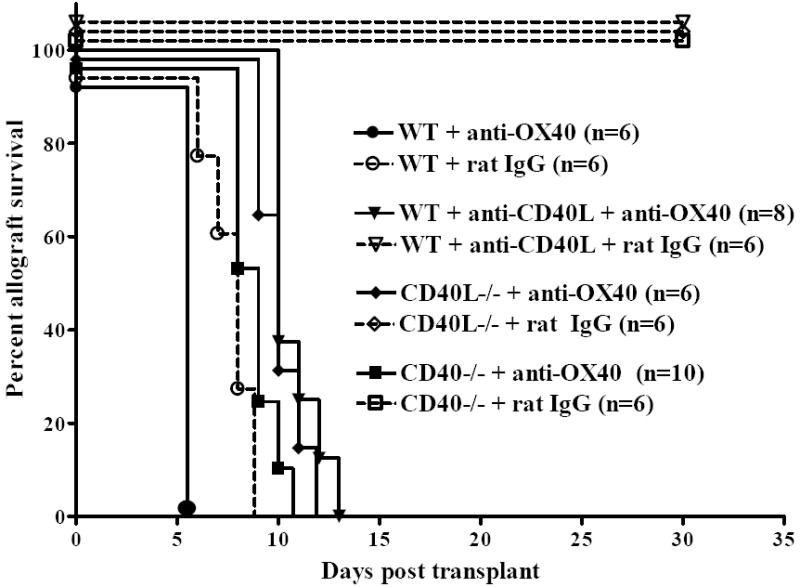
C57BL/6 WT recipients, WT recipients treated with anti-CD40L mAb (1 mg i.p. days 0, 1 and 2 relative to transplant), CD40L-/-, or CD40-/- C57BL/6 mice were transplanted with BALB/c cardiac allografts and treated with agonistic anti-OX40 mAb (closed symbols, solid line) or a control rat IgG (open symbols, dashed line) and grafts were monitored for function.
Unconventional pathology induced by OX40 stimulation
OX40-induced rejection in allograft recipients following CD40-CD40L blockade was characterized by thrombosis of arteries (Fig. 4A). Arterial thrombosis is not a feature of unmodified acute rejection in WT mice (Fig. 4A, lower left panel). OX40-induced rejection was additionally characterized by a granulocytic infiltration of the graft (Fig. 4B) that is also not a feature of unmodified rejection (Fig. 4B, (31)). Indeed, the few graft infiltrating cells (GIC) that were recovered from control mice following CD40-CD40L blockade were primarily mononuclear in nature (Fig. 4B, bottom right panel). The absolute GIC yield recovered from grafts in anti-OX40 treated recipients was significantly greater when compared to control recipients following CD40-CD40L blockade (Fig. 4D).
Figure 4. Pathology of acute rejection induced by OX40 stimulation following CD40-CD40L blockade.

(A) Allografts from C57BL/6 WT recipients, WT recipients treated with anti-CD40L mAb, CD40L-/-, or CD40-/- recipients treated with either anti-OX40 mAb (top row) or control rat IgG (bottom row) were harvested at time of rejection (anti-OX40 mAb treated) or 10 days post transplant (rat IgG treated) and stained by H&E. Note the perivascular infiltrate and thrombosed vessels (black arrows) in rejecting grafts following OX40 stimulation (magnification = 400x). GIC (B) and splenocytes (C) were harvested from recipients treated with anti-OX40 mAb or control rat IgG and Wright stained. One hundred cells were counted in at least 4 different fields and bars (mean + S.E.M.) represent the percentage of granulocytic cells present in either the graft (B) or spleen (C) (n ≥3 mice). Representative fields from CD40-/- recipients treated with anti-OX40 show both granulocytes (red arrows) and mononuclear cells that appear activated (characterized by diffuse nuclei and abundant cytoplasm, black arrows, center panels, 1000x). Representative fields of control CD40-/- recipients receiving rat IgG are depicted in bottom right of Panels (B) and (C). (D) Absolute number of cells recovered from the spleens and allografts of recipients treated with either anti-OX40 (shaded bars) or rat IgG (open bars) are depicted. The number of cells present in the spleens of allograft recipients was not dramatically increased, whereas the number of cells present within the allografts of recipients in which CD40-CD40L interactions were ablated was significantly increased after treatment with anti-OX40 mAb (all p values < 0.05). Bars represent the mean (+ S.E.M) of cells recovered from at least six recipients per group.
A significantly higher percentage of granulocytes were also present in the spleens of anti-OX40 treated allograft recipients (Fig. 4C, top panel). Further, splenocytes from anti-OX40 treated recipients exhibited morphology suggestive of cell activation (abundant cytoplasm and diffuse nuclei) (Fig. 4C, bottom left panel). It should be noted that these alterations in both graft pathology (thrombosed vessels and granulocyte accumulation) and splenocyte composition were not observed in syngeneic graft recipients that were treated with anti-OX40 (data not shown). Thus, these OX40-induced pathologies were antigen dependent.
OX40 stimulation induces donor-reactive Th1 and Th2 responses
ELISPOT analysis for primed, donor-reactive Th1 and Th2 responses revealed that OX40 stimulation induced significant IFNγ (Fig. 5A) and IL-4 (Fig. 5B) responses relative to those observed in control recipients. It should be noted that donor-reactive Th2 are not characteristic of unmodified acute rejection in this model (Fig. 5B, (31)), yet OX40 stimulation induced IL-4 production in all groups regardless of if or how CD40-CD40L interactions were disrupted. OX40-induced donor-reactive Th17 responses were not remarkable (data not shown).
Figure 5. OX40 stimulation induces donor-reactive Th1 and Th2 responses following CD40-CD40L blockade.
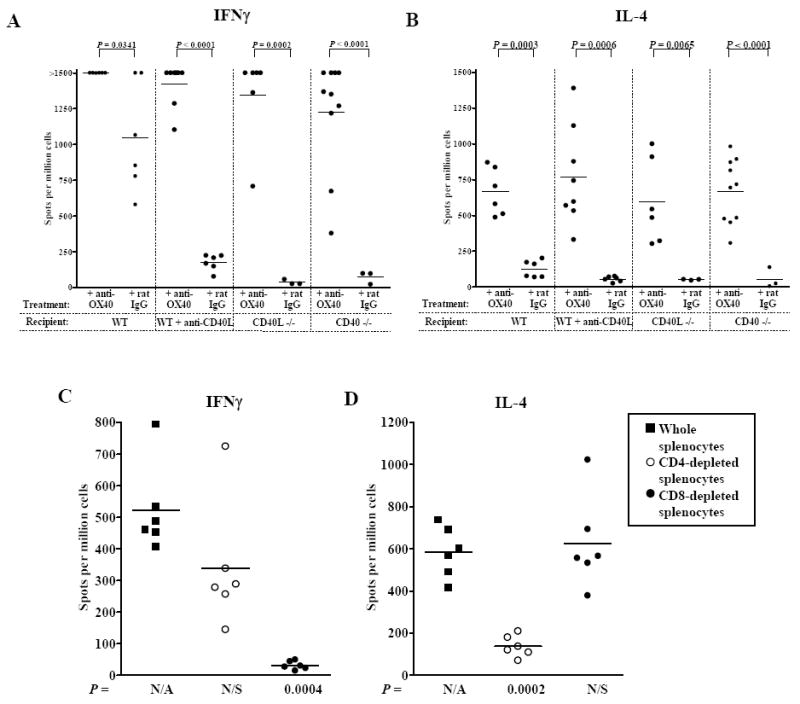
C57BL/6 WT recipients, WT recipients treated with anti-CD40L mAb, CD40L-/-, or CD40-/- recipients were transplanted with cardiac allografts and were treated with either anti-OX40 mAb or control rat IgG. Splenocytes were harvested at the time of rejection (recipients treated with anti-OX40 and WT + rat IgG) or 30 days after transplantation (rat IgG treated recipients in which CD40-CD40L interactions were ablated). Donor-reactive Th1 (IFNγ, A) and Th2 (IL-4, B) responses were quantified by ELISPOT. To determine the cell populations producing these cytokines, either CD4+ (open circles) or CD8+ (closed circles) cells were depleted from whole splenocyte (closed squares) populations prior to addition to ELISPOT Th1 (IFNγ, C) or Th2 (IL-4, D) assays. P values are in comparison to data from whole splenocyte populations. N/A; non-applicable, N/S; not significant.
To determine which T cell subsets were responsible for OX40-induced cytokine production, splenocytes from OX40 stimulated allograft recipients were depleted of CD4+ or CD8+ cells prior to addition to ELISPOT cultures (Fig. 5C & D). Depicted are results obtained from anti-OX40 treated CD40-/- allograft recipients and similar results were generated in WT recipients treated with anti-CD40L and CD40L-/- recipients. Depletion of CD8+ cells eliminated the Th1 response (Fig. 5C) while depleting CD4+ cells abrogated the Th2 response (Fig. 5D). These data are consistent with reports that CD8+ cells are the principle source of IFNγ in this model (31, 43) and that OX40 stimulation of CD4+ cells induces a Th2 response (8, 9, 28). As we were unable to detect OX40 expression on the surface of CD8+ T cells (data not shown), and depletion of CD8+ T cells concurrent with OX40 stimulation failed to prolong graft survival (Fig. 2A), these data suggest that OX40 stimulation induces CD4+ T cells to provide help for the primed CD8+ T cell response. This notion is further supported by the fact that anti-OX40 treatment of recipients depleted of CD4+ T cells did not induce allograft rejection or either Th1 or Th2 responses (Fig. 2A & 2B).
Delayed OX40 stimulation does not induce acute rejection of accepted allografts but promotes chronic rejection following CD40-CD40L disruption
In the previous experiments, our OX40 stimulation protocol was initiated at the time of transplantation. We next asked if stimulation through OX40 would reverse established allograft acceptance following CD40-CD40L blockade. To this end, WT recipients treated with anti-CD40L mAb, or CD40L-/- or CD40-/- recipients were transplanted with cardiac allografts and were left otherwise untreated. After 30 days these allograft recipients were treated with either anti-OX40 mAb or a control rat IgG. OX40 stimulation did not induce acute rejection of established allografts (Fig. 6A). Although grafts continued to function in mice that received delayed anti-OX40, contractions of these grafts were noticeably weaker when compared to their rat IgG treated counterparts as assessed by palpation. Histologically, an increased density of GIC resulted from delayed OX40 stimulation relative to rat IgG treated controls (Fig. 6B).
Figure 6. Delayed OX40 stimulation promotes chronic, rather than acute rejection of accepted grafts following CD40-CD40L blockade.
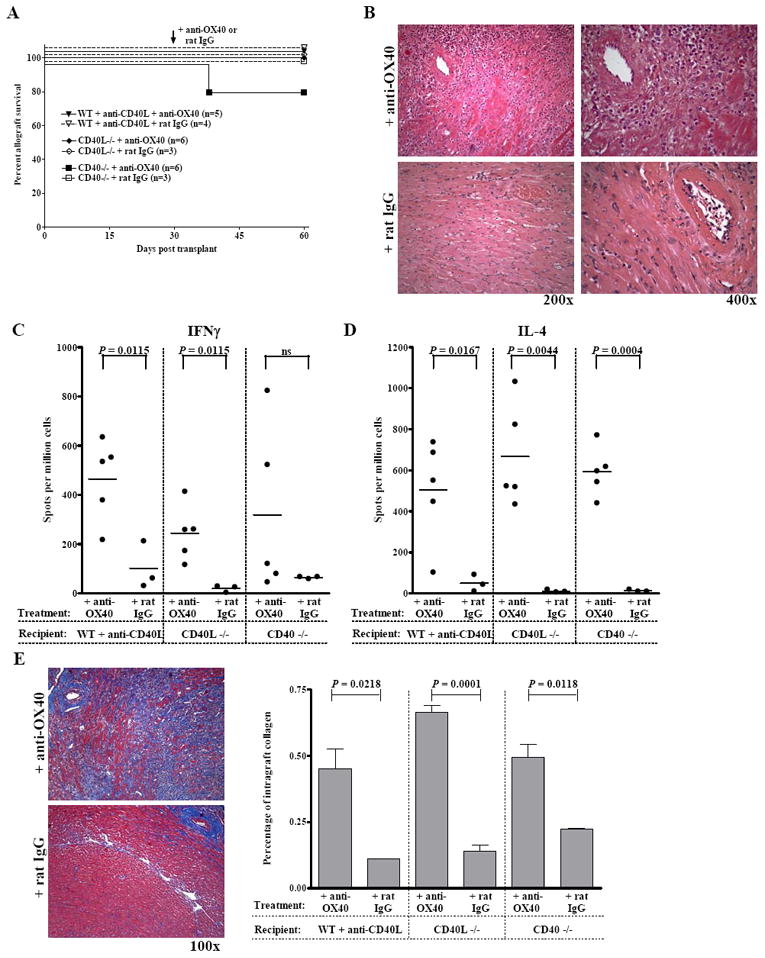
(A) WT recipients treated with anti-CD40L mAb, CD40L-/-, or CD40-/- recipients were transplanted with cardiac allografts and were treated with delayed anti-OX40 mAb (closed symbols, solid line) or control rat IgG (open symbols, dashed line) (0.25 mg i.p. days 30, 31, 33 and 40 relative to transplant). (B) Grafts from recipients described in (A) were harvested 60 days post transplantation and stained with H&E. In the anti-OX40 mAb treated groups areas of increased infiltrate were apparent, particularly around the arteries. Splenocytes from recipients described in Panel A were assessed for donor-reactive Th1 (IFNγ, C) and Th2 (IL-4, D) by ELISPOT. (E) Sections of grafts from recipients described in Panel A were stained with Masson trichrome stain, which stains collagen blue. Frames in both (B) and (E) are of grafts from CD40-/- recipients and are representative of n = 3 – 4 control rat IgG treated mice and n = 5 - 6 anti-OX40 treated recipients in which recipient CD40-CD40L interactions were disrupted. The areas of grafts staining positive for collagen deposition were quantified by morphometric analysis (Panel E, right). Bars represent the average percentage + S.E.M. of graft area positive for collagen in n = 3 – 4 rat IgG treated recipients and n = 5 – 6 anti-OX40 treated recipients.
We also assessed Th1 and Th2 responses by ELISPOT to determine whether delayed anti-OX40 treatment resulted in donor-reactive T cell priming (Fig. 6C & D). At the termination of the experiment (day 60 post transplant), both donor-reactive Th1 and Th2 were readily detectable in recipients receiving delayed OX40 stimulation but not in the control groups. Hence, despite the presence of primed, donor-reactive T cell responses allografts were not acutely rejected.
Additionally, delayed OX40 stimulation resulted in increased collagen deposition within the grafts as detected by Masson trichrome staining (Fig. 6E, left panel). Morphometric analysis was used to quantify the percent of the graft containing collagen deposition (stained blue) and revealed significantly more collagen in the grafts of mice receiving delayed OX40 stimulation relative to rat IgG treated controls (Fig. 6E, right panel). Since collagen deposition within the graft is a hallmark of chronic allograft rejection, these data indicate that delayed anti-OX40 treatment of mice bearing established allografts promotes the progression of chronic, rather than acute rejection.
OX40 stimulation induces donor-reactive IgM and IgG responses in the absence of CD40
Since CD40 engagement is reportedly necessary in stimulating an IgG class switch response (44), we assessed the ability of OX40 stimulation in CD40-/- allograft recipients to induce donor alloantigen-reactive IgM and IgG responses (Fig. 7). OX40 stimulation in CD40-/- allograft recipients induced a significant increase in donor-reactive IgM production when compared to rat IgG treated control recipients (Fig. 7A). Importantly, OX40 stimulation induced a donor-reactive IgG response in CD40-/-allograft recipients (Fig. 7B) indicating that OX40 stimulation overrides the reported requirement for CD40 in inducing the IgG isotype switch (44). These data were confirmed by a mouse IgG-specific sandwich ELISA that revealed significant differences in total IgG levels in the sera of anti-OX40 as compared to rat IgG control treated CD40-/- allograft recipients (Fig. 7C).
Figure 7. OX40 stimulation induces production of donor-reactive alloantibodies in the absence of CD40.
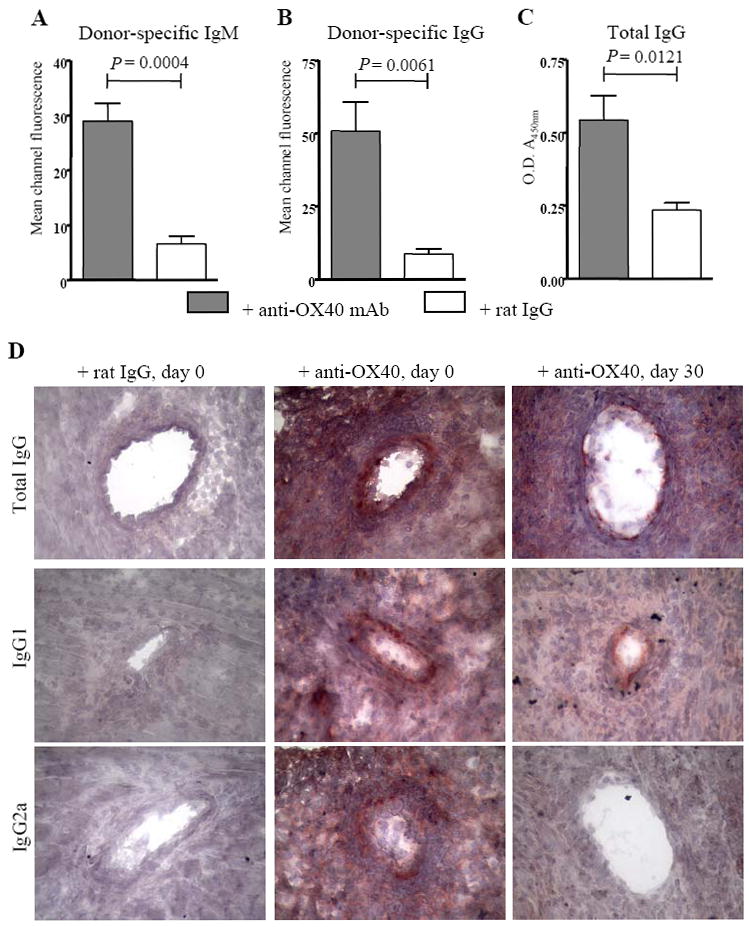
Sera from C57BL/6 CD40-/- recipients of cardiac allografts treated with anti-OX40 mAb (shaded bars) or rat IgG (open bars) were harvested at either time of rejection or 10 days after transplant. P815 (H-2d) cells were then incubated with a 1:50 dilution of sera, and bound donor-reactive antibody was detected by incubation with FITC-tagged anti-IgM (A) or anti-IgG (B) antibodies. The mean channel fluorescence is indicative of the relative amount of donor-reactive antibodies. Bars represent the average mean channel fluorescence of n = 3 control rat IgG treated recipient and n = 5 anti-OX40 treated recipient samples + S.E.M. (C) Sera of CD40-/- allograft recipients treated with anti-OX40 mAb or control rat IgG were diluted 10-3 and the relative amounts of total mouse IgG present were quantified by sandwich ELISA. Shaded bars (anti-OX40 mAb treated) and open bars (rat IgG control treated) represent the average O.D. (A450nm) of n = 3 control rat IgG treated recipient and n = 5 anti-OX40 treated recipient sera samples + S.E.M. (D) C57BL/6 CD40-/- allograft recipients were treated with control rat IgG (left column) or anti-OX40 mAb at time of transplant (inductive, middle column) or 30 days after transplantation (delayed, right column) and grafts were harvested at either time of rejection or functioning grafts were harvested 60 days after transplantation. Depicted are grafts from recipients treated with rat IgG at the time of transplantation, which is representative of results obtained from both inductive and delayed treatment with rat IgG. Graft sections were fixed and incubated with goat anti-mouse IgG, rabbit anti-mouse IgG1, or rabbit anti-mouse IgG2a followed by development with AEC to visualize mouse antibody deposition. Results are representative of grafts from n = 4 – 10 recipients. 400X magnification.
In addition to donor-reactive antibody circulating in the recipient serum, mouse IgG was also detectable by immunohistochemistry within the grafts of CD40-/- recipients receiving anti-OX40 at the time of transplantation (+ anti-OX40, day 0, Fig. 7D). Interestingly, although donor-reactive antibody was not detectable in the serum of CD40-/- recipients treated with anti-OX40 30 days after transplantation (data not shown), and these grafts were not acutely rejected (Fig. 6A), mouse IgG was also detectable within the grafts of these recipients (+ anti-OX40, day 30, Fig. 7D). This finding suggests that the donor-reactive antibody that is produced is absorbed by the graft. Further, this observation indicates that differential levels of donor-reactive antibody are produced following OX40 stimulation, dependent upon the timing of OX40 stimulation relative to transplantation. In addition, the isotypes of IgG deposited within the grafts of recipients treated with anti-OX40 at day 0 vs. day 30 differed. While both IgG1 and IgG2a were readily detectable within vessels of grafts from recipients treated with anti-OX40 at the time of transplantation, only IgG1 was detectable within vessels of grafts from recipients treated with anti-OX40 30 days after transplantation. Hence, the presence of complement-fixing IgG2a (45) correlated with acute rejection, while the presence of non-complement-fixing IgG1 (46) correlated with prolonged graft function and the progression of chronic rejection.
Discussion
In the current study, we present data that demonstrate that OX40 costimulation overrides cardiac allograft acceptance induced by CD40-CD40L blockade (Fig. 3) and results in the induction of both donor-reactive Th1 and Th2 responses (Fig. 5). Unmodified rejection in this model is associated with a Th1, but not Th2 response (31, 32, 47). However, the induction of Th2 by OX40 perturbation in our model correlates with reports from other systems that OX40 stimulation is associated with a Th2 response (8, 9). In transplantation the induction of a Th2 response results in an alternative pathology of rejection (31, 32, 47-49) that includes the vascular involvement and granulocytic infiltration as is induced by OX40 stimulation (Fig. 4). Hence, we observed a biologic effect of OX40 mediated Th2 induction. In the absence of CD4+ T cells OX40 stimulation failed to induce this allograft pathology in WT recipients (Fig. 2) suggesting that OX40 stimulation acts directly on CD4+ T cells. Cell selection studies revealed that these CD4+ cells were the principle source of IL-4 in this system (Fig. 5D) while CD8+ cells were the main source of IFNγ (Fig. 5C). Interestingly, OX40 stimulation also induced donor-reactive IgG responses in the complete absence of CD40 (Fig. 7). To our knowledge, this role for OX40 in B cell responses has not been described.
OX40 is expressed preferentially by activated CD4+ T cells (Fig. 1, (3)) and it is likely that in our experiments OX40 stimulation induces CD4+ T cells to provide help to CD8+ T cells (50, 51) to promote their activation. Indeed, following CD4+ T cell depletion, OX40 stimulation failed to result in T cell priming and/or allograft rejection (Fig. 2). While OX40 stimulation has been reported to promote CD8+ T cell effector functions (52), we were unable to detect CD8+OX40+ cells by flow cytometry either in the spleen or in the graft (data not shown) suggesting that OX40 expression may not be induced on CD8+ T cells in this system. Alternatively, CD8+OX40+ cells may localize in a different compartment in vivo. However, WT recipients transiently depleted of CD4+ T cells and treated with delayed agonistic anti-OX40 do reject their allografts. This rejection is likely dependent upon the return of CD4+ T cells (30 days post transplant, approximately 5% of splenic cells are CD4+ as detected by flow cytometry, data not shown). Indeed, after CD4+ T cell repopulation, OX40 stimulation induced both IFNγ and IL-4 producing cells (Fig. 2). Hence, our data indicate that OX40 stimulation affects not only CD4+ cells expressing detectable OX40 on their surface, but also induces these CD4+ T cells to provide help for CD8+ T cell responses generated in conjunction with alloantigen exposure.
Stimulation of OX40 through OX40L on APC is believed to play a critical role in driving antigen-specific T cell responses in vivo (53). In our study, deliberate stimulation of OX40 may be necessary to observe these effects as both CD4+ T cells and CD11c+ dendritic cells from C57BL/6 mice express lower levels of OX40 and OX40L as compared to their BALB/c counterparts (Fig. 1 and data not shown). OX40L has a broad tissue distribution, including B cells, dendritic cells, and endothelial cells (3, 54, 55). Further, CD40 engagement upregulates OX40L surface expression on both B cells (53) and dendritic cells (53, 56), suggesting that OX40L expression may be dependent upon CD40-CD40L interactions. In our experiments, T cell OX40 expression does not appear to limit OX40-OX40L interactions as agonistic anti-OX40 mAb is sufficient to drive acute allograft rejection (Fig. 3) and induce donor-reactive cytokine priming (Fig. 5). Rather, low levels of OX40L expression may impede contributions of the OX40-OX40L costimulatory pathway to graft pathology under conditions where the CD40-CD40L pathway is disrupted. Indeed, Jenkins et. al. (57) demonstrated that stimulation via OX40 restored production of both Th1 and Th2 responses to Schistosoma mansoni-soluble egg antigen presented by CD40-/- dendritic cells, suggesting that the T cell response was limited by APC OX40L expression as opposed to a lack of T cell OX40 expression. Hence, endogenous OX40-OX40L interactions may not override CD40-CD40L blockade due to limited OX40L, but not OX40 expression, resulting from the absence of CD40 stimulation of APC.
Disruption of CD40-CD40L interactions is highly effective in promoting allograft acceptance (reviewed in (2, 12)),(13-17). However, it is likely that OX40 stimulation mediates multiple effects on CD4+ cells in the absence of CD40-CD40L interactions that culminate in allograft rejection. After TCR engagement OX40 is upregulated on the CD4+ T cell surface (4, 58). OX40 stimulation enhances both CD4+ T cell cytokine production and proliferation (59), and can prevent the induction of tolerance to peptide (52). Further, OX40 stimulation can promote the expansion of a pool of antigen-reactive T cells (60, 61). Thus, in our model inductive OX40 stimulation may serve as the costimulatory signal needed to activate and expand graft-reactive T cells in the absence of CD40-CD40L interactions. Maxwell et. al. (60) have established that when initiated concurrent with antigen and “danger signals” such as LPS, OX40 stimulation results in both clonal expansion and survival of antigen-specific T cells. In transplantation, heat-shock proteins are induced by both ischemia/reperfusion injury and surgery and provide a “danger signal” (62), which in addition to the antigen supplied by the graft could synergize with OX40 stimulation to result in the expansion of the graft-reactive T cell pool. In addition, OX40 stimulation in the presence of cytokines can reverse the non-responsiveness of anergic cells and induce further cytokine production and effector functions (63). While the actions of anti-CD40L mAb are not fully understood (12, 13, 64-67), it is generally thought that deficiency in either CD40L (16) or CD40 (reviewed in (68)) disrupts a crucial step in T cell activation and renders these cells anergic. These cells could now provide a target for OX40 stimulation, as anergic cells upregulate OX40 on their surface (52). Thus, OX40 stimulation of T cells may reverse anergy induced by CD40-CD40L blockade thereby allowing the emergence of otherwise silenced donor-reactive effector cells resulting in anti-graft responses. Our findings are in keeping with these possibilities and represent the first report of OX40 stimulation in the absence of CD40-CD40L interactions in the transplant setting.
The findings discussed above were generated when OX40 stimulation occurred at the onset of CD40-CD40L blockade. Hence, we assessed the consequences of delayed OX40 stimulation once allografts had been accepted following CD40-CD40L blockade. Inductive CD40-CD40L blockade allowed for continued graft function and primed donor-reactive cytokine-producing cells were rare (Fig. 3, 5A & 5B). However, delayed OX40 stimulation induced graft pathology and Th1 and Th2 cell priming yet did not induce acute graft rejection (Fig. 6). Thus, these findings support previous observations that following CD40-CD40L blockade a number of recipient lymphocytes retain graft reactivity despite graft acceptance (14). As OX40 is only expressed following T cell activation (Fig. 1, (3)), naive cells should fail to respond to OX40 stimulation while anergic cells should be responsive. Indeed, anti-CD40L treatment has been shown to maintain graft-reactive cells in a quiescent precursor state and these cells can be activated in vitro (14). Inhibition of CD40-CD40L interactions favors the generation of regulatory T cells (69-71), and these regulatory T cells may be adequate to prevent donor-reactive cells from acutely rejecting accepted grafts following delayed OX40 stimulation. Recent data suggest that after OX40 stimulation regulatory T cells are less efficient suppressors of effector cell proliferation and cytokine production (72, 73), and that OX40 stimulation may reverse regulatory T cell function (74).
Differential induction of antibody isotypes by initial vs. delayed OX40 stimulation correlated with acute vs. chronic graft rejection. Interestingly, donor-reactive cytokine production was induced in both settings. Indeed, initial vs. delayed OX40 stimulation was associated with differential antibody deposition within the allografts of CD40-/- recipients (Fig. 7D). Hence, deliberate OX40 stimulation is sufficient to induce a donor-reactive isotype switch in the absence of CD40 interactions. The isotypes of these antibodies are consistent with the types of cytokines produced. Inductive OX40 stimulation induced both IFNγ and IL-4 (Fig. 5) and both IgG2a and IgG1 donor-reactive antibodies (Fig. 7, (75)). In contrast, delayed OX40 stimulation induced a strong Th2 and dampened Th1 response as compared to the inductively treated recipients (Fig. 6D) and IgG1 production (Fig. 7D). While IgG2 is a potent activator of complement, IgG1 is not (46). It has been previously reported that IgG2b, but not IgG1 alone is sufficient to induce acute graft rejection (76), and that together IgG2a and IgG1 can synergize to induce acute graft rejection and complement deposition (45, 77). Additionally, IgG1 alone does not induce acute graft rejection but does induce chemokine release by the endothelial cells (77). Hence, inductive OX40 stimulation may lead to the production of both IgG1 and IgG2a, synergistically contributing to acute graft rejection by activating complement, whereas delayed OX40 stimulation may lead to the production of IgG1 alone and contribute to chronic rejection. This possibility remains to be formally tested.
Taken together, data herein suggest that OX40 stimulation may have multiple effects on immune responses following CD40-CD40L blockade. These effects may depend on both cell types affected and timing of OX40 stimulation relative to both antigen exposure and “danger signal” presence. Inductive OX40 stimulation may affect the cytokine profile of activated cells, prevent the development of regulatory cells, and/or rescue graft-reactive cells from anergy induced by CD40-CD40L blockade. Conversely, after the “danger signals” associated with transplantation have been resolved, delayed OX40 stimulation may affect the suppressive activities of OX40+ regulatory T cells. Further, inductive, but not delayed OX40 stimulation correlated with thrombosed arteries in grafts undergoing acute, but not chronic rejection. Our findings should be of interest clinically as they demonstrate redundancy and key functional differences in costimulatory molecule function and should be considered when developing anti-rejection regimens.
Acknowledgments
The authors would like to thank Dr. Wesley A. Dunnick for insightful discussions regarding antibody responses in CD40-/- mice.
Footnotes
This work was supported by R01 AI061469 (DKB), R01 HL070613 (DKB), and R01 AI070315 (XCL) from the National Institutes of Health, and The Herman and Dorothy Miller Fund Award for Innovative Immunology Research (BEB).
Abbreviations used in this paper: CD40-/-, CD40 deficient; CD40L-/-, CD40L deficient; GIC, graft-infiltrating cells; OX40L, OX40 ligand; WT, wild type
Disclosures The authors declare no conflict of interest.
References
- 1.Rothstein DM, Sayegh MH. T-cell costimulatory pathways in allograft rejection and tolerance. Immunol Rev. 2003;196:85–108. doi: 10.1046/j.1600-065x.2003.00088.x. [DOI] [PubMed] [Google Scholar]
- 2.Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol. 2004;22:307–328. doi: 10.1146/annurev.immunol.22.012703.104533. [DOI] [PubMed] [Google Scholar]
- 3.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 4.Gramaglia I, Weinberg AD, Lemon M, Croft M. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol. 1998;161:6510–6517. [PubMed] [Google Scholar]
- 5.Humphreys IR, Edwards L, Walzl G, Rae AJ, Dougan G, Hill S, Hussell T. OX40 ligation on activated T cells enhances the control of Cryptococcus neoformans and reduces pulmonary eosinophilia. J Immunol. 2003;170:6125–6132. doi: 10.4049/jimmunol.170.12.6125. [DOI] [PubMed] [Google Scholar]
- 6.O’Sullivan B, Thomas R. CD40 and dendritic cell function. Crit Rev Immunol. 2003;23:83–107. doi: 10.1615/critrevimmunol.v23.i12.50. [DOI] [PubMed] [Google Scholar]
- 7.Grewal IS, Flavell RA. The role of CD40 ligand in costimulation and T-cell activation. Immunol Rev. 1996;153:85–106. doi: 10.1111/j.1600-065x.1996.tb00921.x. [DOI] [PubMed] [Google Scholar]
- 8.Akiba H, Miyahira Y, Atsuta M, Takeda K, Nohara C, Futagawa T, Matsuda H, Aoki T, Yagita H, Okumura K. Critical contribution of OX40 ligand to T helper cell type 2 differentiation in experimental leishmaniasis. J Exp Med. 2000;191:375–380. doi: 10.1084/jem.191.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roos A, Schilder-Tol EJ, Weening JJ, Aten J. Strong expression of CD134 (OX40), a member of the TNF receptor family, in a T helper 2-type cytokine environment. J Leukoc Biol. 1998;64:503–510. doi: 10.1002/jlb.64.4.503. [DOI] [PubMed] [Google Scholar]
- 10.Kaleeba JA, Offner H, Vandenbark AA, Lublinski A, Weinberg AD. The OX-40 receptor provides a potent co-stimulatory signal capable of inducing encephalitogenicity in myelin-specific CD4+ T cells. Int Immunol. 1998;10:453–461. doi: 10.1093/intimm/10.4.453. [DOI] [PubMed] [Google Scholar]
- 11.Williams CA, Murray SE, Weinberg AD, Parker DC. OX40-mediated differentiation to effector function requires IL-2 receptor signaling but not CD28, CD40, IL-12Rbeta2, or T-bet. J Immunol. 2007;178:7694–7702. doi: 10.4049/jimmunol.178.12.7694. [DOI] [PubMed] [Google Scholar]
- 12.Kirk AD, Blair PJ, Tadaki DK, Xu H, Harlan DM. The role of CD154 in organ transplant rejection and acceptance. Philos Trans R Soc Lond B Biol Sci. 2001;356:691–702. doi: 10.1098/rstb.2001.0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larsen CP, Alexander DZ, Hollenbaugh D, Elwood ET, Ritchie SC, Aruffo A, Hendrix R, Pearson TC. CD40-gp39 interactions play a critical role during allograft rejection. Suppression of allograft rejection by blockade of the CD40-gp39 pathway. Transplantation. 1996;61:4–9. doi: 10.1097/00007890-199601150-00002. [DOI] [PubMed] [Google Scholar]
- 14.Nathan MJ, Yin D, Eichwald EJ, Bishop DK. The immunobiology of inductive anti-CD40L therapy in transplantation: allograft acceptance is not dependent upon the deletion of graft-reactive T cells. Am J Transplant. 2002;2:323–332. doi: 10.1034/j.1600-6143.2002.20406.x. [DOI] [PubMed] [Google Scholar]
- 15.Niimi M, Pearson TC, Larsen CP, Alexander DZ, Hollenbaugh D, Aruffo A, Linsley PS, Thomas E, Campbell K, Fanslow WC, Geha RS, Morris PJ, Wood KJ. The role of the CD40 pathway in alloantigen-induced hyporesponsiveness in vivo. J Immunol. 1998;161:5331–5337. [PubMed] [Google Scholar]
- 16.Shimizu K, Schonbeck U, Mach F, Libby P, Mitchell RN. Host CD40 ligand deficiency induces long-term allograft survival and donor-specific tolerance in mouse cardiac transplantation but does not prevent graft arteriosclerosis. J Immunol. 2000;165:3506–3518. doi: 10.4049/jimmunol.165.6.3506. [DOI] [PubMed] [Google Scholar]
- 17.Larsen CP, Knechtle SJ, Adams A, Pearson T, Kirk AD. A new look at blockade of T-cell costimulation: a therapeutic strategy for long-term maintenance immunosuppression. Am J Transplant. 2006;6:876–883. doi: 10.1111/j.1600-6143.2006.01259.x. [DOI] [PubMed] [Google Scholar]
- 18.Nathan MJ, Mold JE, Wood SC, Csencsits K, Lu G, Eichwald EJ, Bishop DK. Requirement for donor and recipient CD40 expression in cardiac allograft rejection: induction of Th1 responses and influence of donor-derived dendritic cells. J Immunol. 2004;172:6626–6633. doi: 10.4049/jimmunol.172.11.6626. [DOI] [PubMed] [Google Scholar]
- 19.Larsen CP, Elwood ET, Alexander DZ, Ritchie SC, Hendrix R, Tucker-Burden C, Cho HR, Aruffo A, Hollenbaugh D, Linsley PS, Winn KJ, Pearson TC. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434–438. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 20.Williams MA, Trambley J, Ha J, Adams AB, Durham MM, Rees P, Cowan SR, Pearson TC, Larsen CP. Genetic characterization of strain differences in the ability to mediate CD40/CD28-independent rejection of skin allografts. J Immunol. 2000;165:6849–6857. doi: 10.4049/jimmunol.165.12.6849. [DOI] [PubMed] [Google Scholar]
- 21.Demirci G, Amanullah F, Kewalaramani R, Yagita H, Strom TB, Sayegh MH, Li XC. Critical role of OX40 in CD28 and CD154-independent rejection. J Immunol. 2004;172:1691–1698. doi: 10.4049/jimmunol.172.3.1691. [DOI] [PubMed] [Google Scholar]
- 22.Vu MD, Amanullah F, Li Y, Demirci G, Sayegh MH, Li XC. Different costimulatory and growth factor requirements for CD4+ and CD8+ T cell-mediated rejection. J Immunol. 2004;173:214–221. doi: 10.4049/jimmunol.173.1.214. [DOI] [PubMed] [Google Scholar]
- 23.Sacks D, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev. 2002;2:845–858. doi: 10.1038/nri933. [DOI] [PubMed] [Google Scholar]
- 24.Sadick MD, Heinzel FP, Holaday BJ, Pu RT, Dawkins RS, Locksley RM. Cure of murine leishmaniasis with anti-interleukin 4 monoclonal antibody. Evidence for a T cell-dependent, interferon gamma-independent mechanism. J Exp Med. 1990;171:115–127. doi: 10.1084/jem.171.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang ZE, Reiner SL, Zheng S, Dalton DK, Locksley RM. CD4+ effector cells default to the Th2 pathway in interferon gamma-deficient mice infected with Leishmania major. J Exp Med. 1994;179:1367–1371. doi: 10.1084/jem.179.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 27.Campbell KA, Ovendale PJ, Kennedy MK, Fanslow WC, Reed SG, Maliszewski CR. CD40 ligand is required for protective cell-mediated immunity to Leishmania major. Immunity. 1996;4:283–289. doi: 10.1016/s1074-7613(00)80436-7. [DOI] [PubMed] [Google Scholar]
- 28.Ishii N, Ndhlovu LC, Murata K, Sato T, Kamanaka M, Sugamura K. OX40 (CD134) and OX40 ligand interaction plays an adjuvant role during in vivo Th2 responses. Eur J Immunol. 2003;33:2372–2381. doi: 10.1002/eji.200324031. [DOI] [PubMed] [Google Scholar]
- 29.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- 30.al-Shamkhani A, Birkeland ML, Puklavec M, Brown MH, James W, Barclay AN. OX40 is differentially expressed on activated rat and mouse T cells and is the sole receptor for the OX40 ligand. Eur J Immunol. 1996;26:1695–1699. doi: 10.1002/eji.1830260805. [DOI] [PubMed] [Google Scholar]
- 31.Chan SY, DeBruyne LA, Goodman RE, Eichwald EJ, Bishop DK. In vivo depletion of CD8+ T cells results in Th2 cytokine production and alternate mechanisms of allograft rejection. Transplantation. 1995;59:1155–1161. [PubMed] [Google Scholar]
- 32.Bishop DK, Chan Wood S, Eichwald EJ, Orosz CG. Immunobiology of allograft rejection in the absence of IFN-gamma: CD8+ effector cells develop independently of CD4+ cells and CD40-CD40 ligand interactions. J Immunol. 2001;166:3248–3255. doi: 10.4049/jimmunol.166.5.3248. [DOI] [PubMed] [Google Scholar]
- 33.Bishop DK, Shelby J, Eichwald EJ. Mobilization of T lymphocytes following cardiac transplantation. Evidence that CD4-positive cells are required for cytotoxic T lymphocyte activation, inflammatory endothelial development, graft infiltration, and acute allograft rejection. Transplantation. 1992;53:849–857. [PubMed] [Google Scholar]
- 34.Chen M, Xiao X, Demirci G, Li XC. OX40 controls islet allograft tolerance in CD154 deficient mice by regulating FOXP3+ Tregs. Transplantation. 2008;85:1659–1662. doi: 10.1097/TP.0b013e3181726987. [DOI] [PubMed] [Google Scholar]
- 35.Piccotti JR, Li K, Chan SY, Eichwald EJ, Bishop DK. Cytokine regulation of chronic cardiac allograft rejection: evidence against a role for Th1 in the disease process. Transplantation. 1999;67:1548–1555. doi: 10.1097/00007890-199906270-00008. [DOI] [PubMed] [Google Scholar]
- 36.Matesic D, Lehmann PV, Heeger PS. High-resolution characterization of cytokine-producing alloreactivity in naive and allograft-primed mice. Transplantation. 1998;65:906–914. doi: 10.1097/00007890-199804150-00008. [DOI] [PubMed] [Google Scholar]
- 37.Hogaboam CM, Gallinat CS, Bone-Larson C, Chensue SW, Lukacs NW, Strieter RM, Kunkel SL. Collagen deposition in a non-fibrotic lung granuloma model after nitric oxide inhibition. Am J Pathol. 1998;153:1861–1872. doi: 10.1016/S0002-9440(10)65700-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bishop DK, Li W, Chan SY, Ensley RD, Shelby J, Eichwald EJ. Helper T lymphocyte unresponsiveness to cardiac allografts following transient depletion of CD4-positive cells. Implications for cellular and humoral responses. Transplantation. 1994;58:576–584. doi: 10.1097/00007890-199409150-00009. [DOI] [PubMed] [Google Scholar]
- 39.Snapper CM, Paul WE. B cell stimulatory factor-1 (interleukin 4) prepares resting murine B cells to secrete IgG1 upon subsequent stimulation with bacterial lipopolysaccharide. J Immunol. 1987;139:10–17. [PubMed] [Google Scholar]
- 40.Zelazowski P, Collins JT, Dunnick W, Snapper CM. Antigen receptor cross-linking differentially regulates germ-line CH ribonucleic acid expression in murine B cells. J Immunol. 1995;154:1223–1231. [PubMed] [Google Scholar]
- 41.Wood SC, Lu G, Burrell BE, Bishop DK. Transplant acceptance following anti-CD4 vs. anti-CD40L therapy: evidence for differential maintenance of graft-reactive T cells. Am J Transplant. 2008;8:2037–2048. doi: 10.1111/j.1600-6143.2008.02372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bishop DK, Chan S, Li W, Ensley RD, Xu S, Eichwald EJ. CD4-positive helper T lymphocytes mediate mouse cardiac allograft rejection independent of donor alloantigen specific cytotoxic T lymphocytes. Transplantation. 1993;56:892–897. doi: 10.1097/00007890-199310000-00023. [DOI] [PubMed] [Google Scholar]
- 43.Piccotti JR, Li K, Chan SY, Ferrante J, Magram J, Eichwald EJ, Bishop DK. Alloantigen-reactive Th1 development in IL-12-deficient mice. J Immunol. 1998;160:1132–1138. [PubMed] [Google Scholar]
- 44.Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, Kikutani H. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1994;1:167–178. doi: 10.1016/1074-7613(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 45.Murata K, Fox-Talbot K, Qian Z, Takahashi K, Stahl GL, Baldwin WM, 3rd, Wasowska BA. Synergistic deposition of C4d by complement-activating and non-activating antibodies in cardiac transplants. Am J Transplant. 2007;7:2605–2614. doi: 10.1111/j.1600-6143.2007.01971.x. [DOI] [PubMed] [Google Scholar]
- 46.Klaus GG, Pepys MB, Kitajima K, Askonas BA. Activation of mouse complement by different classes of mouse antibody. Immunology. 1979;38:687–695. [PMC free article] [PubMed] [Google Scholar]
- 47.Piccotti JR, Chan SY, Goodman RE, Magram J, Eichwald EJ, Bishop DK. IL-12 antagonism induces T helper 2 responses, yet exacerbates cardiac allograft rejection. Evidence against a dominant protective role for T helper 2 cytokines in alloimmunity. J Immunol. 1996;157:1951–1957. [PubMed] [Google Scholar]
- 48.Le Moine A, Flamand V, Demoor FX, Noel JC, Surquin M, Kiss R, Nahori MA, Pretolani M, Goldman M, Abramowicz D. Critical roles for IL-4, IL-5, and eosinophils in chronic skin allograft rejection. J Clin Invest. 1999;103:1659–1667. doi: 10.1172/JCI5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Le Moine A, Surquin M, Demoor FX, Noel JC, Nahori MA, Pretolani M, Flamand V, Braun MY, Goldman M, Abramowicz D. IL-5 mediates eosinophilic rejection of MHC class II-disparate skin allografts in mice. J Immunol. 1999;163:3778–3784. [PubMed] [Google Scholar]
- 50.Dannull J, Nair S, Su Z, Boczkowski D, DeBeck C, Yang B, Gilboa E, Vieweg J. Enhancing the immunostimulatory function of dendritic cells by transfection with mRNA encoding OX40 ligand. Blood. 2005;105:3206–3213. doi: 10.1182/blood-2004-10-3944. [DOI] [PubMed] [Google Scholar]
- 51.Biagi E, Dotti G, Yvon E, Lee E, Pule M, Vigouroux S, Gottschalk S, Popat U, Rousseau R, Brenner M. Molecular transfer of CD40 and OX40 ligands to leukemic human B cells induces expansion of autologous tumor-reactive cytotoxic T lymphocytes. Blood. 2005;105:2436–2442. doi: 10.1182/blood-2004-07-2556. [DOI] [PubMed] [Google Scholar]
- 52.Bansal-Pakala P, Jember AG, Croft M. Signaling through OX40 (CD134) breaks peripheral T-cell tolerance. Nat Med. 2001;7:907–912. doi: 10.1038/90942. [DOI] [PubMed] [Google Scholar]
- 53.Murata K, Ishii N, Takano H, Miura S, Ndhlovu LC, Nose M, Noda T, Sugamura K. Impairment of antigen-presenting cell function in mice lacking expression of OX40 ligand. J Exp Med. 2000;191:365–374. doi: 10.1084/jem.191.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Imura A, Hori T, Imada K, Ishikawa T, Tanaka Y, Maeda M, Imamura S, Uchiyama T. The human OX40/gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. J Exp Med. 1996;183:2185–2195. doi: 10.1084/jem.183.5.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor L, Bachler M, Duncan I, Keen S, Fallon R, Mair C, McDonald TT, Schwarz H. In vitro and in vivo activities of OX40 (CD134)-IgG fusion protein isoforms with different levels of immune-effector functions. J Leuko Biol. 2002;72:522–529. [PubMed] [Google Scholar]
- 56.Ohshima Y, Tanaka Y, Tozawa H, Takahashi Y, Maliszewski C, Delespesse G. Expression and function of OX40 ligand on human dendritic cells. J Immunol. 1997;159:3838–3848. [PubMed] [Google Scholar]
- 57.Jenkins SJ, Perona-Wright G, Worsley AG, Ishii N, MacDonald AS. Dendritic cell expression of OX40 ligand acts as a costimulatory, not polarizing, signal for optimal Th2 priming and memory induction in vivo. J Immunol. 2007;179:3515–3523. doi: 10.4049/jimmunol.179.6.3515. [DOI] [PubMed] [Google Scholar]
- 58.Calderhead DM, Buhlmann JE, van den Eertwegh AJ, Claassen E, Noelle RJ, Fell HP. Cloning of mouse Ox40: a T cell activation marker that may mediate T-B cell interactions. J Immunol. 1993;151:5261–5271. [PubMed] [Google Scholar]
- 59.Weinberg AD, Vella AT, Croft M. OX-40: life beyond the effector T cell stage. Semin Immunol. 1998;10:471–480. doi: 10.1006/smim.1998.0146. [DOI] [PubMed] [Google Scholar]
- 60.Maxwell JR, Weinberg A, Prell RA, Vella AT. Danger and OX40 receptor signaling synergize to enhance memory T cell survival by inhibiting peripheral deletion. J Immunol. 2000;164:107–112. doi: 10.4049/jimmunol.164.1.107. [DOI] [PubMed] [Google Scholar]
- 61.Ruby CE, Montler R, Zheng R, Shu S, Weinberg AD. IL-12 is required for anti-OX40-mediated CD4 T cell survival. J Immunol. 2008;180:2140–2148. doi: 10.4049/jimmunol.180.4.2140. [DOI] [PubMed] [Google Scholar]
- 62.Pockley AG. Heat shock proteins, anti-heat shock protein reactivity and allograft rejection. Transplantation. 2001;71:1503–1507. doi: 10.1097/00007890-200106150-00001. [DOI] [PubMed] [Google Scholar]
- 63.Lathrop SK, Huddleston CA, Dullforce PA, Montfort MJ, Weinberg AD, Parker DC. A signal through OX40 (CD134) allows anergic, autoreactive T cells to acquire effector cell functions. J Immunol. 2004;172:6735–6743. doi: 10.4049/jimmunol.172.11.6735. [DOI] [PubMed] [Google Scholar]
- 64.Lechler RI, Garden OA, Turka LA. The complementary roles of deletion and regulation in transplantation tolerance. Nat Rev. 2003;3:147–158. doi: 10.1038/nri1002. [DOI] [PubMed] [Google Scholar]
- 65.Monk NJ, Hargreaves RE, Marsh JE, Farrar CA, Sacks SH, Millrain M, Simpson E, Dyson J, Jurcevic S. Fc-dependent depletion of activated T cells occurs through CD40L-specific antibody rather than costimulation blockade. Nat Med. 2003;9:1275–1280. doi: 10.1038/nm931. [DOI] [PubMed] [Google Scholar]
- 66.Sanchez-Fueyo A, Domenig C, Strom TB, Zheng XX. The complement dependent cytotoxicity (CDC) immune effector mechanism contributes to anti-CD154 induced immunosuppression. Transplantation. 2002;74:898–900. doi: 10.1097/01.TP.0000028779.24725.CF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Csencsits K, Burrell BE, Lu G, Eichwald EJ, Stahl GL, Bishop DK. The classical complement pathway in transplantation: unanticipated protective effects of C1q and role in inductive antibody therapy. Am J Transplant. 2008;8:1622–1630. doi: 10.1111/j.1600-6143.2008.02295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vogel LA, Noelle RJ. CD40 and its crucial role as a member of the TNFR family. Semin Immunol. 1998;10:435–442. doi: 10.1006/smim.1998.0145. [DOI] [PubMed] [Google Scholar]
- 69.Oderup C, Malm H, Ekberg H, Qi Z, Veress B, Ivars F, Corbascio M. Costimulation blockade-induced cardiac allograft tolerance: inhibition of T cell expansion and accumulation of intragraft CD4(+)Foxp3(+) T cells. Transplantation. 2006;82:1493–1500. doi: 10.1097/01.tp.0000244064.66136.04. [DOI] [PubMed] [Google Scholar]
- 70.Taylor PA, Lees CJ, Waldmann H, Noelle RJ, Blazar BR. Requirements for the promotion of allogeneic engraftment by anti-CD154 (anti-CD40L) monoclonal antibody under nonmyeloablative conditions. Blood. 2001;98:467–474. doi: 10.1182/blood.v98.2.467. [DOI] [PubMed] [Google Scholar]
- 71.Ochando JC, Yopp AC, Yang Y, Garin A, Li Y, Boros P, Llodra J, Ding Y, Lira SA, Krieger NR, Bromberg JS. Lymph node occupancy is required for the peripheral development of alloantigen-specific Foxp3+ regulatory T cells. J Immunol. 2005;174:6993–7005. doi: 10.4049/jimmunol.174.11.6993. [DOI] [PubMed] [Google Scholar]
- 72.Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, Killeen N, Ishii N, Li XC. OX40 costimulation turns off Foxp3+ TREGS. Blood. 2007;110:2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kroemer A, Xiao X, Vu MD, Gao W, Minamimura K, Chen M, Maki T, Li XC. OX40 controls functionally different T cell subsets and their resistance to depletion therapy. J Immunol. 2007;179:5584–5591. doi: 10.4049/jimmunol.179.8.5584. [DOI] [PubMed] [Google Scholar]
- 74.Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205:825–839. doi: 10.1084/jem.20071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Snapper CM, Paul WE. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science. 1987;236:944–947. doi: 10.1126/science.3107127. [DOI] [PubMed] [Google Scholar]
- 76.Wasowska BA, Qian Z, Cangello DL, Behrens E, Van Tran K, Layton J, Sanfilippo F, Baldwin WM., 3rd Passive transfer of alloantibodies restores acute cardiac rejection in IgKO mice. Transplantation. 2001;71:727–736. doi: 10.1097/00007890-200103270-00007. [DOI] [PubMed] [Google Scholar]
- 77.Rahimi S, Qian Z, Layton J, Fox-Talbot K, Baldwin WM, 3rd, Wasowska BA. Non-complement- and complement-activating antibodies synergize to cause rejection of cardiac allografts. Am J Transplant. 2004;4:326–334. doi: 10.1111/j.1600-6143.2004.00334.x. [DOI] [PubMed] [Google Scholar]