

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder that impairs memory and cognition. One of the major neuropathological hallmarks is the accumulation of the extracellular senile plaques that are mainly composed of amyloid beta (Aβ) protein. Plaques are associated with synapse loss, dystrophic neurites and altered neurite trajectories. A reversal of such morphological changes has been observed days after single dose anti Aβ immunotherapy. In this study we investigated the extended effects of a single dose of passive anti Aβ immunotherapy on morphological changes associated with senile plaques. We found that although plaque burden was not reduced 30 days after immunotherapy, there were fewer dystrophic neurites around each plaque, a recovery of synapse density, and normalization of neurite curvature near plaques. Taken together these results suggest that single dose immunotherapy is sufficient to cause lasting benefits to the morphology of cortical neurons, implying substantial plasticity of neural circuits despite the continued presence of plaques.
Keywords: Alzheimer’s, immunotherapy, amyloid, senile plaque, synaptic plasticity
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder affecting more than 4 million Americans and >30 million individuals worldwide (Kawas, et al., 2003). It results in memory loss, personality changes and cognitive decline. The main neuropathological hallmarks of AD are intracellular neurofibrillary tangles consisting of hyperphosphorylated tau and extracellular senile plaque deposition consisting mainly of amyloid beta (Aβ) peptide. Plaques have been shown to disrupt synaptic integration in mice (Stern, et al., 2004) hence possibly affecting cognition. In addition, plaques are also associated with altered neuritic trajectories, dystrophic neurites and dendritic spine loss (D’Amore, et al., 2003, Knowles, et al., 1998, Knowles, et al., 1999, Spires, et al., 2005, Tsai, et al., 2004). Neurites have been shown to be more curved inside and close to plaques compared to plaques and that has been suggested to interrupt neuronal networks (Knowles, et al., 1999, Le, et al., 2001). Swollen dystrophic neurites appear around plaques in AD and are may interfere with cell transport (Spires and Hyman, 2004). Work in several mouse models has shown that dendritic spine loss is most profound around plaques (Moolman, et al., 2004, Spires, et al., 2005, Spires-Jones, et al., 2007). The cause for this spine loss could be the concentration of toxic oligomeric Aβ species (or other toxic molecules) around plaques (Koffie, et al., 2009, Meyer-Luehmann, et al., 2008, Shankar, et al., 2008).
Removing Aβ plaques and oligomeric species could prove to be a valuable treatment (Schenk, et al., 1999). So far it has been shown that with passive immunotherapy it is possible to remove Aβ plaques in mice (Bacskai, et al., 2001, Bard, et al., 2000, Lombardo, et al., 2003, Morgan, et al., 2000). Along with clearing plaques, immunotherapy restores neurite architecture, increases synapse density and improves behavior (Dodart, et al., 2002, Kotilinek, et al., 2002, Lombardo, et al., 2003, Wilcock, et al., 2006, Thakker, et al., 2009). These events have been shown to occur over several days. Indeed, early improvements in neuritic morphology occur even prior to amyoloid clearance, within 24 hours (Brendza, et al., 2005, Spires-Jones, et al., 2008). However, no extended effects of plaque associated neuritic changes have yet been investigated. It is essential to observe how long the recovery of neuritic morphological changes after a single dose of anti Aβ treatment persists. In this study we performed a single dose passive immunotherapy and quantified morphological changes 30 days later. We hypothesize that single-dose immunotherapy can have lasting beneficial effects on plaque-associated anatomical degeneration.
2. Results
After acute anti Aβ immunotherapy treatment it has been shown that Aβ plaques have been cleared within a few days (Bacskai, et al., 2001). To determine whether Aβ plaques are present 30 days after a single dose of anti-Aβ antibody treatment, we labeled plaques with R1282 immunostaining and calculated the plaque burden in treated and untreated areas of cortex. We found that plaques were present in Tg2576 cortex but not non-transgenic controls as expected. Treatment with anti-Aβ antibody 3D6 did not lead to a statistically significant reduction in plaques observed 30 days post-treatment (Fig 1). Plaque burden was not significantly reduced in anti-Aβ treated mice (mean plaque burden = 0.99% ± 0.99 in region treated with antibody) either in comparison to animals treated with an irrelevant antibody (mean plaque burden=1.42% ± 1.54 in treated area) or when compared to a region within the anti-Aβ treated brain that was not exposed to antibody treatment (mean plaque burden in untreated area of anti-Aβ treated mice=1.01% ± 1.06 (p>0.05)). These data indicate that despite early clearance of plaques with passive immunotherapy as has been observed previously, there is a probable re-deposition within 30 days after a single dose of monoclonal antibody in this model.
Fig 1.

Plaque burden 30 days after anti Aβ immunotherapy treatment. Tg2576 and age-matched non-transgenic animals were treated with topical application of either anit-Aβ antibody or anti human tau antibody. The area treated with antibody is outlined in red in panel (A) and is referred to in text as the treated cortex. (B) Plaques were immunolabeled with R1282 primary antibody and visualized with Alexa488. (C) There was no difference in cortical plaque burden between animals treated with an anti Aβ antibody 3D6 and those treated with control antibody 12E8 (p<0.05). Control non-transgenic animals had no plaques as expected. Scale bar 50 μm. Data presented as mean ± SD. Panel with the coronal section was modified from the high resolution mouse brain atlas (http://www.hms.harvard.edu/research/brain/atlas.html).
Dystrophic neurites are often seen around plaques in AD and are an indication of neurodegeneration (Spires and Hyman, 2004). A reduction in the number of dystrophies has been observed in the first several days after anti Aβ immunotherapy treatment (Brendza, et al., 2005). We wanted to investigate whether the effect would persist 30 days after a single dose of passive immunotherapy treatment. In the antibody treated area there was a reduction in the number of axonal dystrophies per plaque in mice treated with anti Aβ (3D6) antibody (mean number of dystrophies per plaque=2.85 ± 0.64), compared to the control anti human tau antibody (mean number of dystrophies per plaque=4.51 ± 1.75, t-test p<0.001) (Fig 2). The effect was only seen in the area treated with anti Aβ antibody. There was a reduction in axonal dystrophies when anti Aβ treated cortex was compared to the area of cortex untreated with antibody in the same animals (p<0.01). No such effect was seen in Tg2576 animals treated with control antibody. These results together with previous findings suggest that dystrophic swellings can be rapidly reversed and the recovery can persist for at least 30 days despite the presence of Aβ plaques with a single dose of anti Aβ treatment.
Fig 2.
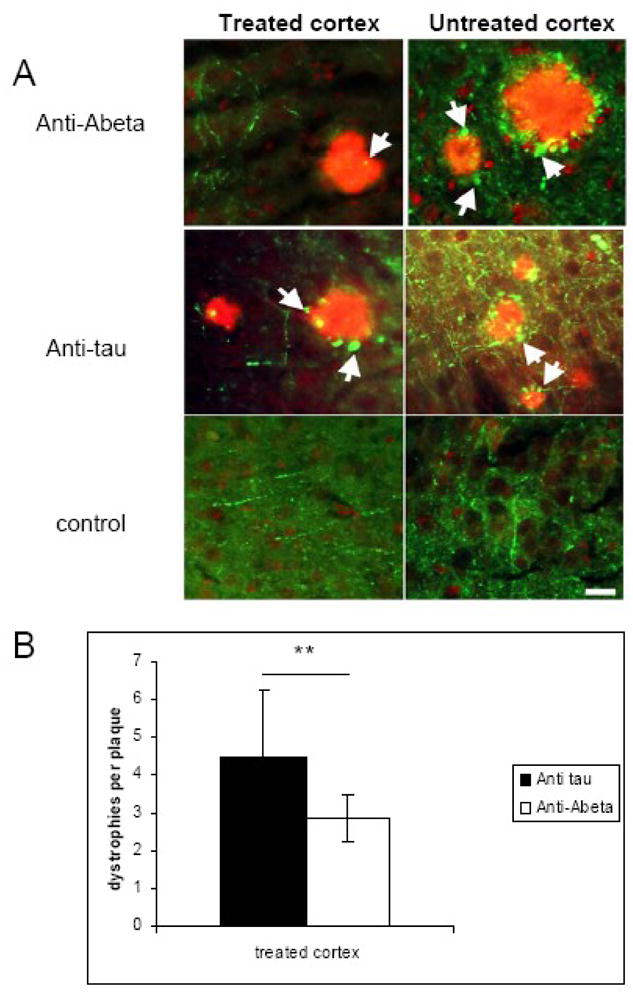
Effects of passive immunotherapy on axonal dystrophies. (A) Plaque-associated axonal dystrophies (arrows) were analyzed on sections stained with SMI312 (green) and R1282 to label plaques (red). (B) Quantification shows that a single topical treatment with an antibody to Aβ caused a reduction in the average number of dystrophic axonal swellings per plaque 30 days after the treatment (** p<0.001). The effect on axons was observed only in the area treated with anti Aβ antibody and was not seen in the untreated area or in animals treated with irrelevant anti human tau antibody (12E8). Scale bar 20 μm. Data presented as mean ± SD.
Synapse loss is the strongest correlate of cognitive decline in Alzheimer’s disease (Terry, et al., 1991). We wanted to determine whether improvements in synapse density with a single dose of anti Aβ immunotherapy treatment persisted for an extended period (30 days). Untreated Tg2576 cortex had a trend toward synapse loss compared to controls (Mann-Whitney test p=0.05) of 55% loss in synaptophysin immunoreactivity. We detected a significant 56% increase of presynaptic terminal (synaptophysin) optical density in Tg2576 cortex treated with anti Aβ antibody compared to the control treatment, so that synaptophysin optical density in the treated area is the same as in non-tg animals (p<0.01 Mann-Whitney test, Fig 3). In non-Tg animals, treatment with anti Aβ antibody had no effect compared to treatment with control antibody. These results suggest that synapse loss can be rescued by single dose passive immunotherapy and persist 30 days after the treatment despite the presence of fibrillar Aβ.
Fig 3.

Presynaptic terminal density increases 30 days after a single dose of passive immunotherapy treatment. (A) SY38 synaptophysin staining (green) shows presynaptic terminal loss in the area of cortex occupied by R1282 labeled (red) plaques (arrows). (B) A box plot of synaptophysin optical density data shows that there is a trend towards 55% presynaptic terminal loss in animals treated with the control anti tau antibody compared to non-transgenic animals (# Mann-Whitney test p=0.0527). Presynaptic terminal density recovered in tissue treated with anti Aβ antibody (3D6) compared to tissue treated with anti tau antibody (* Mann-Whitney test p<0.01). The recovery occurred despite persistent plaque pathology. Scale bar 50 μm. Optical density data were not normally distributed thus are presented in box plots with median, maxima, and minima shown.
It has been shown that neurites have altered trajectories around plaques in AD (D’Amore, et al., 2003, Knowles, et al., 1998, Knowles, et al., 1999, Le, et al., 2001, Lombardo, et al., 2003) and with passive immunotherapy this can be rescued in several days (Lombardo, et al., 2003). In Tg2576 cortex treated with the control antibody, axons have an average curvature ratio of 1.026 ± 0.027. Anti-Aβ treated areas average curvature ratio was 1.034 ± 0.038 showing no effect of treatment on average curvature when distance from a plaque was not taken into account (t-test p>0.05). When phantom plaques were created and distance to axons measured we found no difference between axons close (<50 μm) and far (>50 μm) from phantom plaques as expected. We calculated the ratio of close to far and found it to be 1.0057 and used that as a control baseline (Fig. 4). However, in transgenic animals treated with control antibody, axons were more curved close (<50 μm) to plaques compared to far (>50 μm) from plaques (curvature ratio 1.05 ± 0.065 and 1.034 ± 0.042, respectively, ratio close to far 1.011 t-test, p<0.05) (Fig. 4). 30 days after the treatment with anti-Aβ antibody there was no difference between axons that were close (curvature ratio 1.056 ± 0.042) to those that were far (curvature ratio 1.045 ± 0.057 ratio close to far 1.007) from plaques (t-test, p>0.05) (Fig. 4), indicating some normalization of axon trajectory with anti-Aβ treatment, although the curvature does not approach control levels.
Fig 4.

Effects of single dose passive immunotherapy treatment on axon curvature ratios. (A) Axon curvature ratios were measured on SMI312 labeled axons (red) in proximity to Thioflavin S positive plaques (blue). (B) After single dose topical application of antibody to Aβ (3D6) axons close (<50 μm) to plaque were as curved as axons far (>50 μm) from plaque (p>0.05), however without reaching the level of non-transgenic animals. Axons of animals treated with anti tau antibody were significantly more curved close to plaques compared to far from plaques (* p<0.05). Control animals had significantly straighter axons compared to transgenic animals (p<0.05). Scale bar 10 μm. Data presented as mean ± SD.
Together, these data show lasting morphological benefits of a single dose of passive anti-Aβ immunotherapy.
3. Discussion
The amyloid hypothesis of AD suggests that the onset and progression of pathogenesis of AD is influenced by the accumulation of Aβ in the brain (Hardy and Selkoe, 2002). However, plaques have been shown to poorly correlate with the progression of the disease (Arriagada, et al., 1992). A much better indicator of cognitive decline is synaptic density, neurofibrillary tangles and dystrophic neurites around plaques (Ingelsson, et al., 2004, McKee, et al., 1991, Terry, et al., 1991), indicating that prolonged recovery of these markers is possibly more important than plaque removal which could theoretically release more synaptotoxic soluble species of Aβ.
In our study we showed that plaques were present 30 days after a single treatment with anti-Aβ antibody similar to findings by Oddo et al. in another mouse model of AD (Oddo, et al., 2004). Since immunotherapy is a well established way of clearing plaques (Bacskai, et al., 2001, Bard, et al., 2000, Schenk, et al., 1999), it is likely that in our mice, plaques were cleared quickly, then re-emerged during the 30 days post-treatment as seen in the triple transgenic model (Oddo, et al., 2004).
We find that despite the persistent Aβ plaques 30 days after treatment, the density of presynaptic markers was increased. This indicates that it is possible to reverse synapse loss - one of the strongest correlates with cognitive decline. It has been recently shown that dendritic spine plasticity can be improved after 1 hour of immunotherapy treatment (Spires-Jones, et al., 2008) and here we have shown that similar effects in presynaptic markers can persist for at least 30 days.
Another important correlate with stages of dementia and a pathological hallmark of AD are dystrophic neuritic swellings (McKee, et al., 1991, Spires and Hyman, 2004). It has been shown that dystrophic neurites can recover 3 days after an anti Aβ treatment (Brendza, et al., 2005). In the current study we showed that axonal dystrophies around plaques remain reduced 30 days after the treatment despite the presence of Aβ plaques. This suggests that the presence of fibrillar aggregates of Aβ does not preclude the recovery of either synapses or axonal dystrophies. These results taken together with previous results indicate that dystrophic swellings can appear within a matter of days (Meyer-Luehmann, et al., 2008), clear with passive immunotherapy within several days (Brendza, et al., 2005), and the current study suggests that this recovery can persist for 30 days.
It has been reported that dendrites and axons are more curved close compared to far from Aβ plaques (D’more, et al., 2003, Garcia-Alloza, et al., 2006, Knowles, et al., 1998, Knowles, et al., 1999) and that dendrites (labeled with SMI32) can straighten within 4 days after treatment, an effect which persists for 32days (Lombardo, et al., 2003, McKee, et al., 1991). In this study we found that, similar to the results with dendrites, 30 days after treatment, the curvature of axons near plaques was the same as those farther away, while in animals treated with an irrelevant antibody, curvature was still more pronounced near plaques.
It can be inferred from our study that anti Aβ treatment is sufficient to induce the recovery of synapses and neurite morphology persists for at least 30 days despite the presence of fibrillar plaques at this time point. It is possible that toxic oligomeric species being produced in the Tg2576 animals are being sequestered into new plaques after an initial clearance of both soluble and fibrillar species explaining the lingering beneficial effects on neurite morphology. We presume that once the pre-treatment equilibrium of fibrillar and soluble species is reached, another dose anti Aβ antibody would be necessary to continue the beneficial effects. This could prove beneficial in patient immunotherapy trials as only one dose passive immunotherapy treatment could provide lasting recovery of neurite morphology and by extension neural system integrity.
4. Experimental Procedures
Animals and Surgery
All animal work conformed to National Institutes of Health and institutional guidelines. Tg2576 mice expressing human amyloid precursor protein with the AD-associated Swedish mutation (Hsiao, et al., 1996) between ages 18 to 26 month and non-transgenic littermate controls were used for this study. A craniotomy was performed as described previously (Bacskai, et al., 2002, Klunk, et al., 2002, Lombardo, et al., 2003, Skoch, et al., 2004). After dura resection animals were treated with topical application of (25 μL volume) of either anti Aβ antibody (3D6) (1mg/mL concentration) (Elan Pharmaceuticals) or an irrelevant human tau antibody (12E8 or 16B5) (1 mg/mL concentration) (Elan Pharmaceuticals). Following the treatment, the craniotomy was sealed with an 8mm diameter glass coverslip and animals were left to recover. 30 days post treatment animals were euthanized using an overdose of Avertin (400mg/kg, i.p.). Brains were fixed using 4% Paraformaldehyde and 15% glycerol for cryoprotection for 48h then one hemisphere was sectioned at 50 μm using freezing microtome while the other was paraffin embedded and sectioned at 16 μm, both were sectioned coronally.
Plaque burden
Every 15th section (selected on systematic random basis) from the paraffin-embedded hemisphere was deparafinized and blocked in 5% Normal Goat Serum (NGS) for 1h. Sections were then immunostained with Aβ antibody R1282 (1: 500 in 1 % NGS in Tris-buffered saline, gift from Dr. D. Selkoe, Brigham and Women’s Hospital, Boston, MA) and Alexa488 (Invitrogen, Eugene, OR) was used as a secondary antibody (1:100 in 1 % NGS in Tris-buffered saline, Invitrogen, Eugene, OR). Aβ fluorescent immunostaining was detected by scanning stained sections in a scan array reader (Perkin Elmer, Downers Growe, IL) using a 488 laser for excitation. Scanned images were analyzed using ImageJ software. Regions of interest were outlined in the superficial 200 μm of cortex under the craniotomy (the area that was exposed to antibody treatment) and in the superficial 200 μm of an untreated region (piriform cortex). The total area occupied by plaques was divided by the total area of the cortex sampled to obtain the percentage of cortex covered by Aβ immunoreactivity (plaque burden).
Axon curvature
Every 10th PFA fixed (free floating) section (first section selected on systematic random basis) was permeabilized using 0.5% TritionX-100 in TBS for 20 minutes followed by 1h blocking in 5% NGS. To visualize axons the tissue was incubated with a SMI 312 (1:1000 in 1 % NGS in Tris-buffered saline, Covance Research Products, Inc., Berkely, CA) antibody that recognizes pan axonal neurofilament and conveniently labels axons and axonal dystrophies associated with plaques. A secondary antibody anti-mouse IgG conjugated to Cy3 dye (1:200 in 1 % NGS in Tris-buffered saline, Jackson ImmunoResearch, West Grove, PA) was used and then sections were counterstained with Thioflavine-S (0.05% in 50% ethanol) (Sigma, St. Louis, MO).
Images for axon analysis were taken on upright Olympus BX51 (Olympus, Denmark) fluorescence microscope with a DP70 camera sampling 100% of the 200 um of the area treated with the antibody. Only axons longer than 20 μm were chosen for analysis and their curvature ratio was calculated in Image J by dividing the curvilinear length of the axon by the straight line length of the process (D’more, et al., 2003, Garcia-Alloza, et al., 2006, Knowles, et al., 1999, Lombardo, et al., 2003). Distance from the measured axon segment to the closest senile plaque (if present) was measured at three points – from each end and the midpoint of the segment and the average distance was taken from these three measurements. Axons that were closer than 50 μm to the plaque were defined as being close to the plaque and others as far from the plaque.
Dystrophies per plaque
Every 15th section (selected on systematic random basis) was immunolabled with SMI312 antibody as described above using Alexa488 (Invitrogen, Eugene, OR) to visualize axons and dystrophies. Plaques were immunolabled using Amyloid beta antibody R1282 (gift from Dr. D. Selkoe, Brigham and Women’s Hospital, Boston, MA) as described above and visualized using Cy3 (Jackson ImmunoResearch, West Grove, PA). Smi312 positive dystrophies within the plaque area that were larger than 2.5 μm2 were classified as dystrophic neurites and counted for each plaque using an upright Olympus BX51 (Olympus, Denmark) fluorescence microscope. The average number of dystrophies per plaque was calculated.
Synapse quantification
Every 15th paraffin section (selected on systematic random basis) was deparaffinized and incubated in 1 mg/ml pepsin in 0.2N HCl for 10 min and blocked in 5% NGS for 1h. Sections were immunolabled with presynaptic marker antibody Sy38 (1:10 in 1 % NGS in Tris-buffered saline, Synaptic Systems) with an anti-mouse Alexa 546 secondary antibody (1:100 in 1 % NGS in Tris-buffered saline, Invitrogen, Eugene, OR) and plaques were labeled with R1282 and an anti-rabbit Alexa 488 secondary antibody as described earlier. Optical density of fluorescent immunostaining was determined using a Scan Array Express (Perkin Elmer, Boston MA). A 568nm laser was used to excite the fluorophore and scans of each section were acquired at 3 focal planes. In Image J software (free from the NIH), the three images of each section were projected into one composite. Regions of interest were outlined in the superficial 200 μm of cortex under the craniotomy (the area that was exposed to antibody treatment) and in the superficial 200 μm of an untreated region (piriform cortex). Mean grayvalues for each group were calculated.
Acknowledgments
This work was supported by NIH grants AG08487, a John D French Foundation Fellowship, the Alzheimer’s Disease Drug Discovery Foundation, and Alzheimer’s Association Pioneer Award and grant EB00768. We also thank Elan for providing antibodies (3D6 and 12E8).
Abbreviations
- AD
Alzheimer’s disease
- Aβ
amyloid-beta peptide
- NGS
normal goat serum
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arriagada P, Growdon J, Hedley-Whyte E, Hyman B. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–39. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–72. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Klunk WE, Mathis CA, Hyman BT. Imaging amyloid-beta deposits in vivo. J Cereb Blood Flow Metab. 2002;22:1035–41. doi: 10.1097/00004647-200209000-00001. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–9. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Brendza RP, Bacskai BJ, Cirrito JR, Simmons KA, Skoch JM, Klunk WE, Mathis CA, Bales KR, Paul SM, Hyman BT, Holtzman DM. Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest. 2005;115:428–33. doi: 10.1172/JCI23269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amore JD, Kajdasz ST, McLellan ME, Bacskai BJ, Stern EA, Hyman BT. In vivo multiphoton imaging of a transgenic mouse model of Alzheimer disease reveals marked thioflavine-S-associated alterations in neurite trajectories . J Neuropathol Exp Neurol. 2003;62:137–45. doi: 10.1093/jnen/62.2.137. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci. 2002;5:452–7. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Dodwell SA, Meyer-Luehmann M, Hyman BT, Bacskai BJ. Plaque-derived oxidative stress mediates distorted neurite trajectories in the Alzheimer mouse model. J Neuropathol Exp Neurol. 2006;65:1082–9. doi: 10.1097/01.jnen.0000240468.12543.af. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–31. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- Kawas CH, Corrada MM, Brookmeyer R, Morrison A, Resnick SM, Zonderman AB, Arenberg D. Visual memory predicts Alzheimer’s disease more than a decade before diagnosis. Neurology. 2003;60:1089–93. doi: 10.1212/01.wnl.0000055813.36504.bf. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61:797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- Knowles RB, Gomez-Isla T, Hyman BT. Abeta associated neuropil changes: correlation with neuronal loss and dementia. J Neuropathol Exp Neurol. 1998;57:1122–30. doi: 10.1097/00005072-199812000-00003. [DOI] [PubMed] [Google Scholar]
- Knowles RB, Wyart C, Buldyrev SV, Cruz L, Urbanc B, Hasselmo ME, Stanley HE, Hyman BT. Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer’s disease. Proc Natl Acad Sci U S A. 1999;96:5274–9. doi: 10.1073/pnas.96.9.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie R, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva K, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0811698106. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH. Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J Neurosci. 2002;22:6331–5. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le R, Cruz L, Urbanc B, Knowles RB, Hsiao-Ashe K, Duff K, Irizarry MC, Stanley HE, Hyman BT. Plaque-induced abnormalities in neurite geometry in transgenic models of Alzheimer disease: implications for neural system disruption. J Neuropathol Exp Neurol. 2001;60:753–8. doi: 10.1093/jnen/60.8.753. [DOI] [PubMed] [Google Scholar]
- Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, Hyman BT. Amyloid-beta antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J Neurosci. 2003;23:10879–83. doi: 10.1523/JNEUROSCI.23-34-10879.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Kosik KS, Kowall NW. Neuritic pathology and dementia in Alzheimer’s disease. Ann Neurol. 1991;30:156–65. doi: 10.1002/ana.410300206. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–4. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML. Dendrite and dendritic spine alterations in alzheimer models. J Neurocytol. 2004;33:377–87. doi: 10.1023/B:NEUR.0000044197.83514.64. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–5. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–32. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoch J, Hickey GA, Kajdasz ST, Hyman BT, Bacskai BJ. In vivo imaging of amyloid-beta deposits in mouse brain with multiphoton microscopy. In: Sigurdsson EM, editor. Amyloid proteins: methods and protocols. Humana Press; Totowa: 2004. pp. 349–364. [DOI] [PubMed] [Google Scholar]
- Spires TL, Hyman BT. Neuronal structure is altered by amyloid plaques. Rev Neurosci. 2004;15:267–78. doi: 10.1515/revneuro.2004.15.4.267. [DOI] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–87. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, Meyer-Luehmann M, Osetek JD, Jones PB, Stern EA, Bacskai BJ, Hyman BT. Impaired Spine Stability Underlies Plaque-Related Spine Loss in an Alzheimer’s Disease Mouse Model. Am J Pathol. 2007;171:1304–1311. doi: 10.2353/ajpath.2007.070055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, Mielke M, Rozkalne A, Meyer-Luehmann M, de Calignon A, Bacskai B, Schenk D, Hyman BT. Passive immunotherapy rapidly increases structural plasticity in a mouse model of Alzheimer disease. Neurobiol Dis. 2008 doi: 10.1016/j.nbd.2008.10.011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern EA, Bacskai BJ, Hickey GA, Attenello FJ, Lombardo JA, Hyman BT. Cortical synaptic integration in vivo is disrupted by amyloid-beta plaques. J Neurosci. 2004;24:4535–40. doi: 10.1523/JNEUROSCI.0462-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Thakker DR, Weatherspoon MR, Harrison J, Keene TE, Lane DS, Kaemmerer WF, Stewart GR, Shafer LL. Intracerebroventricular amyloid-{beta} antibodies reduce cerebral amyloid angiopathy and associated micro-hemorrhages in aged Tg2576 mice. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0813404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–3. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Alamed J, Gottschall PE, Grimm J, Rosenthal A, Pons J, Ronan V, Symmonds K, Gordon MN, Morgan D. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5340–6. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]