

Abstract
Astrocytes in the hippocampus release calcium (Ca2+) from intracellular stores intrinsically and in response to activation of Gq-linked G-protein-coupled receptors (GPCRs) through the binding of inositol 1,4,5-trisphosphate (IP3) to its receptor (IP3R). Astrocyte Ca2+ has been deemed necessary and sufficient to trigger the release of gliotransmitters, such as ATP and glutamate, from astrocytes to modulate neuronal activity. Several lines of evidence suggest that IP3R type 2 (IP3R2) is the primary IP3R expressed by astrocytes. To determine whether IP3R2 is the primary functional IP3R responsible for astrocytic Ca2+ increases, we conducted experiments using an IP3R2 knock-out mouse model (IP3R2 KO). We show, for the first time, that lack of IP3R2 blocks both spontaneous and Gq-linked GPCR-mediated increases in astrocyte Ca2+. Furthermore, neuronal Gq-linked GPCR Ca2+ increases remain intact, suggesting that IP3R2 does not play a major functional role in neuronal calcium store release or may not be expressed in neurons. Additionally, we show that lack of IP3R2 in the hippocampus does not affect baseline excitatory neuronal synaptic activity as measured by spontaneous EPSC recordings from CA1 pyramidal neurons. Whole-cell recordings of the tonic NMDA receptor-mediated current indicates that ambient glutamate levels are also unaffected in the IP3R2 KO. These data show that IP3R2 is the key functional IP3R driving Gq-linked GPCR-mediated Ca2+ increases in hippocampal astrocytes and that removal of astrocyte Ca2+ increases does not significantly affect excitatory neuronal synaptic activity or ambient glutamate levels.
Keywords: astrocyte; calcium; inositol 1,4,5-trisphosphate; IP3 receptor; hippocampus; gliotransmitter
Introduction
Astrocytes have been reported recently to have a functional role in neuronal excitability (Araque et al., 1998a; Haydon and Carmignoto, 2006), heterosynaptic depression (Pascual et al., 2005; Serrano et al., 2006; Andersson et al., 2007), cerebrovascular dynamics (Zonta et al., 2003; Straub and Nelson, 2007), and pathological states such as epilepsy (Kang et al., 2005; Tian et al., 2005; Fellin et al., 2006a). One unifying feature of these findings is that astrocyte modulation of these processes occurs via a Ca2+-dependent release of “gliotransmitters,” including ATP (which is converted to adenosine by ectonucleotidases) and glutamate (Montana et al., 2006). Astrocytes primarily use spatially and temporally encoded increases in Ca2+ as an intracellular signaling mechanism (Cornell-Bell et al., 1990; Jensen and Chiu, 1990; Scemes and Giaume, 2006). Astrocytes display Ca2+ increases both spontaneously (Parri et al., 2001; Nett et al., 2002; Hirase et al., 2004) and in response to neuronal stimulation (Porter and McCarthy, 1996; Aguado et al., 2002; Perea and Araque, 2005) and have been reported to modulate synaptic transmission through activation of metabotropic glutamate receptors (mGluRs), ionotropic glutamate receptors (iGluRs), and adenosine receptors (Haydon and Carmignoto, 2006).
Calcium increases in astrocytes are elicited predominantly by Gq-linked G-protein-coupled receptor (GPCR) activation, driving the production of IP3 and the activation of IP3 receptors (IP3Rs) coupled to endoplasmic reticulum (ER) Ca2+ stores. IP3Rs are a family of genes expressing three isoforms (types 1–3) of an ER Ca2+ release channel that are found in nearly every cell type (Foskett et al., 2007). Immunohistochemical studies aimed at identifying the expression profile of IP3Rs in the brain suggest that hippocampal astrocytes express primarily IP3R2. Evidence for IP3R2 expression in neurons is inconclusive (Sharp et al., 1999; Holtzclaw et al., 2002; Hertle and Yeckel, 2007). These data point to IP3R2 as a potential key mediator of astrocyte intracellular Ca2+ release and Ca2+-dependent signaling cascades, but functional evidence for IP3R2 in astrocytes is limited and has not been demonstrated in situ (Sheppard et al., 1997; Weerth et al., 2007).
We used an IP3R2 knock-out (KO) mouse model to determine whether IP3R2 has a functional role in Ca2+ increases of hippocampal astrocytes and neurons. We present the novel finding that genetic deletion of IP3R2 results in complete loss of spontaneous and agonist-evoked IP3R-dependent Ca2+ increases in astrocytes but leaves intact agonist-evoked IP3R-dependent Ca2+ increases in neurons. These data indicate that IP3R2 is the primary functional IP3R in astrocytes, and that IP3R2 does not play a demonstrated role in CA1 pyramidal neurons. We performed electrophysiological recordings of CA1 pyramidal neuron spontaneous EPSCs (sEPSCs) to determine the effect of eliminating astrocytic Ca2+ responses on baseline neuronal excitatory synaptic activity. No significant changes were found in any of the AMPA receptor (AMPAR) and NMDA receptor (NMDAR) sEPSC parameters of mice lacking astrocytic Ca2+ responses compared with littermate controls. Our results indicate that astrocytic Ca2+ responses are not important in modulating basal neuronal excitatory synaptic activity, contrary to the current state of the literature. We also found no differences in activation of NMDARs by ambient glutamate, providing additional evidence that ambient glutamate of glial origin is not released in a Ca2+-dependent manner (Jabaudon et al., 1999; Cavelier and Attwell, 2005). This study provides the first functional evidence that IP3R2 is the only IP3R isoform expressed by astrocytes, IP3R2 is not required for neuronal Gq GPCR-mediated Ca2+ elevations, and removal of astrocyte Ca2+ increases has no effect on basal neuronal excitatory activity.
Materials and Methods
Generation of IP3R2 KO mice.
IP3R2 KO mice were generated as described previously (Li et al., 2005). Briefly, a 539 bp fragment of exon 3 of IP3R2 (116 bp) was inserted into a targeting vector between two loxP sites. Mice were bred to heterozygosity for the floxed allele (IP3R2+/flox) and crossed to Pro-Cre mice. Pro-Cre, IP3R2+/flox were crossed to generate germline heterozygous null mutant offspring (IP3R2+/−), which were interbred to generate homozygous full mutant mice (IP3R2−/−) and littermate controls (IP3R2+/+ and IP3R2+/−). Mice were genotyped by PCR analysis using genomic DNA with IP3R2 wild-type (WT) and mutant allele-specific primers as given in the study by Li et al. (2005).
Hippocampal slice preparation.
All procedures followed the guidelines of the Institutional Animal Care and Use Committee of University of North Carolina at Chapel Hill. Littermate control and IP3R2 knock-out mice 10–16 d of age [postnatal day 10 (P10) to P16] were anesthetized by isoflurane inhalation. The brains were rapidly removed after decapitation and submerged into 4°C slicing buffer containing the following (in mm): 125 NaCl, 10 glucose, 1.25 NaH2PO4, 26 NaCHO3, 2.5 KCl, 3.8 MgCl2, and 0.1 kynurenic acid and bubbled with 95% O2 and 5% CO2. Brains were cut sagittally at a thickness of 300 μm on a Leica (Bannockburn, IL) vibratome. During sectioning, brains were kept submerged in 4°C oxygenated slicing buffer. Hippocampi were dissected out of each brain slice and incubated 45 min in artificial CSF (ACSF) warmed to 35–37°C and bubbled continuously with 95% O2 and 5% CO2. The ACSF contained the following (in mm): 125 NaCl, 10 glucose, 1.25 NaH2PO4, 26 NaCHO3, 2.5 KCl, 2.5 CaCl2, and 1.3 MgCl2.
Calcium imaging.
Astrocytes were bulk loaded with either the Ca2+ indicator Calcium Green-1 A.M. or Fluo-4 A.M. as described previously (Nett et al., 2002). Slices were incubated for 45 min at 35–37°C in oxygenated ACSF that included either 16 μm Fluo-4 A.M. or 11 μm Calcium Green 1-AM ester dye and 0.07% pluronic acid (final DMSO concentration, 0.4%). For measuring neuronal Ca2+ increases, CA1 pyramidal neurons were patch clamped with 200 μm Alexa 568 and 400 μm Fluo-4 calcium indicator dye made up in neuronal internal solution (see below). The pipette was then removed, and the neuron was allowed to recover for 10 min. Regions of interest were placed over the cell bodies of astrocytes and over the cell body and primary dendrite of CA1 pyramidal neurons. Increases in average fluorescence in regions of interest indicate an increase in Ca2+ concentration. Fold increase over baseline was calculated for each trace and reported as ΔF/F 0.
Neuronal patch-clamp recordings.
For AMPAR sEPSC recordings, CA1 pyramidal neurons were patch clamped using pipettes (4.0–6.0 MΩ resistance), and a gap-free recording was performed for 10 min in ACSF as described previously (Fiacco and McCarthy, 2004). For NMDAR sEPSCs, neurons were voltage clamped at −70 mV and superfused with ACSF containing 5 μm Mg2+ and 10 μm CNQX to block AMPA responses. For the ambient glutamate recordings, neurons were patch clamped at a holding potential of +40 mV in normal ACSF with an internal solution containing the following (in mm): 100 cesium methanesulfonate (CH3CsO3S), 10 tetraethylammonium-Cl, 4 NaCl, 1 MgCl2, 10 HEPES, 10 BAPTA, 5 phosphocreatine, 2 ATP, 0.3 GTP, pH adjusted to 7.3 with CsOH. Ambient glutamate current was recorded in ACSF containing 1 μm tetrodotoxin (TTX) and 100 μm picrotoxin.
Data collection and analysis.
Membrane currents were recorded using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). Traces were analyzed for sEPSCs in Clampfit 10.2 software (Molecular Devices) using a template constructed from four to six sEPSCs intrinsic to each recording. The event statistics were taken for each individual event and then averaged. The averaged event statistics from each cell were then averaged together and reported as mean ± SEM. For tonic NMDA currents from ambient glutamate recordings, membrane currents were normalized to the amplitude of the currents blocked by a saturating concentration of d-AP-5. Statistical differences between two samples were evaluated using Student's t test performed using Prism 4 software (Graphpad, San Diego, CA).
Histology.
Littermate control and IP3R2 KO brains were fixed with formalin and embedded sagittally in paraffin. Brains were sectioned at 6 μm thickness with a Leica (Nussloch, Germany) microtome from P30 littermate controls and IP3R2 KOs. Sections were stained with hematoxylin and eosin and imaged using a Zeiss (Oberkochen, Germany) Axioscope light microscope.
Reagents.
d-AP-5, TTX, CNQX, (RS)-3,5-dihydroxyphenylglycine (DHPG), histamine, carbachol, thapsigargin, dl-threo-β-benzyloxyaspartic acid (dl-TBOA), and picrotoxin were obtained from Tocris Bioscience (Bristol, UK). Calcium Green-1 A.M., Fluo-4, Fluo-4 A.M., and Alexa 568 were obtained from Invitrogen (Carlsbad, CA).
Results
Histological analysis of IP3R2 KO mouse brains
It has been reported previously that mice homozygous for the IP3R2 KO allele are viable and fertile and that the mice display no overt behavioral abnormalities (Li et al., 2005). We performed histological staining of paraffin-embedded sections from adult IP3R2 KO mice to determine whether there were any obvious abnormalities in brain cytoarchitecture indicative of improper proliferation or neurite outgrowth during development. Hematoxylin and eosin staining of P30 littermate control (n = 3) and IP3R2 KO (n = 3) brains revealed that lack of IP3R2 does not significantly affect brain cytoarchitecture. The hippocampus, cortex, and cerebellum of IP3R2 KO mice were examined and did not show any obvious structural abnormalities (Fig. 1 A). These findings suggest that IP3R2-mediated release of Ca2+ is not critical to the overall development of the brain or that alternate IP3Rs may be expressed in astrocytes during development.
Figure 1.
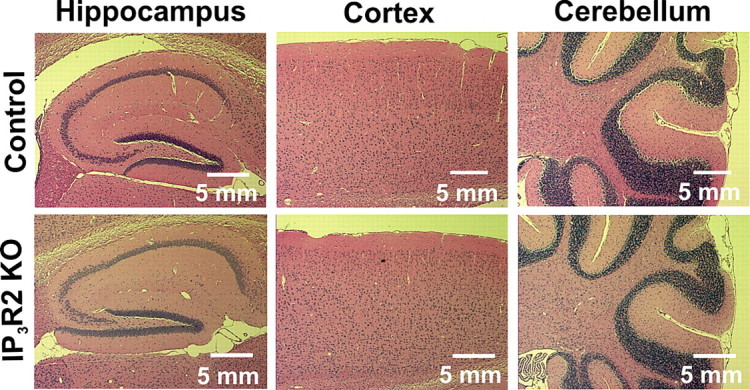
Histological analysis of IP3R2 KO brains reveals no obvious abnormalities. Histological staining of brain sections were taken from littermate control (n = 3) and IP3R2 KO (n = 3) mice. Six-micrometer-thick paraffin-embedded sections were cut and stained with hematoxylin and eosin to visualize brain cytoarchitecture. No difference in the gross overall morphology or in the general cell layering was apparent between the IP3R2 KO mice and littermate controls in any brain region; data from hippocampus, cortex, and cerebellum are shown.
IP3R2 KO hippocampal astrocytes lack spontaneous and Gq-linked GPCR Ca2+ increases
Astrocytes are known to respond to a wide variety of Gq GPCR agonists with Ca2+ elevations (Verkhratsky and Kettenmann, 1996). To address the functional role of IP3R2 in hippocampal astrocytes, we performed Ca2+ imaging experiments on bulk-loaded slices from littermate control and IP3R2 KO mice. Bath application of a Gq-linked GPCR agonist cocktail (DHPG, histamine, carbachol; 10 μm each) to hippocampal slices taken from littermate controls elicited robust increases in astrocyte intracellular Ca2+ (65% of 144 cells from 17 slices; five animals total) (Fig. 2 A,B). In striking contrast, the GPCR cocktail failed to elicit Ca2+ increases in IP3R2 KO hippocampal astrocytes (0% of 104 cells from 11 slices; five animals total) (Fig. 2 A,B). Similar results were obtained in experiments using 100 μm ATP, with 80% of 40 astrocytes from littermate controls (four slices total from one animal) and 0% of 38 astrocytes from IP3R2 KOs (three slices total from one animal) responding with Ca2+ increases (Fig. 2 A,B). In experiments where thapsigargin (2 μm) was applied as a control for Ca2+ increases, both littermate control (45% of 40 cells; four slices total from one mouse) and IP3R2 KO (45% of 38 cells, three slices total from one mouse) astrocytes responded to thapsigargin with increases in intracellular Ca2+, indicating that the Ca2+ stores themselves were intact (Fig. 2 A,B).
Figure 2.
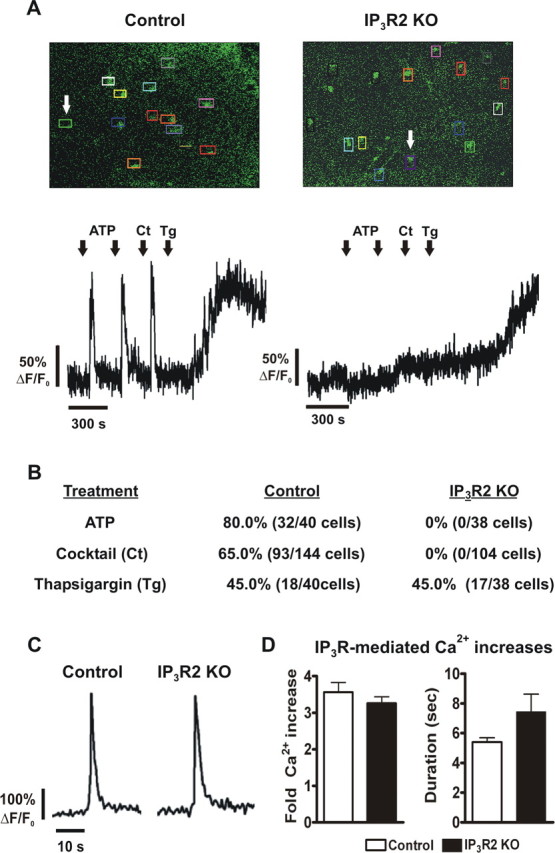
Knock-out of IP3R2 affects astrocyte but not neuronal GPCR-mediated Ca2+ increases. A, Representative Ca2+ traces from astrocytes of Calcium Green AM-loaded hippocampal slices. Regions of interest were placed over the cell bodies of bulk-loaded hippocampal astrocytes to measure Ca2+ increases in response to agonist application (top). Application of ATP (100 μm) or a Gq-linked GPCR agonist cocktail (Ct, 10 μm DHPG, 10 μm histamine, and 10 μm carbachol) elicited Ca2+ responses in astrocytes from littermate control but not IP3R2 KO hippocampal slices. The arrows indicate the astrocyte Ca2+ traces shown in the bottom panels. Thapsigargin (Tg; 2 μm) was used as a control and increased Ca2+ in astrocytes of both littermate control and IP3R2 KOs. Data are presented as fold increases over baseline. B, Percentage of astrocytes responding to application of ATP or the Gq-linked GPCR agonist cocktail from all experiments. C, Representative Ca2+ traces from CA1 pyramidal neurons patch clamped with internal solution containing Fluo-4 Ca2+ indicator in response to application of a Gq-linked GPCR cocktail (50 μm DHPG, 10 μm histamine, 10 μm carbachol) in the presence of 1 μm TTX to block action potentials. D, Amplitude and duration of IP3R-mediated Ca2+ responses in CA1 pyramidal neurons (control, n = 8; IP3R2 KO, n = 6). There were no significant differences for amplitude (left; p = 0.39) or duration (right; p = 0.08). Error bars indicate SEM.
Astrocytes have also been reported to exhibit spontaneous Ca2+ oscillations in the absence of neuronal activity (Nett et al., 2002). To determine whether IP3R2 is necessary for spontaneous Ca2+ oscillations, Ca2+ measurements were analyzed in astrocytes in the absence of agonist application. In bulk-loaded slices from littermate controls, spontaneous Ca2+ increases were observed in 21% of 144 total astrocytes (17 slices from five animals) but were absent in IP3R2 KO astrocytes (0% of 104 total cells, 11 slices from five animals). Together, these data indicate that astrocytes lacking IP3R2 are incapable of releasing Ca2+ from internal stores either spontaneously or in response to agonists to Gq-linked GPCRs.
Neuronal Ca2+ increases are intact in IP3R2 KO CA1 pyramidal neurons
To address the question of a potential functional role of IP3R2 in hippocampal neurons, we conducted Ca2+ imaging experiments on CA1 pyramidal neurons loaded with Ca2+ indicator via a patch pipette. Application of a Gq-linked GPCR agonist cocktail (in μm: 50 DHPG, 10 histamine, 10 carbachol) in 1 μm TTX to block action potentials elicited Ca2+ transients in IP3R2 KO neurons (six cells from six slices, five animals total) not significantly different in amplitude (Fig. 2 D, left) (p = 0.39) or duration (Fig. 2 D, right) (p = 0.08) from those measured in littermate controls (eight cells from eight slices, five animals total). These results indicate that lack of IP3R2 does not significantly alter IP3R-dependent Ca2+ signaling in CA1 pyramidal neurons.
Lack of spontaneous Ca2+ oscillations in astrocytes does not affect CA1 pyramidal neuron sEPSCs
It has been reported that spontaneous and evoked astrocyte Ca2+ elevations lead to gliotransmitter release, which modulates basal neuronal excitatory and inhibitory synaptic activity via the activation of neuronal mGluRs and iGluRs (Hassinger et al., 1995; Araque et al., 1998b; Kang et al., 1998; Parri et al., 2001; Fiacco and McCarthy, 2004; Liu et al., 2004a,b). The effect of blocking astrocyte Ca2+ elevations and therefore Ca2+-dependent gliotransmitter release on basal neuronal excitatory activity has not been addressed thoroughly. Therefore, we performed whole-cell patch-clamp experiments on CA1 pyramidal neurons and found that basic neuronal properties such as resting membrane potential (control, −61.7 ± 1.1 mV; IP3R2 KO, −60.3 ± 0.8 mV; p = 0.33), membrane resistance (control, 274.5 ± 26.2 M IP3R2 KO, 249.1 ± 15.5 MÙ; p = 0.40), and membrane capacitance (control, 89.6 ± 5.5 pF; IP3R2 KO, 96.4 ± 4.8 pF; p = 0.36) were not significantly different between littermate control and IP3R2 KO neurons (control, 21 cells from 20 slices, 10 animals total; IP3R2 KO, 22 cells from 17 slices, 10 animals total). In addition, there were no significant differences found between littermate control and IP3R2 KO AMPAR sEPSC peak amplitude (p = 0.62), 10–90% rise time (p = 0.94), decay tau (p = 0.66), and event frequency (p = 0.90) (Fig. 3 B) (control, 15 cells from 14 slices, nine animals total; IP3R2 KO, 16 cells from 11 slices, nine animals total). Additionally, no significant differences were found in NMDAR-mediated sEPSCs in peak amplitude (p = 0.39), 10–90% rise time (p = 0.92), decay tau (p = 0.94), and event frequency (p = 0.70) (Fig. 3 D) (control, 15 cells from 13 slices, six animals total; IP3R2 KO, nine cells from eight slices, three animals total). Together, these results indicate that lack of IP3R-dependent Ca2+ increases in astrocytes has no significant effect on either spontaneous AMPAR- or NMDAR-mediated excitatory synaptic currents in CA1 pyramidal neurons.
Figure 3.

Spontaneous EPSCs from CA1 pyramidal neurons are unchanged in IP3R2 KO mice. A, Representative AMPAR sEPSC traces from littermate control (n = 15) and IP3R2 KO (n = 16) CA1 pyramidal neurons. B, AMPAR sEPSC peak amplitude (p = 0.62), 10–90% rise times (p = 0.94), decay tau (p = 0.66), and event frequency (p = 0.90) were not significantly different between littermate control and IP3R2 KO CA1 pyramidal neurons as determined by Student's t test. C, Representative NMDAR sEPSC traces from littermate control (n = 15) and IP3R2 KO (n = 9) CA1 pyramidal neurons. D, NMDAR sEPSC peak amplitude (p = 0.39), 10–90% rise times (p = 0.92), decay tau (p = 0.94), and event frequency (p = 0.70) were not significantly different between littermate control and IP3R2 KO CA1 pyramidal neurons as determined by Student's t test. Error bars indicate SEM.
Ambient glutamate of astrocyte origin is not released in a Ca2+-dependent manner
Ambient glutamate levels in the hippocampus have been the focus of several recent studies (Herman and Jahr, 2007; Le Meur et al., 2007). Findings in this area suggest that the majority of ambient glutamate present in the hippocampus is of glial origin and might be released in a Ca2+-independent manner (Jabaudon et al., 1999; Cavelier and Attwell, 2005). To address whether ambient glutamate release occurs in a Ca2+-independent manner, whole-cell currents were recorded from CA1 pyramidal neurons held at +40 mV in ACSF containing 1 μm TTX and 100 μm picrotoxin to isolate NMDAR-mediated currents. Similar to previous reports (Herman and Jahr, 2007; Le Meur et al., 2007), application of the NMDAR antagonist d-AP-5 (50 μm) revealed a tonic NMDAR current of 33.7 ± 6.0 pA in littermate control neurons (six cells from six slices, four animals total) (Fig. 4 A,B). Recordings done in IP3R2 KO neurons found a d-AP-5-sensitive current that was not significantly different from that found in littermate control neurons (35.7 ± 9.2 pA; p = 0.8; five cells from five slices, three animals total) (Fig. 4 A,B).
Figure 4.

Ambient glutamate is unaffected by the lack of Ca2+ increases in astrocytes. A, Bath application of 50 μm d-AP-5 blocked a tonic NMDAR current in CA1 pyramidal neurons held at +40 mV in ACSF from both littermate control and IP3R2 KOs. B, The amplitude of the d-AP-5-sensitive NMDAR current was not significantly different (p = 0.8) in IP3R2 KO neurons (n = 5) versus littermate controls (n = 6). C, Representative traces showing that bath application of 100 μm TBOA induced a large fold increase of the tonic current in CA1 pyramidal neurons held at +40mV from both littermate control and IP3R2 KOs. D, The fold increase over baseline induced by TBOA is not significantly different (p = 0.4) between IP3R2 (n = 5 cells) and littermate controls (n = 6 cells). Error bars indicate SEM.
To determine whether removal of astrocytic Ca2+ increases affects ambient glutamate accumulation during elevated extracellular glutamate, glutamate transporters were blocked using 100 μm TBOA. Application of TBOA caused a 7.0 ± 1.3-fold change in the tonic NMDAR current of littermate control neurons (six cells from six slices, four animals total) and a similar fold change in KO neurons of 8.6 ± 1.1 (five cells from five slices, three animals total; p = 0.4) (Fig. 4 C,D). Additionally, there was no significant difference in the level of synaptic noise during TBOA application in the IP3R2 KOs (55.4 ± 4.1 pA; six cells from six animals; four slices total) versus littermate controls (66.8 ± 5.4 pA; p = 0.14; five cells from five slices; three animals total). Overall, these data indicate that astrocyte Ca2+ elevations do not play a significant role in regulating the ambient extracellular concentration of glutamate.
Discussion
Astrocytes have been reported to display both spontaneous and evoked intracellular Ca2+ increases using a variety of stimulation protocols (Montana et al., 2006). Astrocyte Ca2+ increases are caused by the release of Ca2+ from internal stores after activation of IP3Rs (Sheppard et al., 1997; Scemes, 2000). In this communication, we show that knock-out of IP3R2 abolishes both spontaneous and Gq-linked GPCR agonist evoked IP3R-dependent Ca2+ increases in astrocytes. To our knowledge, the findings presented here are the first demonstration that astrocyte Ca2+ release in situ is functionally reliant on IP3R2. In contrast, IP3R-dependent Ca2+ increases in CA1 pyramidal neurons remain intact, indicating that IP3R2 is not necessary for neuronal IP3R-mediated Ca2+ increases, or that IP3R2 may not be expressed by CA1 pyramidal neurons. This is supported by immunostaining data showing that IP3R2 is not expressed in neurons (Sharp et al., 1999; Hertle and Yeckel, 2007). It is somewhat surprising that in astrocytes, in which IP3R-dependent intracellular Ca2+ signals are thought to modulate an increasingly large number of key processes in brain (neuronal excitability, synaptic plasticity, and cerebrovascular control), that deletion of IP3R2 should result in the complete loss of Ca2+ activity without any apparent form of compensation. It is even more surprising that these mice: (1) are not embryonic lethal, (2) do not show early mortality, and (3) do not exhibit any obvious histological or behavioral abnormalities. They appear healthy, breed well, and live normal lifespans. This is in stark contrast to the IP3R1 KO mouse model, which displays tonic–clonic seizures, ataxia, and either die in utero or by weaning age (P21) (Matsumoto et al., 1996). Additional behavioral testing of the IP3R2 KO mouse model will provide valuable insight into the role of astrocyte Ca2+-dependent signaling in specific animal behaviors such as learning and memory.
Astrocytes have been implicated as a major source of ambient glutamate in the hippocampus (Jabaudon et al., 1999; Cavelier and Attwell, 2005; Herman and Jahr, 2007; Le Meur et al., 2007). In the present study, removal of astrocyte Ca2+ increases has no effect on the amplitude of the tonic NMDA-R mediated current activated by ambient glutamate. Furthermore, use of TBOA to block glutamate transporters revealed that the extent to which ambient glutamate accumulates during transporter block is unaffected by removal of IP3R2 in astrocytes (Fig. 4 B). These findings are in agreement with the hypothesis that ambient glutamate release from astrocytes occurs in a Ca2+-independent manner, possibly through nonvesicular release mechanisms such as connexin hemichannels, P2X channels or anion channels (Cavelier and Attwell, 2005; Malarkey and Parpura, 2008).
A substantial literature has developed in the field of astrocyte biology concerning the role of Ca2+-dependent release of gliotransmitters such as ATP (which is converted to adenosine by ectonucleotidases) and glutamate on neuronal activity (for review, see Carmignoto and Fellin, 2006; Fellin et al., 2006b; Fiacco and McCarthy, 2006). These findings led to the development of the tripartite synapse model, in which astrocytes are active participants in synaptic transmission through Ca2+-dependent gliotransmitter release (Araque et al., 1999). Although the majority of studies have focused on the outcome of pharmacologically evoking astrocyte Ca2+, very few studies have directly described the effect of blocking IP3R-dependent Ca2+ release on basal excitatory neuronal activity. Furthermore, it has been reported that spontaneous Ca2+ increases and subsequent glutamate release from astrocytes directly evoke NMDAR-mediated currents in neurons (Parri et al., 2001). Additionally, astrocyte Ca2+ increases have been associated with neuronal Ca2+ increases mediated by iGluRs (Hassinger et al., 1995; Pasti et al., 2001; Fellin et al., 2004). A mathematical model incorporating data from numerous studies, including our own (Fiacco and McCarthy, 2004), on the role of astrocytes at the tripartite synapse predicts that astrocytes enhance synaptic release (Nadkarni and Jung, 2007). This is reflected by an increase in spontaneous postsynaptic events during and immediately after astrocyte Ca2+ elevations that trigger astrocytic release of glutamate compared with synapses lacking an associated astrocyte (Nadkarni and Jung, 2007). According to this model, a lack of astrocyte Ca2+ increases would produce a reduced event frequency, reflecting reduced synaptic release. It has also been reported that heterosynaptic depression caused by Ca2+-dependent ATP release from astrocytes suppresses glutamate release at CA3–CA1 synapses (Pascual et al., 2005). Based on these findings, it is reasonable to speculate that abolishment of Ca2+ increases in IP3R2 KO astrocytes would lead to significant changes in basal excitatory neuronal activity and perhaps long-term changes in brain activity and behavior. In recordings of sEPSCs from IP3R2 KO and littermate control CA1 pyramidal neurons, we found no significant differences in peak amplitude, 10–90% rise time, and decay tau of both AMPAR- and NMDAR-mediated synaptic currents. Furthermore, we found no change in the frequency of AMPAR- and NMDAR-mediated sEPSCs, suggesting that lack of Ca2+ increases and Ca2+-dependent gliotransmitter release may not significantly affect baseline release probability from neuronal synaptic terminals.
The IP3R2 KO mouse model offers a compliment to another mouse model developed in our laboratory that enables selective stimulation of Gq-GPCR signaling cascades in astrocytes (Fiacco et al., 2007). The findings presented here support our recent discovery using the MrgA1 transgenic mice that selective, widespread astrocyte Ca2+ elevations have no effect on baseline neuronal excitatory synaptic activity. It has been reported previously by a number of labs (including our own) that mechanical stimulation or uncaging Ca2+ or IP3 in astrocytes leads to gliotransmitter release and changes in neuronal excitatory activity (Parpura and Haydon, 2000; Fiacco and McCarthy, 2004), neuronal inhibitory activity (Kang et al., 1998; Liu et al., 2004a,b), and cerebrovascular tone (Zonta et al., 2003; Straub et al., 2006). Although use of these pharmacological tools may elicit such responses, they may represent a nonphysiological level of stimulation that does not occur in vivo and therefore does not accurately recapitulate endogenous IP3 generating signaling pathways. There may be important regulatory mechanisms activated in GPCR signaling that work downstream of Ca2+ to inhibit vesicular release of glutamate by astrocytes. The IP3R2 KO and MrgA1 mouse models together fully corroborate the concept that stimulation of astrocytic Gq GPCRs and spontaneous astrocyte Ca2+ activity are not sufficient to cause vesicular release of glutamate from astrocytes, and that astrocyte Ca2+ elevations are not necessary for normal neuronal excitatory synaptic activity in hippocampal CA1 pyramidal neurons.
The IP3R2 mouse model also affords significant improvement over previously used techniques to examine the necessity of evoked IP3R-dependent Ca2+ increases to changes in neuronal synaptic activity. Calcium chelators such as BAPTA or the bulk loadable BAPTA-AM have been used to block astrocyte Ca2+ increases. There are technical issues with the use of BAPTA and Ca2+ chelators that are eliminated by the use of the IP3R2 KO mouse model. In the IP3R2 KO, release of Ca2+ from astrocyte internal stores is removed specifically, without causing a global change in the resting cytoplasmic Ca2+ concentration.
In conclusion, the IP3R2 KO mouse model represents a significant step forward in our ability to study the astrocyte Ca2+ contribution to key physiological processes. Use of this model has the potential to clarify and further define the role of astrocytes in physiology and pathology without the use of pharmacological manipulations to block astrocyte Ca2+ increases. This model can be used to identify Ca2+-dependent and Ca2+-independent mechanisms and their influence on astrocyte–neuronal communication, as well as re-evaluate the many important brain functions to which Ca2+-dependent gliotransmitter release has been reported to play a significant role.
Footnotes
This work was supported by National Institutes of Health Grants NS033938 and NS020212. We thank Dr. Cendra Agulhon for considerable discussion and feedback on previous versions of this manuscript. We also thank Dr. Ju Chen (University of California, San Diego, San Diego, CA) for the mice used in this study.
References
- Aguado F, Espinosa-Parrilla JF, Carmona MA, Soriano E. Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ . J Neurosci. 2002;22:9430–9444. doi: 10.1523/JNEUROSCI.22-21-09430.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson M, Blomstrand F, Hanse E. Astrocytes play a critical role in transient heterosynaptic depression in the rat hippocampal CA1 region. J Physiol (Lond) 2007;585:843–852. doi: 10.1113/jphysiol.2007.142737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci. 1998a;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci. 1998b;18:6822–6829. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Fellin T. Glutamate release from astrocytes as a non-synaptic mechanism for neuronal synchronization in the hippocampus. J Physiol (Paris) 2006;99:98–102. doi: 10.1016/j.jphysparis.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Cavelier P, Attwell D. Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J Physiol (Lond) 2005;564:397–410. doi: 10.1113/jphysiol.2004.082131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247:470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Fellin T, Gomez-Gonzalo M, Gobbo S, Carmignoto G, Haydon PG. Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices. J Neurosci. 2006a;26:9312–9322. doi: 10.1523/JNEUROSCI.2836-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Sul JY, D'Ascenzo M, Takano H, Pascual O, Haydon PG. Bidirectional astrocyte-neuron communication: the many roles of glutamate and ATP. Novartis Found Symp. 2006b;276:208–217. doi: 10.1002/9780470032244.ch16. discussion 217–221, 233–237, 275–281. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Astrocyte calcium elevations: properties, propagation, and effects on brain signaling. Glia. 2006;54:676–690. doi: 10.1002/glia.20396. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, Taves SR, Petravicz J, Casper KB, Dong X, Chen J, McCarthy KD. Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron. 2007;54:611–626. doi: 10.1016/j.neuron.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassinger TD, Atkinson PB, Strecker GJ, Whalen LR, Dudek FE, Kossel AH, Kater SB. Evidence for glutamate-mediated activation of hippocampal neurons by glial calcium waves. J Neurobiol. 1995;28:159–170. doi: 10.1002/neu.480280204. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Herman MA, Jahr CE. Extracellular glutamate concentration in hippocampal slice. J Neurosci. 2007;27:9736–9741. doi: 10.1523/JNEUROSCI.3009-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertle DN, Yeckel MF. Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience. 2007;150:625–638. doi: 10.1016/j.neuroscience.2007.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirase H, Qian L, Bartho P, Buzsaki G. Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol. 2004;2:E96. doi: 10.1371/journal.pbio.0020096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzclaw LA, Pandhit S, Bare DJ, Mignery GA, Russell JT. Astrocytes in adult rat brain express type 2 inositol 1,4,5-trisphosphate receptors. Glia. 2002;39:69–84. doi: 10.1002/glia.10085. [DOI] [PubMed] [Google Scholar]
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gahwiler BH, Gerber U. Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci USA. 1999;96:8733–8738. doi: 10.1073/pnas.96.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AM, Chiu SY. Fluorescence measurement of changes in intracellular calcium induced by excitatory amino acids in cultured cortical astrocytes. J Neurosci. 1990;10:1165–1175. doi: 10.1523/JNEUROSCI.10-04-01165.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Kang N, Xu J, Xu Q, Nedergaard M, Kang J. Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;94:4121–4130. doi: 10.1152/jn.00448.2005. [DOI] [PubMed] [Google Scholar]
- Le Meur K, Galante M, Angulo MC, Audinat E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol (Lond) 2007;580:373–383. doi: 10.1113/jphysiol.2006.123570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zima AV, Sheikh F, Blatter LA, Chen J. Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res. 2005;96:1274–1281. doi: 10.1161/01.RES.0000172556.05576.4c. [DOI] [PubMed] [Google Scholar]
- Liu QS, Xu Q, Kang J, Nedergaard M. Astrocyte activation of presynaptic metabotropic glutamate receptors modulates hippocampal inhibitory synaptic transmission. Neuron Glia Biol. 2004a;1:307–316. doi: 10.1017/S1740925X05000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QS, Xu Q, Arcuino G, Kang J, Nedergaard M. Astrocyte-mediated activation of neuronal kainate receptors. Proc Natl Acad Sci USA. 2004b;101:3172–3177. doi: 10.1073/pnas.0306731101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkey EB, Parpura V. Mechanisms of glutamate release from astrocytes. Neurochem Int. 2008;52:142–154. doi: 10.1016/j.neuint.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Nakagawa T, Inoue T, Nagata E, Tanaka K, Takano H, Minowa O, Kuno J, Sakakibara S, Yamada M, Yoneshima H, Miyawaki A, Fukuuchi Y, Furuichi T, Okano H, Mikoshiba K, Noda T. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature. 1996;379:168–171. doi: 10.1038/379168a0. [DOI] [PubMed] [Google Scholar]
- Montana V, Malarkey EB, Verderio C, Matteoli M, Parpura V. Vesicular transmitter release from astrocytes. Glia. 2006;54:700–715. doi: 10.1002/glia.20367. [DOI] [PubMed] [Google Scholar]
- Nadkarni S, Jung P. Modeling synaptic transmission of the tripartite synapse. Phys Biol. 2007;4:1–9. doi: 10.1088/1478-3975/4/1/001. [DOI] [PubMed] [Google Scholar]
- Nett WJ, Oloff SH, McCarthy KD. Hippocampal astrocytes in situ exhibit calcium oscillations that occur independent of neuronal activity. J Neurophysiol. 2002;87:528–537. doi: 10.1152/jn.00268.2001. [DOI] [PubMed] [Google Scholar]
- Parpura V, Haydon PG. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci USA. 2000;97:8629–8634. doi: 10.1073/pnas.97.15.8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci. 2001;4:803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Pasti L, Zonta M, Pozzan T, Vicini S, Carmignoto G. Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J Neurosci. 2001;21:477–484. doi: 10.1523/JNEUROSCI.21-02-00477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J Neurosci. 2005;25:2192–2203. doi: 10.1523/JNEUROSCI.3965-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci. 1996;16:5073–5081. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scemes E. Components of astrocytic intercellular calcium signaling. Mol Neurobiol. 2000;22:167–179. doi: 10.1385/MN:22:1-3:167. [DOI] [PubMed] [Google Scholar]
- Scemes E, Giaume C. Astrocyte calcium waves: what they are and what they do. Glia. 2006;54:716–725. doi: 10.1002/glia.20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano A, Haddjeri N, Lacaille JC, Robitaille R. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci. 2006;26:5370–5382. doi: 10.1523/JNEUROSCI.5255-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AH, Nucifora FC, Jr, Blondel O, Sheppard CA, Zhang C, Snyder SH, Russell JT, Ryugo DK, Ross CA. Differential cellular expression of isoforms of inositol 1,4,5-triphosphate receptors in neurons and glia in brain. J Comp Neurol. 1999;406:207–220. [PubMed] [Google Scholar]
- Sheppard CA, Simpson PB, Sharp AH, Nucifora FC, Ross CA, Lange GD, Russell JT. Comparison of type 2 inositol 1,4,5-trisphosphate receptor distribution and subcellular Ca2+ release sites that support Ca2+ waves in cultured astrocytes. J Neurochem. 1997;68:2317–2327. doi: 10.1046/j.1471-4159.1997.68062317.x. [DOI] [PubMed] [Google Scholar]
- Straub SV, Nelson MT. Astrocytic calcium signaling: the information currency coupling neuronal activity to the cerebral microcirculation. Trends Cardiovasc Med. 2007;17:183–190. doi: 10.1016/j.tcm.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub SV, Bonev AD, Wilkerson MK, Nelson MT. Dynamic inositol trisphosphate-mediated calcium signals within astrocytic endfeet underlie vasodilation of cerebral arterioles. J Gen Physiol. 2006;128:659–669. doi: 10.1085/jgp.200609650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Kettenmann H. Calcium signalling in glial cells. Trends Neurosci. 1996;19:346–352. doi: 10.1016/0166-2236(96)10048-5. [DOI] [PubMed] [Google Scholar]
- Weerth SH, Holtzclaw LA, Russell JT. Signaling proteins in raft-like microdomains are essential for Ca2+ wave propagation in glial cells. Cell Calcium. 2007;41:155–167. doi: 10.1016/j.ceca.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]