

Abstract
The UDP-3-O-(R-3-hydroxyacyl)-N-acetylglucosamine deacetylase LpxC catalyzes the committed reaction of lipid A (endotoxin) biosynthesis in Gram-negative bacteria and is a validated antibiotic target. Although several previously described compounds bind to the unique acyl chain binding passage of LpxC with high affinity, strategies to target the enzyme's UDP-binding site have not been reported. Here the identification of a series of uridine-based LpxC inhibitors is presented. The most potent examined, 1-68A, is a pH-dependent, two-step, covalent inhibitor of E. coli LpxC that competes with UDP to bind the enzyme in the first step of inhibition. Compound 1-68A exhibits a KI of 54 μM and a maximal rate of inactivation (kinact) of 1.7 min-1 at pH 7.4. Dithiothreitol, glutathione and the C207A mutant of E. coli LpxC prevent the formation of a covalent complex by 1-68A, suggesting a role for Cys-207 in inhibition. The inhibitory activity of 1-68A and a panel of synthetic analogs identified moieties necessary for inhibition. 1-68A and a 2-dehydroxy analog, 1-68Aa, inhibit several purified LpxC orthologs. These compounds may provide new scaffolds for extension of existing LpxC-inhibiting antibiotics to target the UDP binding pocket.
With the emergence of multidrug-resistant bacterial infections, there is a critical need for the identification of antibiotics with novel mechanisms of action (1, 2) to complement or supplant existing drugs that inhibit protein, nucleic acid or cell wall biosynthesis (3).
The biosynthesis of lipid A (endotoxin), the membrane anchor of lipopolysaccharide, is a promising target for antibiotic design because lipid A is an essential molecule in most Gram-negative bacteria (4). Lipid A is synthesized in the cytoplasm and on the inner surface of the inner membrane by nine unique enzymes (5). The first step is acylation at the 3-OH group of UDP-N-acetylglucosamine (Figure 1), followed by the deacetylation of the product UDP-3-O-(R-3-hydroxyacyl)-N-acetylglucosamine (UDP-3-O-acyl-GlcNAc), catalyzed by the zinc-dependent enzyme LpxC (6).
Figure 1.
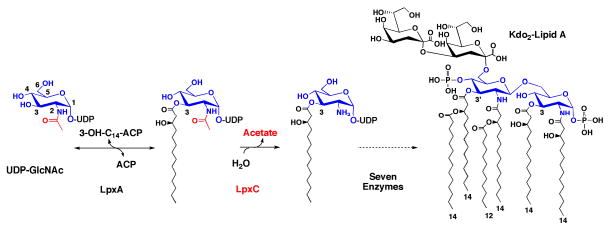
Reaction catalyzed by LpxC and the structure of E. coli lipid A. The biosynthesis of lipid A begins with the 3-O-acylation of UDP-N-acetylglucosamine by the cytosolic enzyme LpxA (5). In the first irreversible (committed) reaction of the pathway, the deacetylase LpxC unblocks the nitrogen at the 2 position of the glucosamine ring for subsequent acylation by LpxD (5). Seven downstream reactions produce Kdo2-lipid A, the hydrophobic membrane anchor of lipopolysaccharide (5).
Substrate binding by LpxC has been extensively studied using enzyme kinetics, NMR and X-ray crystallography. These studies suggest that the acyl chain of UDP-3-O-acyl-GlcNAc binds to LpxC within an unusual hydrophobic passage (7-10). The passage is open at the protein surface, and therefore can accommodate different acyl chain lengths (11, 12). The UDP moiety of UDP-3-O-acyl-GlcNAc is also critical to substrate binding (13), and a structure of LpxC with bound UDP has revealed the nucleotide binding site (10). Unlike the hydrophobic passage, the UDP binding site has not been exploited in the development of potent LpxC inhibitors.
Low to sub-nanomolar inhibitors targeting LpxC display antibiotic activity (14-17). CHIR-090, the most potent LpxC inhibitor currently available, inhibits a diverse range of LpxC orthologs, generally by a two-step, slow, tight-binding mechanism (14, 16). For E. coli LpxC the first step of binding is competitive with respect to substrate and exhibits a Ki of 4.0 nM, followed by isomerization to a complex with a Ki* of 0.5 nM (14). Rhizobium leguminosarum LpxC is orders of magnitude (680-fold) less sensitive to CHIR-090 (14), indicating that CHIR-090 does not inhibit all LpxCs with equal potency and thus could be improved to extend the spectrum of inhibition.
The most potent LpxC inhibitors interact with the hydrophobic passage, including CHIR-090 which coordinates the catalytic zinc ion and occupies the hydrophobic passage of Aquifex aeolicus LpxC (18). Similarly, the less potent, narrow-spectrum inhibitor L-161,240 (17) also utilizes the hydrophobic passage of E. coli LpxC (18). Recent crystallographic evidence suggests another potent LpxC inhibitor, BB-78485 (15), does not penetrate the hydrophobic passage but rather deforms this passage and the active site to cradle the two naphthalene moieties of this compound (19). Neither CHIR-090, BB-78485 nor L-161,240 interact with the UDP-binding site.
To explore the UDP-binding site as a target of future drug design, a uridine-based library was screened for novel LpxC inhibitors. Despite the low probability that such a compound would be an effective drug, the identification of a uridine-containing compound will provide a basis for the development of analogs with more favorable drug properties and provide a new direction for increasing the avidity of existing inhibitors. From this screen compound 1-68A was identified as a two-step covalent inhibitor of E. coli LpxC that competes with UDP when binding. Additionally, the inhibition of many purified LpxC orthologs by this compound and a small group of analogs is reported.
Experimental Procedures
Materials, Strains and Reagents
All chemicals, unless otherwise noted, were obtained from Sigma-Aldrich, St. Louis, MO. [α-32P]-UTP was purchased from PerkinElmer Life and Analytical Sciences, Inc., Waltham, MA. Plasmid miniprep kits were purchased from Qiagen, Valencia, CA. Primers were purchased from MWG Biotech, High Point, NC. The LpxC inhibitors CHIR-090, L-161,240 and BB-78485 were prepared according to published procedures (18). The uridine-based inhibitor library, 1-68A and 2-68A were synthesized as previously described (20). In aqueous solution 1-68A decomposes in air over the course of 1-2 days, and was stored at -80°C (alternatively, this compound may be stabilized by the addition of DTT or pyruvate).
Assay of LpxC Activity
E. coli LpxC (14), UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine, and [α-32P]UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine were prepared as previously described (21). Assays of LpxC activity were performed at pH 7.4 with 5 μM substrate, except where noted. In experiments including inhibitors dissolved in DMSO, the final concentration of DMSO was 10% (v/v) in each assay. In all experiments, the concentration of the enzyme was at least 10-fold below the concentration of either the inhibitor or the substrate.
Library Screening Conditions
The 1338-membered uridine-based library was compiled into 112 pools, each containing 500 μM of 12 compounds in 60% DMSO. Pools were diluted 1:10 into an assay of E. coli LpxC containing 25 mM NaPO4, pH 7.4, 1 mg / mL bovine serum albumin (BSA), and 5 μM [α-32P]UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine. Each reaction was initiated with 0.1 nM enzyme with a final volume of 5 μL and incubated for 30 min at 23 °C. Reactions were quenched with 2 μL of 1.25 M NaOH and incubated for 15 min at 23 °C to hydrolyze the 3-O-linked acyl chain of the substrate and product. Reactions were subsequently neutralized with 2 μL of 1.25 M acetic acid, and protein was precipitated with 1 μL of 5% trichloroacetic acid. UDP-N-acetylglucosamine was separated from UDP-glucosamine using a PEI cellulose thin-layer chromatography plate (EMD Chemicals, Gibbstown, NJ) developed in 0.2 M guanidine hydrochloride. Dried TLC plates were exposed to a PhosphorImager screen and analyzed using a Storm 840 PhosphorImager (GE Healthcare, Chalfont St. Giles, Buckinghamshire, U.K.).
Inhibitor pools showing 40% or greater inhibition of E. coli LpxC activity were rescreened to identify the active compounds. Individual compounds were assayed in concentrations ranging from 1 to 500 μM using the same assay conditions described above, while maintaining 10% DMSO in the assay. Compounds demonstrating favorable dose-response curves were further analyzed by fitting an IC50 value using Equation 1:
| (Eq. 1) |
in which vi is the initial velocity of an inhibited reaction, vo is the initial velocity of an uninhibited reaction, I is the concentration of inhibitor, IC50 is the inhibitor concentration at which 50% inhibition of activity is observed, and H is the Hill slope describing the steepness of the curve.
Rapid Dilution and Dialysis of the E. coli LpxC – 1-68A complex
E. coli LpxC (1 μM) was incubated with 50 μM 1-68A in a buffer containing 1 mg/mL BSA and 25 mM sodium phosphate, pH 7.4 at 30°C for 30 min, then diluted 1:2500 with 1 mg/ml BSA and 25 mM sodium phosphate, pH 7.4. This solution was further diluted 1:4 at timed intervals into an LpxC reaction mixture as described above, where a linear reaction velocity was measured and compared to a control reaction that was incubated and diluted as described above, except with 0 μM 1-68A.
After incubating 5 mL of a mixture containing 2.5 nM E. coli LpxC, 50 μM 1-68A, 1 mg/mL BSA and 25 mM sodium phosphate, pH 7.4 at 30°C for 30 min, this mixture was loaded into a Slyde-A-Lyzer® 10K dialysis cassette (Pierce) and dialyzed against 1 L of 25 mM sodium phosphate, pH 7.4 at 4°C. At timed intervals, aliquots were removed, tested for activity and compared to a control reaction as described for the rapid dilution experiment.
Determination of the Mechanism of 1-68A binding to E. coli LpxC
Product accumulation data with varying concentrations of 1-68A were fitted with the equation describing irreversible time-dependent inhibition (22):
| (Eq. 2) |
in which P is the concentration of product at time t, vi is the initial velocity, kobs is the observed first order exponential term for the formation of the E-I complex and C is a constant.
It was observed that a plot of kobs versus 1-68A concentration was hyperbolic and thus indicative of a two-step mechanism (Scheme 1), therefore these data were fitted with the equation:
Scheme 1.
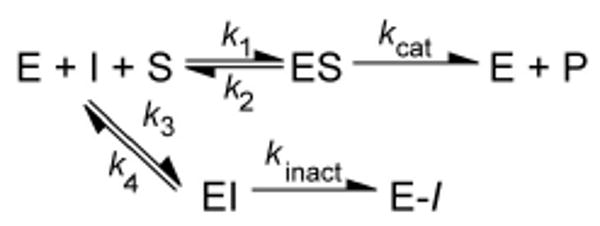
Mechanism for two-step, kinetically irreversible inhibition.
| (Eq. 3) |
in which kinact is the maximal rate of inactivation, KI is a kinetic constant describing the inhibitor concentration at which the formation of the E-I complex occurs at one-half the maximal rate (kinact) (22), and I is the concentration of 1-68A in the assay.
Determining the binding mode of 1-68A
Eq. 2 was fitted to reaction progress curves determined over a range of substrate concentrations (∼0.2 – 5x KM) and 10 or 25 μM 1-68A to extract a kobs value for each condition. These kobs values were plotted against the substrate concentration (S) and fitted with Equation 4:
| (Eq. 4) |
in which KM is the Michaelis constant for E. coli LpxC (14), kinact was determined from Eq. 3, and KI is defined above.
UDP Competition Studies
E. coli LpxC (250 nM) was incubated with 50 μM 1-68A with or without 100 mM UDP at 30 °C in 200 mM sodium phosphate, pH 7.4, and 1 mg / mL BSA. Portions were removed at various time points and immediately diluted 1:400 into a prewarmed LpxC reaction mixture (as described above). The initial reaction velocity (vi) at each time point was compared to that of a reaction containing enzyme diluted from a control incubation (vo) containing E. coli LpxC (250 nM) in 200 mM sodium phosphate, pH 7.4, and 1 mg / mL BSA.
Electrospray Ionization Mass Spectrometry (ESI-MS) analysis
Liquid-chromatography (LC)-MS was performed as previously described (16), except LpxC was analyzed using a Vydac C4 reverse-phase column (2.1 × 50 mm). N-acetylcysteine-methylester and 1-68A were analyzed using a Vydac C8 column. E. coli LpxC and 1-68A were incubated at 15 μM each in a buffer containing 25 mM sodium phosphate, pH 7.4 and 10% DMSO for 1 h at 23 °C. Control samples were prepared that contained each component alone. N-acetylcysteine-methylester (1 mM) and 1-68A (0.6 mM) were incubated in a buffer containing 50 mM TRIS, pH 7.6 for 4 h at 23 °C. Proteolysis utilized Proteomics Grade Trypsin (T6567, Sigma-Aldrich) according to the manufacturer's recommended protocols, followed by LC/MS analysis of the tryptic peptides using the conditions as previously described (16).
Preparation of mutant E. coli LpxC
Mutant E. coli LpxC proteins were prepared with the QuikChange® PCR mutagenesis kit (Stratagene) using the pET-11:E.coli lpxC plasmid and purified as previously described (18). The six cysteines were mutated as follows: C63S, C65A, C125A, C207A, C214N, C250A. The activities of the mutant proteins and wild type E. coli LpxC were assayed in cell-free extracts as previously described (14), except that the cultures were grown at 37 °C and induced with 1 mM isopropyl β-d-thiogalactopyranoside when the A600 reached 0.5, followed by overnight incubation at 18 °C with shaking.
E. coli LpxC C207A was purified as previously described (14), and the kinetic characterization reported herein utilized this purified protein.
Synthesis of 1-68A analogs
All chemical reagents were obtained from commercial suppliers and were used without further purification. Analytical thin-layer chromatography was conducted on Analtech Uniplate silica gel plates with detection by UV light. Reverse phase HPLC was performed on a Varian Prostar system using Microsorb C18 reverse-phase columns (analytical: 4.6 mm ID × 25 cm, 1 mL/min), and UV detection was carried out at 254 nm on a Varian ProStar 325 detector.
3,4-Dihydroxy-benzaldehyde O-[5-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-3,4-dihydroxy-tetrahydro-furan-2-ylmethyl]-oxime (1-68Aa)
3,4-Dihydroxybenzaldehyde (9.9 mg, 0.072 μmol) was added to a solution of aminooxyuridine (25.0 mg, 0.0964 μmol) in 1% acetic acid/DMF (0.6 mL). The resulting solution was stirred in the dark for 4 d at room temperature under nitrogen, the solvent was removed in vacuo, and the crude product mixture was purified by reversed-phase HPLC to yield a white solid (13.5 mg, 50%). Rf: 0.75 (7:2:1 ethyl acetate:methanol:water, UV). 1H NMR (400 MHz, d-methanol) δ 8.02 (1H, s), 7.80 (1H, d, J = 8.0 Hz), 7.10 (1H, d, J = 2.0 Hz), 6.91-6.88 (1H, m), 6.76 (1H, d, J = 8.0 Hz), 5.89 (1H, d, J = 4.8 Hz), 5.60 (1H, d, J = 8.0 Hz), 4.42 (1H, dd, J = 12.4, 2.8 Hz), 4.32 (1H, dd, J = 12.4, 3.6 Hz), 4.23-4.18 (2H, m), 4.13-4.11 (1H, m); 13C NMR (100 MHz, d-methanol) δ 164.67, 150.90, 149.49, 147.64, 145.36, 140.77, 123.54, 120.22, 114.81, 112.42, 101.32, 89.11, 83.26, 74.01, 72.84, 69.92; HRMS (FAB): Calcd for C16H18N3O8 [M+H]+ 380.1094, found 380.1090.
2,4-Dihydroxy-benzaldehyde O-[5-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-3,4-dihydroxy-tetrahydro-furan-2-ylmethyl]-oxime (1-68Ab)
2,4-Dihydroxybenzaldehyde (9.9 mg, 0.072 μmol) was dissolved in a solution aminooxyuridine (25.0 mg, 0.0964 μmol) in 1% acetic acid in DMF (0.6 mL). The resulting solution was stirred for 3 d in the dark at room temperature under nitrogen, the solvent was removed in vacuo, and the crude product mixture was purified by reversed-phase HPLC to yield a white solid (11.6 mg, 43%). Rf: 0.74 (7:2:1 ethyl acetate:methanol:water, UV). 1H NMR (400 MHz, d-methanol) δ 8.28 (1H, s), 7.75 (1H, d, J = 8.0 Hz), 7.18 (1H, d, J = 8.4 Hz), 6.35 (1H, dd, J = 8.4, 2.4 Hz), 6.31 (1H, d, J = 2.4 Hz), 5.88 (1H, d, J = 4.4 Hz), 5.64 (1H, d, J = 8.4 Hz), 4.45 (1H, m), 4.35 (1H, m), 4.23-4.16 (3H, m); 13C NMR (100 MHz, d-methanol) δ 164.68, 160.69, 158.65, 150.83, 150.68, 140.86, 130.96, 108.71, 107.43, 102.22, 101.31, 89.73, 82.82, 73.81, 73.14, 69.76; HRMS (FAB): Calcd for C16H18N3O8 [M+H]+ 380.1094, found 380.1085.
2,3-Dihydroxy-benzaldehyde O-[5-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-3,4-dihydroxy-tetrahydro-furan-2-ylmethyl]-oxime (1-68Ac)
2,3-Dihydroxybenzaldehyde (9.9 mg, 0.072 μmol) was added to a solution of aminooxyuridine (25.0 mg, 0.0964 μmol) in 1% acetic acid/DMF (0.6 mL). The resulting solution was stirred for 4 d in the dark at room temperature, the solvent was removed in vacuo, and the crude product mixture was purified by reversed-phase HPLC to yield a white solid (23.1 mg, 85%). Rf: 0.75 (7:2:1 ethyl acetate:methanol:water, UV). 1H NMR (400 MHz, d-methanol) δ 8.41 (1H, s), 7.74 (1H, d, J = 8.0 Hz), 6.92 (1H, m), 6.87 (1H, m), 6.76 (1H, m), 5.90 (1H, d, J = 4.4 Hz), 5.65 (1H, d, J = 8.4 Hz), 4.53 (1H, m), 4.43 (1H, m), 4.22 (3H, m); 13C NMR (100 MHz, d-methanol) δ 164.65, 150.82, 150.46, 150.45, 145.21, 144.94, 140.83, 119.97, 119.40, 116.86, 101.35, 89.84, 82.74, 73.77, 73.40, 69.72; HRMS (FAB): Calcd for C16H18N3O8 [M+H]+ 380.1094, found 380.1086.
3,4,5-Trihydroxy-benzaldehyde O-[5-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-3,4-dihydroxy-tetrahydro-furan-2-ylmethyl]-oxime (1-68Ad)
3,4,5-Trihydroxybenzaldehyde (11.1 mg, 0.0720 μmol) was dissolved in a solution of aminooxyuridine (25.0 mg, 0.0964 μmol) in 1% acetic acid/DMF (0.6 mL). The resulting solution was stirred for 4 d at room temperature, the solvent was removed in vacuo, and the crude product mixture was purified by reversed-phase HPLC to yield a white solid (6.1 mg, 22%). Rf: 0.70 (7:2:1 ethyl acetate:methanol:water, UV). 1H NMR (300 MHz, d-methanol) δ 7.95 (1H, s), 7.81 (1H, d, J = 8.1 Hz), 6.64 (2H, s), 5.91 (1H, d, J = 4.8 Hz), 5.62 (1H, d, J = 8.1 Hz), 4.43 (1H, dd, J = 12.6, 2.7 Hz), 4.33 (1H, dd, J = 12.6, 3.3 Hz), 4.25-4.18 (2H, m), 4.13 (1H, app t, J = 4.8 Hz); 13C NMR; (100 MHz, d-methanol) δ 161.52, 160.64, 160.65, 150.67, 150.66, 140.84, 130.91, 108.68, 107.40, 102.19, 101.28, 89.77, 82.58, 73.83, 73.11, 69.82; HRMS (ESI): Calcd for C16H16N3O9 [M-H]- 394.0892, found 394.0881.
3,4-dimethoxy-benzaldehyde O-[5-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-3,4-dihydroxy-tetrahydro-furan-2-ylmethyl]-oxime (1-68Ae)
3,4-Dimethoxy benzaldehyde (4.90 mg, 0.0296 μmol) was dissolved in a solution of aminooxyuridine (10.0 mg, 0.0385 μmol) in 1% acetic acid/DMF. The resulting solution was stirred in the dark for 1 d at room temperature under an atmosphere of nitrogen. The crude mixture was then concentrated in vacuo, and purified by reversed-phase HPLC to yield a white solid (3.4 mg, 28 %). 1H NMR (400 MHz, d-methanol) δ 8.16 (1H, s), 7.82 (1H, d, J = 8.4 Hz), 7.32 (1H, s), 7.15 (1H, d, J = 8), 7.0 (1H, d, J = 8.4), 5.92 (1H, d, J = 4.8), 5.64 (1H, d, J = 8.4), 4.5 (1H, dd, J = 12.4, 3.3), 4.41 (1H, dd, J = 12.4, 3.2), 4.24 (2H, m), 4.18 (1H, app t, J = 4.4), 3.88 (3H, s), 3.87 (3H, s); 13C NMR (100 MHz, d-methanol) δ 153.38, 150.10, 148.17, 148.11, 141.17, 139.93, 139.86, 121.22, 110.30, 107.13, 100.03, 89.01, 81.78, 71.98, 71.30, 69.23, 55.79, 55.81; HRMS (ESI): Calcd for C16H16N3O9 [M+H]+ 408.3746, found 408.3822.
2,3,4-Trimethoxy-benzaldehyde O-[5-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-3,4-dihydroxy-tetrahydro-furan-2-ylmethyl]-oxime (1-68Af)
2,3,4-Trimethooxybenzaldehyde (5.80 mg, 0.0296 μmol) was added to a solution of aminooxyuridine (10.0 mg, 0.0385 μmol) in 1% acetic acid/DMF (1 mL). The resulting solution was stirred in the dark for 1 d at room temperature under an atmosphere of nitrogen. The crude product was then concentrated in vacuo and purified by reversed-phase HPLC to yield a white solid (7.3 mg, 56 %). 1H NMR (400 MHz, d-methanol) δ 8.35 (1H, s), 7.84 (1H, d, J = 8.0 Hz), 7.52 (1H, d, J = 8.8), 6.85 (1H, d, J = 9.2), 5.92 (1H, d, J = 4.4), 5.63 (1H, d, J = 8.0 Hz), 4.51 (1H, m), 4.48 (1H, m), 4.41 (1H, d, J = 3.2 Hz), 4.38 (1H, m), 4.25-4.24 (2H, m), 4.18-4.16 (1H, t, J = 4.4), 3.91 (3H, s) 3.90 (3H, s) 3.85 (3H, s); 13C NMR (100 MHz, d-methanol) δ 164.65, 155.62, 152.46, 150.86, 144.89, 141.93, 140.79, 120.91, 117.88, 107.94, 101.19, 89.39, 83.07, 74.02, 72.95, 69.86, 60.84, 59.80, 55.12; HRMS (ESI): Calcd for C16H16N3O9 [M+H]+ 438.4001, found 438.4102.
Results
Identification of a uridine-based LpxC inhibitor
An initial screen of a uridine-based library (20) revealed six compounds that inhibited E. coli LpxC activity by more than 40%. Two related compounds, 1-68A and 2-68A, contain a 2,3,4 tri-hydroxy benzyl moiety appended to uridine through an aminooxy or glycylaminooxy linkage, respectively (Figure 2). These compounds were previously identified as competitive inhibitors of the mucin N-acetyl-α-galactosaminyltransferase family (ppGalNAcT's) (20). Compounds 1-68A and 2-68A exhibited IC50 values against E. coli LpxC of 27 ± 5 μM and 120 ± 30 μM, respectively, using single time-point assays. As depicted in Figure 2C, the best fit of Eq. 1 specified a Hill slope of 0.7 for each curve, indicating a shallower dose-response curve than expected for a rapidly reversible inhibitor.
Figure 2.
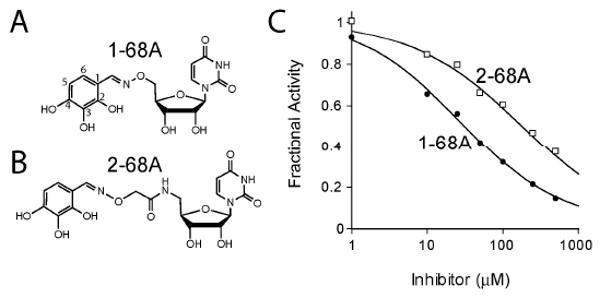
Uridine-based inhibitors of E. coli LpxC. Two compounds, 1-68A (Panel A) and 2-68A (Panel B), were identified that inhibited E. coli LpxC with apparent mid- μM IC50s. (Panel C) Analysis of concentration-response plots fitted with Eq. 1 gave IC50 values of 27 μM and 120 μM for 1-68A (black circles) and 2-68A (open squares), respectively, where H=0.7.
Kinetics of 1-68A inhibition
The reaction velocity of E. coli LpxC in the presence of 1-68A decreased as the reaction progressed, as shown in Figure 3A, indicating time-dependent inhibition. Enzyme inhibition appeared kinetically irreversible because rapid dilution (Figure 3B) or dialysis (Figure 3C) of the LpxC – 1-68A complex failed to restore enzyme activity. Product accumulation data was fitted to a kinetically irreversible, time-dependent inhibition model (Eq. 2, Figure 3A) to derive a kobs value for each concentration of inhibitor; kobs is the pseudo first-order rate constant of enzyme inactivation. The plateau of kobs at high 1-68A concentrations, as seen in Figure 3D, indicates the rate of formation of the terminal complex is saturable at high inhibitor concentrations. The kinetics of 1-68A binding to E. coli LpxC are most consistent with the two-step, kinetically irreversible mechanism shown in Scheme 1 (22). Using non-linear curve fitting, values for the kinetic parameters kinact (the maximal rate of enzyme inactivation) and KI (the inhibitor concentration at which kobs is one-half of kinact) were determined to be 1.7 ± 0.3 min-1 and 54 ± 7 μM, respectively, using Eqs. 3 and 4.
Figure 3.
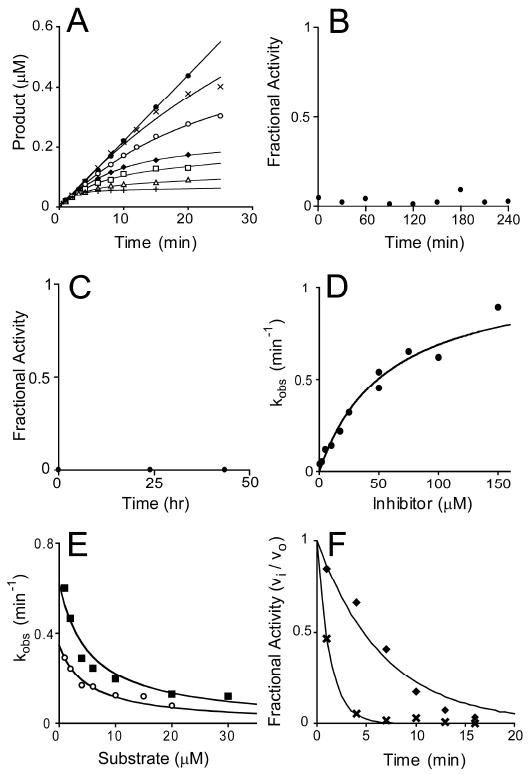
Slow, kinetically irreversible and competitive inhibition of E. coli LpxC by 1-68A. (Panel A) Progress curves for product formation when reactions are initiated by the addition of enzyme in the presence 0 μM (black circles), 0.5 μM (black Xs), 2 μM (open circles), 5 μM (black diamonds), 10 μM (open squares), 17.5 μM (open triangles), or 50 μM (black pluses) 1-68A as fitted with Eq. 2. Data is representative of two separate experiments. (Panel B) Rapid dilution of the E. coli LpxC – 1-68A complex. (Panel C) E. coli LpxC activity following dialysis of the LpxC – 1-68A complex. (Panel D) A plot of the observed pseudo first-order rate constant for the formation of the E-I complex (kobs) as fitted with Eq. 3. Repeat experiments (not shown) gave identical results (within 10 %). (Panel E) A plot showing the effect of increasing substrate concentrations on the observed first order rate constant (kobs) in the presence of 10 μM (open circles) or 25 μM (black squares) 1-68A. Eq. 4 was fitted to these data. These plots include data from three independent experiments. (Panel F) The fractional activity of LpxC after a preincubation in the presence of 50 μM 1-68A (black diamonds) with or (black Xs) without 100 mM UDP. The fit of a first-order, decaying exponential (vi / vo = e-kt) for each data set is shown as a line.
To determine the binding mode of 1-68A, the effect of substrate concentration on kobs at two 1-68A concentrations (10 and 25 μM) was investigated. The diminishing magnitude of kobs as substrate increases, as seen in Figure 3E, is typical of a competitive inhibitor (22).
The uridine-binding site of LpxC (10), which binds the uridine moiety of UDP-3-O-acyl-GlcNAc with low millimolar affinity (13), likely also binds the uridine moiety of 1-68A. If UDP and 1-68A compete for a binding site, millimolar concentrations of UDP would slow the formation of a kinetically-irreversible 1-68A – LpxC complex. Indeed, 100 mM UDP was sufficient to reduce the rate of E. coli LpxC inactivation by 1-68A (in the absence of substrate) more than 5-fold, as shown in Figure 3F.
Wild-type E. coli LpxC displays time-dependent inhibition with CHIR-090 (18). A mutant of E. coli LpxC (Q202W / G210S) loses the time-dependence of CHIR-090 inhibition (18). However, this mutant is still sensitive to inhibition by 1-68A in a time-dependent manner (data not shown). This suggests that despite kinetic similarities, the mechanism of 1-68A inhibition is significantly different than CHIR-090 inhibition.
Role of a thiol in 1-68A inhibition
Given the instability of 1-68A in air and its protection by reducing agents, the effects of reducing agents were explored on the 1-68A inhibition of LpxC. The reducing agents glutathione and dithiothreitol diminished the inhibition of LpxC by 1-68A (Figure 4A). Dithiothreitol is likewise capable of reversing 1-68A inhibition, as seen in Figure 4B, but does not effect the inhibition of the more potent E. coli LpxC inhibitors CHIR-090, BB-78485 and L-161,240 (data not shown).
Figure 4.
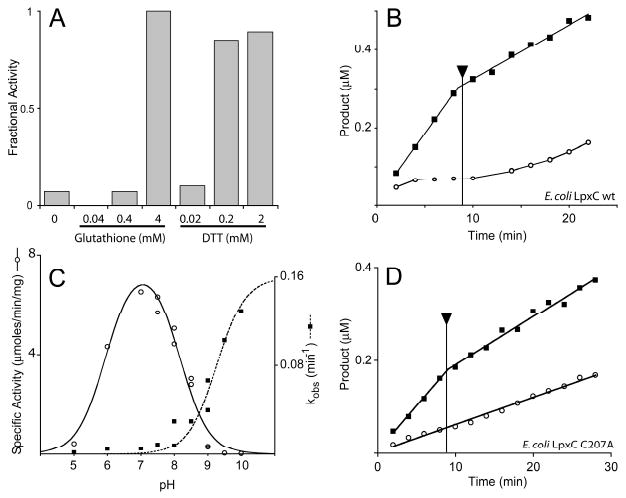
Inhibition of E. coli LpxC by 1-68A depends upon a thiol group. (Panel A) Effect of glutathione or dithiothreitol (DTT) on 1-68A inhibition of E. coli LpxC activity. (Panel B) Reactions containing 0 μM (black squares) or 50 μM (open circles) 1-68A were initiated with 0.1 nM E. coli LpxC. Both reactions progressed for 9 min, at which point dithiothreitol was added (as indicated by the arrow) to a final concentration of 2 mM. The reduced velocity of the 0 μM reaction after 9 min is not due to dilution but rather dithiothreitol is a weak competitive inhibitor E. coli LpxC (data not shown). (Panel C) pH rate profile of wild-type E. coli LpxC specific activity (open circles) and the rate (kobs) of 1-68A inhibition (black squares). The bell-shaped profile is fit with a pK1 of 5.9 and a pK2 of 8.0. The kobs profile is fitted with a curve describing a pKa of at least 9.2. (Panel D) Reactions containing 0 μM (black squares) or 2 mM (open circles) 1-68A were initiated with 0.1 nM E. coli LpxC C207A. Both reactions progressed for 9 min, at which point dithiothreitol was added (as indicated by the arrow) to a final concentration of 2 mM. Data is representative of two separate experiments.
The reactivity of free thiols depends strongly on the pH. Therefore, the pH dependency of kobs at a fixed 1-68A concentration (1 μM) was determined as previously described (8, 23). A plot of these data, shown in Figure 4C, reveals that kobs has a pKa of at least 9.2. The pH-rate profile of LpxC was monitored as a control with 5 μM substrate (24). Both the effect of reductants on complex formation and the pH dependency of kobs are suggestive that a thiolate moiety is involved in 1-68A inhibition.
E. coli LpxC variants with mutated cysteine residues were constructed to test the role of a thiol in 1-68A inhibition. E. coli LpxC contains six, non-conserved cysteine residues. One mutant enzyme, C207A, was much less sensitive to 1-68A inhibition (compare the first 8 min of Figure 4B and D) suggesting Cys-207 is necessary to form the E-I complex. Mutant enzymes containing mutations of the other five cysteines were as sensitive to 1-68A as the wild-type enzyme, and all mutant proteins were nearly as active as the wild type (data not shown). The residual sensitivity of E. coli LpxC C207A may reflect the binding of 1-68A in the active site without the formation of a kinetically irreversible complex, or it may be due to interaction with the remaining five cysteine residues. However, inhibition of LpxC C207A by 1-68A was unaffected by dithiothreitol (Figure 4D) and the inhibition is reversible, strongly suggesting Cys-207 is specifically involved in time-dependent inhibition. Furthermore, the fractional activity (vi / vo) of the mutant enzyme in the presence of 0.5 mM 1-68A was the same at pH 7.4 and 9.0 (data not shown).
1-68A covalently modifies E. coli LpxC
To determine whether the observed kinetic irreversibility of 1-68A inhibition was due to tight binding or covalent modification of LpxC, protein mass spectrometry of the 1-68A – E. coli LpxC complex was performed. The mass of the 1-68A – E. coli LpxC complex was 394 Da greater than that of the uninhibited enzyme as determined by electrospray-ionization mass spectrometry (ESI-MS) (Figure 5A-C). This shift is consistent with the incorporation of one molecule of 1-68A (monoisotopic mass = 395.10 Da) minus one hydrogen atom. The mass spectrum (Figure 5C) demonstrated that a small percentage of E. coli LpxC increased by 788 Da, consistent with the incorporation of two molecules of 1-68A minus two hydrogens. Efforts to locate the site of LpxC modification through proteolytic digestion and MS analysis of the enzyme:inhibitor complex were unsuccessful. No 1-68A-modified tryptic peptides were detected. It is possible that standard proteolytic digestions with trypsin (overnight incubation at 37 °C) result in degradation of the complex.
Figure 5.
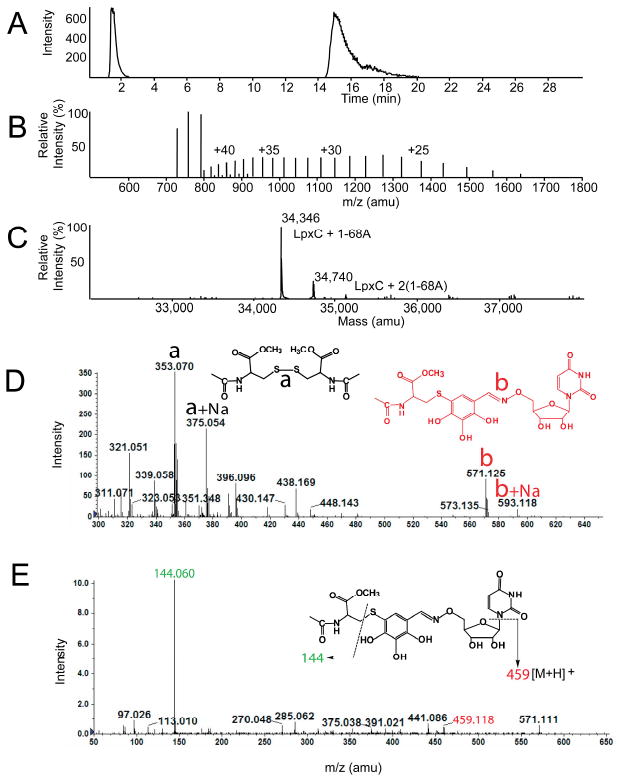
ESI-MS analysis of the E. coli LpxC – 1-68A complex. (Panel A) The LC-elution profile of E. coli LpxC preincubated with equimolar 1-68A, shown as the extracted ion current. Positive ion (Panel B) and deconvoluted (Panel C) mass spectra of E. coli LpxC from the 16-22 min region of the LC elution shown in Panel A. The observed mass of the E. coli LpxC polypeptide was 33,952 Da (data not shown). (Panel D) Mass spectrum of products formed after incubating N-acetylcysteine-methylester and 1-68A at pH 7.4. Note: The proposed structure of b, i.e. covalent complex of N-acetylcysteine-methylester and 1-68A, was proposed based on exact mass measurement and MS/MS, which would not delineate position isomers. (Panel E) MS/MS analysis of the “b” ion in Panel D.
To demonstrate that a covalent complex can form between a thiol-containing agent and 1-68A, a model thiol-containing compound N-acetylcysteine-methylester was incubated with 1-68A. MS studies revealed the formation of a covalent complex between N-acetylcysteine-methylester and 1-68A (Figure 5D). The mass of this complex and its MS/MS analysis are consistent with the structure shown in Figures 5D and 5E. Together, the mass spectral analyses of the E. coli LpxC – 1-68A and N-acetylcysteine-methylester – 1-68A complexes are consistent with a mechanism whereby 1-68A inhibits E. coli LpxC by covalent modification of a cysteine residue.
Sensitivity of LpxC to 1-68A analogs
The inhibitory effects of 1-68A analogs with variations of the 2,3,4-trihydroxy benzyl moiety were tested (Figure 6). Only 1-68Aa, lacking a hydroxyl at the 2 position, inhibited E. coli LpxC as potently as 1-68A. Two compounds that retain the 3 position hydroxyl, 1-68Ac and 1-68Ad, were weak inhibitors. An isomer of 1-68A, 1-68Ad, contains a 5 position hydroxyl and exhibits greatly reduced inhibitory potency. 1-68Ab, a compound that does not contain the 3 position hydroxyl, was ineffective at all concentrations tested. The methyl ester-containing compounds 1-68Ae and 1-68Af were ineffective as well. Based on these data it appears the 3 and 4 position hydroxyls are critical for potent inhibition and removing or blocking them with methyl groups diminished inhibition at the concentrations tested. It was not determined whether these analogs are irreversible inhibitors.
Figure 6. E. coli.
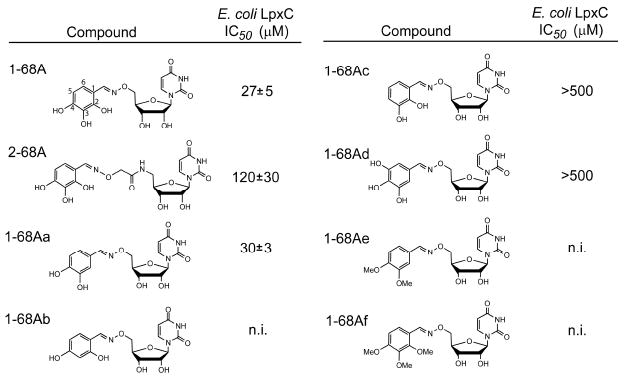
LpxC inhibition by 1-68A analogs. Analogs of 1-68A containing variable substituents on the benzene ring were synthesized and tested for inhibition of E. coli LpxC. Of this series, only 1-68Aa was of comparable potency to 1-68A, and is likewise a time-dependent inhibitor of E. coli and Helicobacter pylori LpxC (data not shown).
By testing the inhibition of a broad range of purified LpxC orthologs by 1-68A and its analogs, it was determined that 1-68Aa is a more effective inhibitor of Helicobacter pylori LpxC and Rhizobium leguminosarum LpxC than the parent compound 1-68A (Table 1). Both E. coli LpxC and H. pylori LpxC are inhibited by 1-68Aa in a time-dependent manner; furthermore, inhibition does not occur in the presence of 2 mM dithiothreitol and 50 μM 1-68Aa (data not shown). Though both R. leguminosarum and H. pylori LpxC contain cysteines, none of these are near Cys-207 of E. coli LpxC based on the primary sequence alignments (Figure S1). Without full structural information, it is not clear whether R. leguminosarum and H. pylori LpxC have cysteine residues proximal to their uridine-binding pockets.
Table 1.
Inhibition of LpxC orthologs* by 50 μM 1-68A or 1-68Aa
| Percent Inhibition | ||
|---|---|---|
| LpxC Source | 1-68A | 1-68Aa |
| Escherichia coli | 80% ±6% | 60% ±14% |
| Pseudomonas aeruginosa | n.i. | n.i. |
| Neisseria meningitidis | 6% ±1% | 9% ±5% |
| Helicobacter pylori | 21% ±1% | 41% ±1% |
| Rhizobium leguminosarum | n.i. | 49% ±10% |
| Aquifex aeolicus | n.i. | 13% ±5% |
n.i. – not inhibited
no inhibition at 50 μM was observed with the remaining 1-68A analogs
Discussion
Inhibitors targeting the UDP pocket
In this study 1-68A was identified as a two-step, covalent inhibitor of E. coli LpxC. 1-68A may be viewed as a competitive inhibitor of E. coli LpxC in that it binds to the UDP-binding site (Figure 3F) and thus represents a new class of LpxC inhibitors. Although 1-68A lacks antibiotic activity (data not shown), it nevertheless demonstrates that the UDP pocket can be targeted for inhibitor discovery. Given that none of the potent LpxC inhibitors with antibiotic activity target the UDP pocket, addition of a UDP analog might enhance binding and specificity. In theory, an inhibitor developed through a multivalency approach may bind to LpxC with a dissociation constant that is the product of the two individual components (25). Addition of a moiety with 1 mM affinity to a tight-binding inhibitor, like CHIR-090, could therefore enhance binding 1000-fold.
Mechanism of inhibition
It is likely that the non-covalent interaction of 1-68A with the UDP-binding site on LpxC is the fast step that forms the EI complex (Scheme 1), followed by a slow step to form the E-I complex. Consistent with this kinetic scheme, E. coli LpxC C207A is much less sensitive to 1-68A when compared to wild-type E. coli LpxC (Figure 4), and does not form an E-I complex. This suggests that the EI complex found with E. coli LpxC C207A is comparable to the initial EI complex found with wild-type E. coli LpxC.
The second step of inhibition, the formation of a covalent E-I complex, requires high pH and Cys-207. Consistent with this notion, the ESI-MS analysis indicated that 1-68A reacts with a cysteine-containing model peptide (N-acetylcysteine-methylester) or with multiple sites on E. coli LpxC to form a covalent complex. However, the predominant form of E. coli LpxC has reacted with only one equivalent of 1-68A (Figure 5). Given that E. coli LpxC C207A is much less sensitive to 1-68A inhibition when compared to wild-type E. coli LpxC (Figure 4), and it does not form an E-I complex, Cys-207 is likely the primary site of 1-68A modification. However, it is not possible to exclude the possibility that, unlike the studies with N-acetylcysteine-methylester, 1-68A does not react with a cysteine residue of LpxC. In this scenario, Cys-207 would be required to form the complex though is itself not permanently modified.
The finding that UDP can delay 1-68A inhibition of LpxC is evidence that 1-68A engages the UDP binding pocket. Comparison of an E. coli LpxC homology model and a crystal structure of the A. aeolicus LpxC – UDP complex (10) indicates that Cys-207 is located <10 Å from C-5 of the ribose ring (Figure 7), a distance that is approximately equal to the length of the aminooxy and tri-hydroxy benzene moieties of 1-68A (Figure 2). Although the role of Cys-207 is not well defined, the model of the E. coli LpxC structure indicates that all of the other cysteine residues are farther from the UDP-binding site and therefore less likely to mediate LpxC inhibition by 1-68A.
Figure 7.
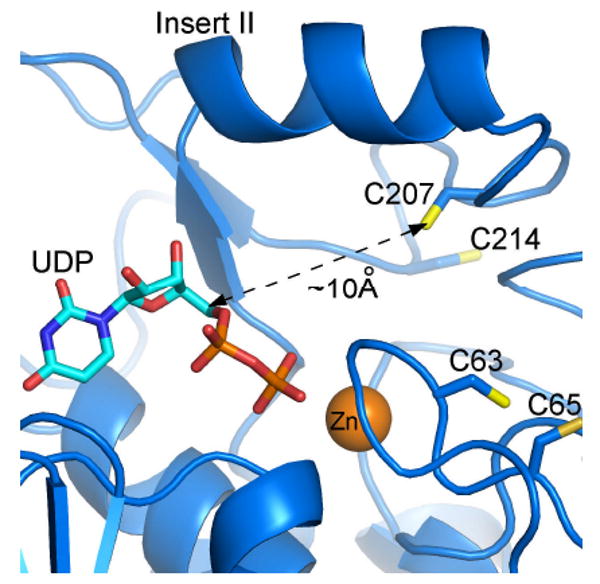
Potential binding mode of UDP and location of cysteine residues in the E. coli LpxC active site. This model is based upon an A. aeolicus LpxC – UDP complex, as determined by x-ray crystallography (2IER). The zinc ion is shown as an orange sphere. This figure was prepared using PyMOL.
In an effort to establish the importance of various substituents for potency, analogs of 1-68A in which the hydroxyls at either positions 3 or 4 are absent (1-68Ab and 1-68Ac) or blocked (1-68Ae and 1-68Af) were assayed and found to be poor inhibitors (Figure 5). However, the absence of the 2 position hydroxyl (1-68Aa) had little effect on inhibition (Figure 5). It is possible the 3 and 4 position hydroxyls are necessary to facilitate the formation of a reactive species. Addition of a hydroxyl at the 5 position (1-68Ad) may prevent the formation of a reactive species. These results suggest the tri-hydroxy benzene moiety, rather than the nucleoside or aminooxy linkage, is responsible for the formation of the covalent E-I complex. A similar covalent inactivator of pyruvic-, α-ketoglutaric-, and succinic-dehydrogenases by γ-l-glutaminyl 3,4-benzoquinone has been described (26).
1-68A inhibits LpxC and ppGalNAcT2 via different mechanisms
Previously 1-68A was identified as an inhibitor of mucin ppGalNAcT2 (20), a glycosyl transferase with no obvious homology to LpxC. However, 1-68A appears to be a fully reversible inhibitor of ppGalNAcT2, and inhibition occurs in the presence of 40 mM β-mercaptoethanol (20). In addition, none of the 1-68A analogs reported here (Figure 6) were inhibitors of ppGalNAcT2 (Leavy and Bertozzi, unpublished results). These data suggest 1-68A inhibits LpxC and ppGalNAcT2 through different mechanisms.
Conclusion
1-68A represents a new class of LpxC inhibitors that exploit the uridine binding site to form a covalent complex. While not antibiotic, it may provide a new scaffold for extension of existing LpxC-inhibiting antibiotics to target the UDP binding pocket.
Supplementary Material
Acknowledgments
We would like to thank Dr. Johannes Rudolph and Dr. K.V. Rajagopalan for helpful discussions and Dr. Craig Bartling for aliquoting the uridine-based inhibitor library.
This research was supported by NIH grants GM-51310 to C. R. H. Raetz and AI-055588 to P. Zhou. A. Barb was supported by the Cellular and Molecular Biology training grant GM-07184 to Duke University. T. Leavy was supported by NIH grant 5R01GM066047.
List of Abbreviations
- UDP-3-O-acyl-GlcNAc
UDP-3-O-(R-3-hydroxyacyl)-N-acetylglucosamine
- DMSO
dimethyl sulfoxide
- ESI-MS
electrospray ionization mass spectrometry
- Kdo
3-deoxy-d-manno-octulosonic acid
- IC50
concentration of half maximal inhibition
- LC
liquid chromatography
- LpxC
UDP-3-O-(R-3-hydroxyacyl)-N-acetylglucosamine deacetylase
- ppGalNAcT2
N-acetyl-α-galactosaminyltransferase
- UDP
uridine diphosphate
Footnotes
Supporting Information Available: A supplementary figure showing the amino acid alignment of the LpxC orthologs tested in Table 1 is provided. This material is available free of charge via the internet at: http://pubs.acs.org
References
- 1.Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature. 2000;406:775–781. doi: 10.1038/35021219. [DOI] [PubMed] [Google Scholar]
- 2.Projan SJ, Youngman PJ. Antimicrobials: new solutions badly needed. Curr Opin Microbiol. 2002;5:463–465. doi: 10.1016/s1369-5274(02)00364-8. [DOI] [PubMed] [Google Scholar]
- 3.Russell AD, Chopra I. Understanding antibacterial action and resistance. E. Horwood; New York: 1990. [Google Scholar]
- 4.Wyckoff TJ, Raetz CRH, Jackman JE. Antibacterial and anti-inflammatory agents that target endotoxin. Trends Microbiol. 1998;6:154–159. doi: 10.1016/s0966-842x(98)01230-x. [DOI] [PubMed] [Google Scholar]
- 5.Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raetz CRH. Escherichia coli and Salmonella: cellular and molecular biology. 2nd. ASM Press; Washington, D.C.: 1996. pp. 1035–1063. [Google Scholar]
- 7.Coggins BE, Li X, McClerren AL, Hindsgaul O, Raetz CRH, Zhou P. Structure of the LpxC deacetylase with a bound substrate-analog inhibitor. Nat Struct Biol. 2003;10:645–651. doi: 10.1038/nsb948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coggins BE, McClerren AL, Jiang L, Li X, Rudolph J, Hindsgaul O, Raetz CRH, Zhou P. Refined solution structure of the LpxC-TU-514 complex and pKa analysis of an active site histidine: insights into the mechanism and inhibitor design. Biochemistry. 2005;44:1114–1126. doi: 10.1021/bi047820z. [DOI] [PubMed] [Google Scholar]
- 9.Gennadios HA, Whittington DA, Li X, Fierke CA, Christianson DW. Mechanistic inferences from the binding of ligands to LpxC, a metal-dependent deacetylase. Biochemistry. 2006;45:7940–7948. doi: 10.1021/bi060823m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gennadios HA, Christianson DW. Binding of Uridine 5′-Diphosphate in the “Basic Patch” of the Zinc Deacetylase LpxC and Implications for Substrate Binding. Biochemistry. 2006;45:15216–15223. doi: 10.1021/bi0619021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williamson JM, Anderson MS, Raetz CRH. Acyl-acyl carrier protein specificity of UDP-GlcNAc acyltransferases from gram-negative bacteria: relationship to lipid A structure. J Bacteriol. 1991;173:3591–3596. doi: 10.1128/jb.173.11.3591-3596.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hyland SA, Eveland SS, Anderson MS. Cloning, expression, and purification of UDP-3-O-acyl-GlcNAc deacetylase from Pseudomonas aeruginosa: a metalloamidase of the lipid A biosynthesis pathway. J Bacteriol. 1997;179:2029–2037. doi: 10.1128/jb.179.6.2029-2037.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hernick M, Fierke CA. Catalytic Mechanism and Molecular Recognition of E. coli UDP-3-O-(R-3-Hydroxymyristoyl)-N-acetylglucosamine Deacetylase Probed by Mutagenesis. Biochemistry. 2006;45:15240–15248. doi: 10.1021/bi061405k. [DOI] [PubMed] [Google Scholar]
- 14.Barb AW, McClerren AL, Karnam S, Reynolds CM, Zhou P, Raetz CRH. Inhibition of Lipid A Biosynthesis as the Primary Mechanism of CHIR-090 Antibiotic Activity in Escherichia coli. Biochemistry. 2007;46:3793–3802. doi: 10.1021/bi6025165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clements JM, Coignard F, Johnson I, Chandler S, Palan S, Waller A, Wijkmans J, Hunter MG. Antibacterial activities and characterization of novel inhibitors of LpxC. Antimicrob Agents Chemother. 2002;46:1793–1799. doi: 10.1128/AAC.46.6.1793-1799.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McClerren AL, Endsley S, Bowman JL, Andersen NH, Guan Z, Rudolph J, Raetz CRH. A Slow, Tight-Binding Inhibitor of the Zinc-Dependent Deacetylase LpxC of Lipid A Biosynthesis with Antibiotic Activity Comparable to Ciprofloxacin. Biochemistry. 2005;44:16574–16583. doi: 10.1021/bi0518186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen MH, Patchett AA, Galloway SM, Hyland SA, Anderson MS, Raetz CRH. Antibacterial agents that inhibit lipid A biosynthesis. Science. 1996;274:980–982. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 18.Barb AW, Jiang L, Raetz CRH, Zhou P. Structure of the deacetylase LpxC bound to the antibiotic CHIR-090: time-dependent inhibition and specificity in ligand binding. Proc Natl Acad Sci U S A. 2007;104:18433–18438. doi: 10.1073/pnas.0709412104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mochalkin I, Knafels JD, Lightle S. Crystal structure of LpxC from Pseudomonas aeruginosa complexed with the potent BB-78485 inhibitor. Protein Sci. 2008;17:450–457. doi: 10.1110/ps.073324108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hang HC, Yu C, Ten Hagen KG, Tian E, Winans KA, Tabak LA, Bertozzi CR. Small molecule inhibitors of mucin-type O-linked glycosylation from a uridine-based library. Chem Biol. 2004;11:337–345. doi: 10.1016/j.chembiol.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 21.Jackman JE, Raetz CRH, Fierke CA. UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine Deacetylase of Escherichia coli Is a Zinc Metalloenzyme. Biochemistry. 1999;38:1902–1911. doi: 10.1021/bi982339s. [DOI] [PubMed] [Google Scholar]
- 22.Copeland RA. Evaluation of enzyme inhibitors in drug discovery : a guide for medicinal chemists and pharmacologists. Wiley-Interscience; Hoboken, N.J.: 2005. [PubMed] [Google Scholar]
- 23.McClerren AL, Zhou P, Guan Z, Raetz CRH, Rudolph J. Kinetic analysis of the zinc-dependent deacetylase in the lipid A biosynthetic pathway. Biochemistry. 2005;44:1106–1113. doi: 10.1021/bi048001h. [DOI] [PubMed] [Google Scholar]
- 24.Hernick M, Gennadios HA, Whittington DA, Rusche KM, Christianson DW, Fierke CA. UDP-3-O-((R)-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase functions through a general acid-base catalyst pair mechanism. J Biol Chem. 2005;280:16969–16978. doi: 10.1074/jbc.M413560200. [DOI] [PubMed] [Google Scholar]
- 25.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 26.Weaver RF, Rajagopalan KV, Handler P, Jeffs P, Byrne WL, Rosenthal D. Isolation of γ-l-glutaminyl 4-hydroxybenzene and γ-l-glutaminyl 3,4-benzoquinone: a natural sulfhydryl reagent, from sporulating gill tissue of the mushroom Agaricus bisporus. Proc Natl Acad Sci U S A. 1970;67:1050–1056. doi: 10.1073/pnas.67.2.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.