

Abstract
Abnormal interaction of β-amyloid 42 (Aβ42) with copper, zinc and iron induce peptide aggregation and oxidation in Alzheimer's disease (AD). However, in health, Aβ degradation is mediated by extracellular metalloproteinases, neprilysin, insulin degrading enzyme (IDE) and matrix metalloproteinases. We investigated the relationship between levels of Aβ and biological metals in CSF. We assayed CSF copper, zinc, other metals and Aβ42 in ventricular autopsy samples of Japanese American men (N= 131) from the population-based Honolulu–Asia Aging Study. There was a significant inverse correlation of CSF Aβ42 with copper, zinc, iron, manganese and chromium. The association was particularly strong in the subgroup with high levels of both zinc and copper. Selenium and aluminum levels were not associated to CSF Aβ42. In vitro, the degradation of synthetic Aβ substrate added to CSF was markedly accelerated by low levels (2 μM) of exogenous zinc and copper. While excessive interaction with copper and zinc may induce neocortical Aβ precipitation in AD, soluble Aβ degradation is normally promoted by physiological copper and zinc concentrations.
Keywords: amyloid, Alzheimer's disease, metalloproteinase, cerebrospinal fluid, zinc, copper, iron, manganese, chromium
1. Introduction
The interaction of β-amyloid (Aβ) with endogenous brain copper, zinc and iron may contribute to the peptide's accumulation and toxicity in the brain in Alzheimer's disease (AD) [2,10,18,20,22,31]. High concentrations of copper (0.4 mM), zinc (1 mM) and iron (1mM) have been found within amyloid plaques [23], but only zinc and copper have been shown to directly coordinate Aβ [14,31]. The toxicity of Aβ in cell culture depends upon the peptide binding to copper [2] or iron [39] through generating radical chemistry (Aβ42>Aβ40) [20,31]. Zinc, copper and iron rapidly precipitate Aβ [2,6,13,19], which then becomes resistant to proteolysis [9].
The brain and CSF are protected from fluctuations in plasma metal concentrations by the blood-brain barrier. The zinc and copper in plaques is likely to have been released by glutamatergic synapses [22,40]. While Aβ interaction with copper and zinc is corrupted in AD, in health a normal amount of interstitial zinc is needed for the degradation of Aβ by zinc-dependent proteinases that are believed to prevent it from accumulating in the interstitium. Such metalloproteinases, which are also present in cerebrospinal fluid (CSF), include neprilysin [24], insulin degrading enzyme (IDE) [35] and matrix metalloproteinases (MMP2 and 3) [42]. Additionally, the normal generation of Aβ may be modulated by zinc since α-secretases, tumor necrosis factor-alpha-converting enzyme (TACE or ADAM-17) and ADAM-10 [11], are members of the cell surface zinc metalloproteinase family of disintegrin and metalloprotease (ADAM) proteins. Abnormal metal homeostasis in the brain in AD [37], may therefore impact upon the processing or catabolism of Aβ.
To test whether there is a biochemical relationship between Aβ and biometal levels in humans, we investigated the association of CSF copper, zinc and other metals to CSF Aβ42 in a large cohort of ventricular fluid autopsy samples.
2. Methods
2.1 Human CSF samples
The autopsy sample is from a cohort of Japanese-American men participating in the population-based Honolulu Asia Aging Study (HAAS) [43]. The HAAS began in 1991 as a supplement to the Honolulu Heart Program Study to investigate the determinants of dementia. All participants were eligible for the autopsy substudy; cases of dementia were preferentially followed up to ensure adequate sample size for comparison of cases to controls. Autopsied demented decedents were similar to the demented men who were not autopsied, and autopsied non-demented men were similar to the non-demented decedents who were not autopsied. The institutional review board of Kuakini Medical Center approved the study, and informed consent for autopsy was obtained from the appropriate surviving person.
2.2 Neuropathology
Neuropathologic methods in the population-based Honolulu Asia Aging Study (HAAS) have been detailed elsewhere [34]. Briefly, 8-micron tissue sections from multiple brain regions were analyzed with several stains. Analyses presented here utilize counts of neurofibrillary tangles, and neuritic plaques, from the left side of the brain for two regions of the hippocampus (CA1 and subiculum) and four areas of the neocortex (middle frontal gyrus, middle temporal gyrus, inferior parietal lobule, and occipital lobe). All plaque and neurofibrillary tangle (NFT) counts were based on modified Bielschowski stains. Neuritic plaques (NPs) were defined as plaques containing silver-positive neurites. Five fields standardized to 1 mm2 were examined for each of the four neocortical and two hippocampal areas. The field with highest count was taken to represent the brain area. Counts for NP and NFT were calculated by averaging across the four neocortical or two hippocampal areas. Results were recorded as NP per square millimeter and were truncated at 17/mm2 [33]. There was no upper limit for NFT counts. Congophilic amyloid angiopathy (CAA) was quantified by Aβ immunohistochemistry of parenchymal arteries and arterioles, from all four neocortical areas (CAA absent = all vessels non-reactive).
Samples were evaluated by one of three neuropathologists who participated in a training session aimed to standardize reading techniques. CERAD criteria were used for pathologic diagnosis of AD [29]. These criteria are based on semiquantitative assessment of NP. Three illustrations representing sparse, moderate, and frequent plaque densities per square millimeter are used as guides for comparison with the microscopic case being assessed. A match with the frequent NP illustration confers neuropathologic diagnosis of definite AD, and the diagnosis of probable AD is made when the case matches the moderate frequency illustration. A maximum NP count of ≥17/mm2 was used to meet CERAD requirements for definite AD, and a count of at least 4/mm2 was used to meet CERAD requirements for probable AD.
2.3 CSF Studies
As the first step in the autopsy, post-mortem CSF was obtained from the cisterna magna by needle aspiration. CSF samples were then chilled in ice and transferred to -70 C for long-term storage. CSF Aβ42 was measured using a special high-sensitivity version of sandwich ELISA [INNOTEST β-amyloid (1-42), Innogenetics, Ghent, Belgium] [1]. The lowest detectable level was 7 ng/L. Levels ranged from 7 ng/L to 858 ng/L with a median of 57 ng/L (interquartile range 12 – 142).
CSF metals were measured by inductively coupled plasma mass spectrometry as previously described [12]. Levels of CSF selenium below 0.04 μM (n=3) and CSF aluminum below 0.008 μM (n=74), which are below the standard calibration range of the spectrometer, were set to the lowest detectable value.
2.4 Analytical sample
At the time of this analysis, 231 men were included in the autopsy study and had CSF data available. Visible blood contamination significantly increased iron, zinc and copper levels (data not shown). Therefore, CSF samples were excluded when visual inspection indicated blood contamination (hemolysis) after centrifugation, leaving 152 individuals with clear CSF considered for this analysis.
The interval between time of CSF collection and death was inversely correlated with Aβ42 levels (Spearman's correlation coefficient = -0.5; p-value <0.001), and positively correlated with CSF metal levels (copper Spearman's correlation coefficient = 0.6; p-value < 0.001; zinc Spearman's rho = 0.5; p-value 0.001). Therefore, we excluded 21 samples with a collection interval > 24 hours, and controlled for the effects of post-mortem interval as a confounder in the final analytical sample of N=131.
Clinical diagnoses were established ante-mortem. The subjects included 82 clinically non-demented individuals (62.1 %). Clinical ante-mortem dementia diagnoses used in this analysis include: 25 cases of Alzheimer's disease (AD), 18 cases of vascular dementia (VD) and 6 cases of other dementias (including Parkinson's disease and Lewy Body dementia, trauma plus dementia and dementia of undetermined cause). Clinical dementia was diagnosed according to DSM IIIR criteria, AD following NINCDS-ADRDA criteria, and VD following criteria of the California Alzheimer's Disease Diagnostic and Treatment Centers guidelines. Among all clinical AD cases, 21 cases met probable and 4 cases met definite pathological CERAD criteria.
2.5 Synthetic Aβ stability in CSF
Human cerebrospinal fluid from neuropathologically normal individuals was obtained from the Victorian Brain Bank Network. Aβ40 (230 μL, 1ng/μL in doubly-distilled deionized water) was added to CSF (30 μL) ± CuCl2 or ZnCl2, TPEN or water (10 μL) and mixed vigorously at 37°C for 4 hours. SDS-sample buffer was then added. The samples were heated (90°C, 5 min) and separated on a 26-well Criterion 10-20% Tris-tricine gel. Blocking was performed for 1 hour in 5% skim milk in TBS-T followed by overnight incubation with primary antibody WO2 (detecting residues 5-8 of Aβ; 1:25 dilution in TBS-T). Following washing the blot was incubated in anti-mouse HRP secondary antibody for one hour, washed in TBS-T and developed with ECL reagent. Imaging was performed using a LAS-3000 system (FujuFilm) and the amounts of Aβ monomer quantified using MultiGauge V3.0 software. All analysis was performed using JMP V.5.01 statistical discovery software (SAS) for Macintosh.
2.6 Statistical analysis
We considered as possible confounders: age at death, education in years, time interval in hours from death to collection of cerebrospinal fluid, and apolipoprotein E4 allele (genotyped with restriction isotyping using a polymerase chain reaction). As the severity of dementia might have changed in the time period from diagnosis to death, we also adjusted for the time interval from diagnosis or last clinical evaluation to death (mean 3.9 years (range 0.3 – 8.7)).
Stata 8.0 (Statacorp, College Station, TX) software was used for analyses. General linear regression was employed to describe CSF Aβ42 level differences among tertiles of metals. Since the distribution of the CSF Aβ42 concentration was skewed, we fit a general linear regression model (GLM) with a log-link and a gamma distribution [27]. Compared to the commonly used ordinary least square regression model with log transformed values, this GLM model preserves the original scale of the variable and hence yields readily interpretable regression coefficients.
To examine the combined effect of both CSF copper and zinc we divided the sample based on their median CSF copper and zinc levels. This gave us four groups: low for both metals (-Zn, -Cu) for participants with CSF-copper and CSF-zinc levels below their median level (n=53); high for both metals (+Zn, +Cu) for participants with both metal levels above their median level (n=52); high for zinc (+Zn, -Cu) for participants with high levels of zinc and low levels of copper (n=13), and low for zinc (-Zn, +Cu) for participants with low zinc and high copper (n=13). The low group (-Zn, -Cu) served as the reference group for the analyses.
Multiple regression analyses based on the over dispersed Poisson model were used to examine the association of NPs and NFTs to tertiles of metal levels. This statistical model yields a ratio of counts; for example a ratio of 0.8, means that the exposed group has 20% fewer maximum counts of NP per mm2 than the reference group.
3. Results
3.1 Inverse correlation between CSF Aβ42 and biometal levels
The relative abundances of elements we surveyed in the CSF were Fe>Zn>Cu>Se>Al>Mn>Cr. Iron had the highest concentration, at a median level of 9.94 μmol/L (range 0.5-31.7), followed by zinc at 1.84 μmol/L (range 0.21-12.45) and copper at 0.52 μmol/L (range 0.01-5.7). Selenium had a mean level of 0.43 μmol/L (range 0.04 – 0.92), aluminum of 0.25 μmol/L (range 0.008 – 5.78), manganese of 0.22 μmol/L (range 0.03 – 0.52), and chromium of 0.20 μmol/L (range 0.03 – 0.43). Baseline characteristics of the sample by copper and zinc tertiles are presented in Table 1.
Table 1. Baseline characteristics of the sample by copper and zinc tertiles: the Honolulu-Asia Aging Autopsy Study.
† Other dementias included Parkinson's disease and Lewy Body disease, trauma plus dementia and dementia of undetermined cause. ‡ Adjusted for age at death and postmortem interval using a logistic regression analysis for dichotomous variables or general linear regression analysis for linear variables. * Adjusted for age at death and postmortem interval using metals as linear independent variables. ^ CERAD cases (n=25) compared to rest of the autopsied samples (n=106).
| Copper μmol/L | Zinc μmol/L | |||||||
|---|---|---|---|---|---|---|---|---|
| Low <0.4 | Mid 0.4-0.75 | High >0.75 | p trend ‡ | Low <1.3 | Mid 1.3-2.5 | High >2.5 | p trend ‡ | |
| Demographic factors | ||||||||
| Age at death (yr ± SD) | 85.8 (5.4) | 85.0 (5.1) | 86.6 (5.6) | 0.5 | 85.5 (5.6) | 84.8 (4.8) | 87.0 (5.6) | 0.2 |
| Postmortem interval (hr ± SD) | 9.1 (5.3) | 12.4 (5.8) | 16.0 (4.5) | <0.001 | 8.8 (5.2) | 12.4 (5.4) | 16.2 (4.7) | <0.001 |
| APOE ε4 allele (%) | 24.4 | 20.9 | 16.3 | 0.7 | 20.5 | 23.3 | 18.2 | 0.7 |
| Clinical dementia (%) | 44.4 | 30.2 | 37.2 | 0.4 | 34.1 | 44.2 | 34.1 | 0.8 |
| Alzheimer's Disease (n=25) | 17.8 | 18.6 | 20.9 | 0.2 * | 18.2 | 20.9 | 18.2 | 0.2 * |
| Vascular dementia (n=18) | 17.8 | 9.3 | 14.0 | 0.7 * | 11.4 | 16.3 | 13.6 | 0.9 * |
| Other dementia† (n=6) | 8.9 | 2.3 | 2.3 | 0.2 * | 4.6 | 7.0 | 2.3 | 0.2 * |
| Time from last clinical exam until death (yr ± SD) | 3.7 (1.6) | 4.2 (1.4) | 4.2 (2.1) | 0.33 | 3.6 (1.6) | 3.9 (1.7) | 4.0 (2.0) | 0.06 |
| Clinico-pathologic groups (%) | ||||||||
| prob/def CERAD̂ | 22.2 | 11.6 | 23.3 | 0.8 | 18.2 | 20.9 | 18.2 | 0.6 |
| Cerebrovascular lesions (% with any) | ||||||||
| Infarct ≥ 1cm | 17.8 | 27.9 | 39.5 | 0.1 | 18.2 | 30.2 | 36.4 | 0.3 |
| Lacune <1 cm | 48.9 | 44.2 | 44.2 | 0.4 | 50 | 46.5 | 40.9 | 0.1 |
| Microinfarct | 69.4 | 62.5 | 54 | 0.1 | 68.6 | 63.2 | 55 | 0.1 |
| CAA | 53.3 | 51.2 | 46.5 | 0.9 | 52.3 | 51.2 | 47.7 | 0.9 |
Increasing biometal concentrations were associated with a decrease in CSF Aβ42 concentration in the postmortem CSF after adjusting for all covariates (Table 2, Figure 1). The association was strongest for copper (β coefficient = -1.3; 95%CI [-1.78, -0.8] p-trend = <0.0001) and zinc in the highest tertile (β coefficient = -1.26; 95%CI [-1.7, -0.8] p-trend= <0.0001), when compared to the lowest tertile. These associations remained significant with a post-mortem interval of less than twelve hours, and equally significant (p-trend= <0.0001) after removing outliers (the six highest Aβ42 and the one highest metal readings) (data not shown).
Table 2. Analysis of the association of Aβ42 and metal levels in cerebrospinal fluid.
Statistical analysis using a general linear regression model with a log-link, adjusted for age at death, education, APOE ε4, post mortem interval until CSF collection, and time from last clinical exam until death.
| Metals (μM) | Mean Aβ42 (ng/L) | Mean difference (95% CI) | p-trend |
|---|---|---|---|
| Copper (range 0.01-5.7) | |||
| Low (<0.4) | 114.9 | 0 (reference) | <0.0001 |
| Mid (0.4-0.75) | 94.7 | -0.19 (-0.62, 0.2) | |
| High (> 0.75) | 32.6 | -1.26 (-1.7, -0.8) | |
| Zinc (range 0.21-12.45) | |||
| Low (<1.3) | 116.7 | 0 (reference) | <0.0001 |
| Mid (1.3-2.5) | 95.2 | -0.2 (-0.6, 0.2) | |
| High (> 2.5) | 33.8 | -1.2 (-1.7, -0.76) | |
| Iron (range 0.53-31.7) | |||
| Low (<7.2) | 118.6 | 0 (reference) | 0.001 |
| Mid (7.2-12.0) | 70.6 | -0.52 (-0.99, -0.05) | |
| High (> 12.0) | 52.2 | -0.83 (-1.4, -0.34) | |
| Manganese (range 0.32-0.52 | |||
| Low (<0.16) | 110.5 | 0 (reference) | 0.003 |
| Mid (0.16-0.24) | 75.1 | -0.39 (-0.9, 0.1) | |
| High (> 0.24) | 53.3 | -0.73 (-1.2, -0.2) | |
| Chromium (range 0.03-0.43) | |||
| Low (<0.157) | 94.3 | 0 (reference) | 0.01 |
| Mid (0.157-0.23) | 102 | 0.08 (-0.4, 0.5) | |
| High (> 0.23) | 43.8 | -0.77 (-1.23, -0.3) | |
| Selenium (range 0.04-0.92) | |||
| Low (<0.32) | 84.2 | 0 (reference) | 0.3 |
| Mid (0.32-0.53) | 89.1 | 0.06 (-0.4, 0.6) | |
| High (> 0.53) | 65.6 | -0.25 (-0.7, 0.2) | |
| Aluminum (range 0.008-5.78) | |||
| Low (≤ 0.008) | 86.3 | 0 (reference) | 0.5 |
| Mid (0.0081-0.096) | 58.9 | -0.38 (-1.05, 0.3) | |
| High (> 0.096) | 74.4 | -0.15 (-0.56, 0.37) |
Figure 1. Relationship of CSF zinc and copper to CSF Aβ42.
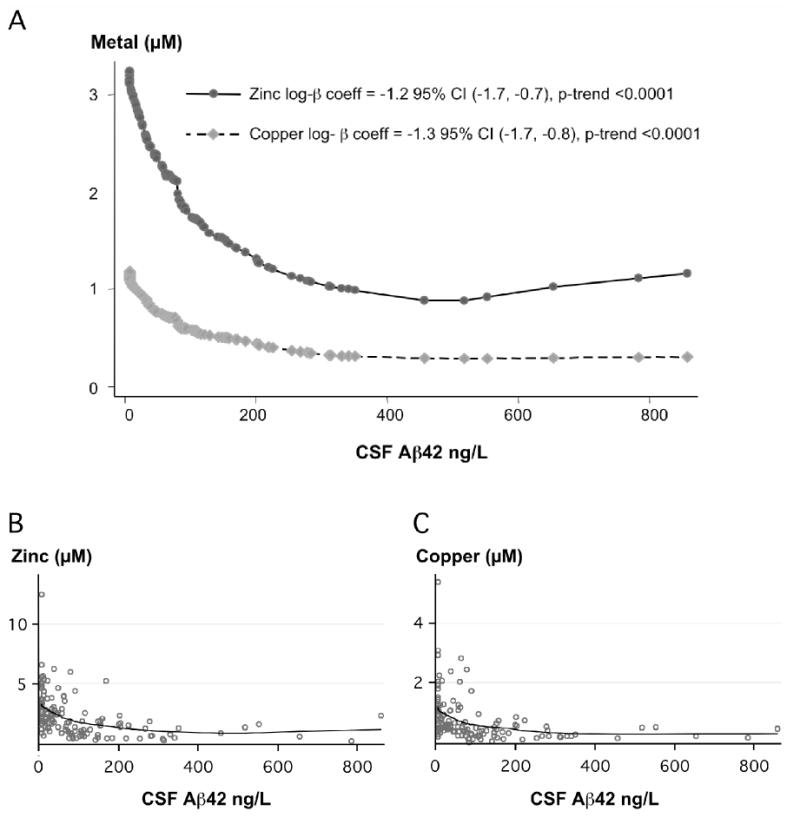
A. Plot of mean adjusted smoothed values achieved by locally weighted scatterplot smoother, bandwidth = 0.8 (Stata 8.0), B. Raw data for zinc levels with mean adjusted smoothed values used for A, C. Raw data for copper levels with mean adjusted smoothed values used for A.
Iron, manganese and chromium were also significantly inversely associated with CSF- Aβ42. There was no relationship of CSF Aβ42 to aluminum and selenium (Table 2).
Copper and zinc were synergistically associated with CSF- Aβ42 levels. Combined high zinc and high copper levels (+Zn, +Cu) were associated with lower CSF- Aβ42 levels (log β coefficient -1.16 [-1.6, -0.7]), when compared to low levels of both CSF metals (-Zn, -Cu) (Figure 2). There was no association with CSF- Aβ42 if only one metal was elevated, i.e. high zinc and low copper (+Zn, -Cu) or low zinc and high copper (-Zn, +Cu).
Figure 2. Combined association of CSF zinc and copper levels to Aβ42 levels.
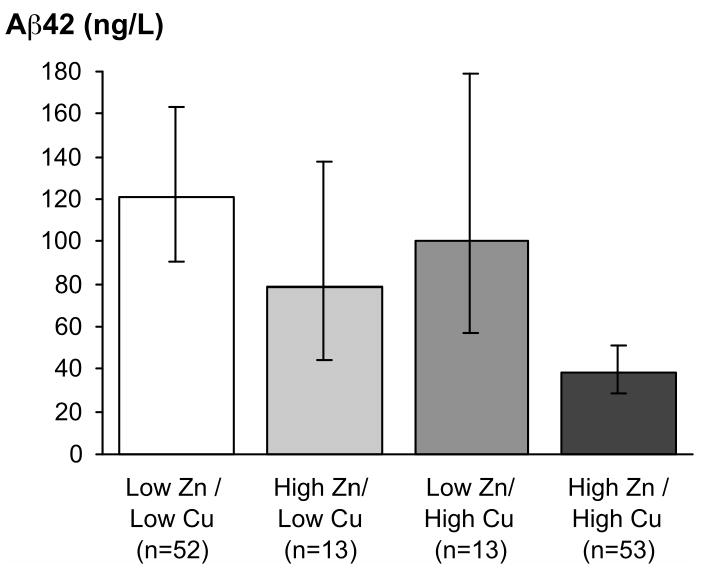
Results represent mean levels adjusted for age at death, post-mortem interval, education, APOE e4 allele, and time from last clinical assessment until death. Error bars are 95% confidence intervals.
3.2 CSF metals, clinical dementia and neuropathology
Prevalence of ante-mortem dementia diagnosis did not vary between postmortem CSF copper, CSF zinc tertiles (Table 1) or other metals. There was a trend of higher CSF copper and zinc levels with fewer neuritic plaque counts in either the neocortex or the hippocampus (Table 3). There was no relationship with other metal levels and neuropathology (data not shown). There were 25 cases of pathologically confirmed AD cases (CERAD). Those cases did not differ by metal levels from other cases (Table 1).
Table 3. Analysis of the association of pathologic hallmarks of AD and CSF metals in cerebrospinal fluid.
| Neocortex | Hippocampus | |||
|---|---|---|---|---|
| Neuritic plaques IRR [95%CI] | Neurofibrillary tangles IRR [95%CI] | Neuritic plaques IRR [95%CI] | Neurofibrillary tangles IRR [95%CI] | |
| Copper | ||||
| Low <0.4 μM | 1 | 1 | 1 | 1 |
| Mid 0.4-0.75 μM | 0.99 [0.5-1.9] | 1.26 [0.5-3.2] | 1.0 [0.5-2.1] | 1.11 [0.6-1.9] |
| High >0.75 μM | 0.95 [0.5-2.0] | 2.23 [0.8-5.9] | 0.85 [0.4-2.0] | 1.08 [0.6-2.0] |
| Zinc | ||||
| Low <1.3 μM | 1 | 1 | 1 | 1 |
| Mid 1.3-2.5 μM | 0.95 [0.48-1.87] | 2.09 [0.8-5.2] | 1.11 [0.5-2.6] | 2.16 [1.2-3.8] |
| High >2.5 μM | 0.82 [0.39-1.74] | 1.43 [0.5-4.2] | 1.14 [0.5-2.8] | 1.38 [0.7-2.7] |
3.3 Degradation of synthetic Aβ by CSF metalloproteinases
To explore whether the inverse correlation between CSF Aβ and Cu or Zn could reflect the modulation of proteolytic activities by the metal ions, we studied the effects of modulating Cu or Zn, the only metal ions shown to directly coordinate Aβ in vivo [14,31], upon the stability of synthetic Aβ added to samples of normal CSF (Figure 3). We incubated synthetic Aβ1-40 added to CSF with the presence of additional Cu or Zn at a physiological concentration (2 μM) or TPEN (150 μM), a zinc/copper-selective chelator. The presence of added Cu or Zn induced the Aβ to degrade markedly after 4 hours incubation, whereas there was only slight loss of Aβ in the absence of the metals or in the presence of TPEN (Fig. 3). The loss of Aβ was not due to precipitation because previous studies have shown that the SDS buffer, which was added to the samples to recover the Aβ, completely dissolves metal-induced Aβ precipitates [8].
Figure 3. Zinc and Copper promote the degradation of Aβ in CSF.
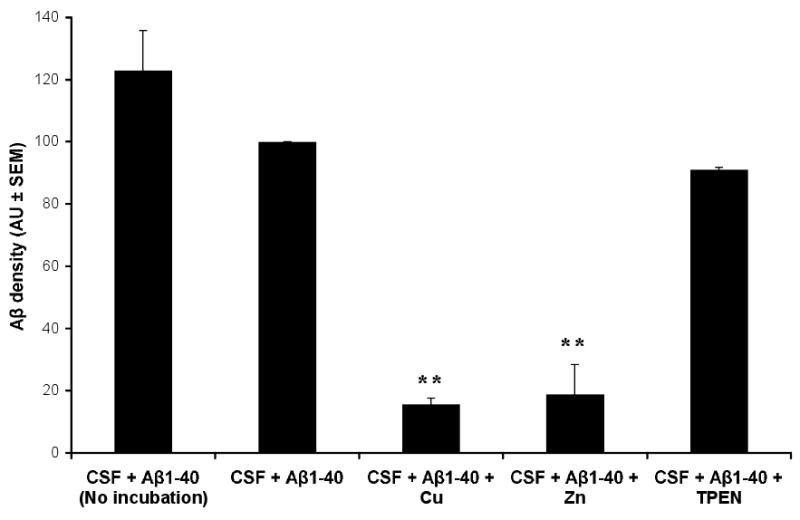
Synthetic Aβ1-40 was added to normal CSF ± Cu, Zn (2 μM) or the Cu/Zn-selective chelator TPEN (150 μM), and incubated for 4 hours. The amount of Aβ remaining in the CSF was then quantified by western blot and densitometry. All data (means + SEM) have been normalized to the density of the 4 kD immunoreactive Aβ density of the CSF + Aβ sample. Samples were assayed in triplicate, and the results are typical of N=6 experiments. ** p<0.01 vs CSF + Aβ sample.
4. Discussion
In this general population autopsy study, we found a strong inverse correlation of CSF-Aβ42 with CSF metals copper, zinc, iron, manganese and chromium. There was no association with selenium or aluminum. There was also a synergistic interaction of elevated copper and zinc being associated with lower CSF Aβ42 levels. These associations were controlled for several confounding variables, including post-mortem interval. Therefore, we conclude that levels of CSF Aβ42 may have a relationship with levels of CSF biometals. This was supported by in vitro data that low concentrations of exogenous Zn and Cu promoted the degradation of synthetic Aβ added to CSF samples (Figure 3).
Aβ is produced in health where it is a normal component of brain and CSF, and is turned over in CSF rapidly as it exits the CNS into the blood [5]. The steady state levels of Aβ in CSF reflect factors that influence amyloid protein precursor (APP) processing, and factors that clear and degrade Aβ. The small amount of Aβ degradation by CSF activities in vitro in the presence of a chelator (Figure 3) indicates that non-metalloproteinase activities may degrade Aβ in CSF. However, the rapid degradation of Aβ by CSF activities upon the addition of 2μM Cu or Zn (Figure 3), is consistent with a major role for metalloproteinases in soluble Aβ degradation. Since the addition of TPEN did not alter Aβ degradation in CSF, we conclude metalloproteinases may be in their inactive apo-forms in CSF, and that adding Cu and Zn activates them (Figure 3). Therefore, the relationship between CSF metals and Aβ that we identified (Figs. 1 & 2, Table 1) probably reflects the interaction of metals with Aβ or with enzymes that cleave Aβ or process APP in the brain interstitium. Cu and Zn could promote non-amyloidogenic APP processing by metalloproteinases such as TACE and ADAM-10 [11], or promote the degradation of Aβ by interstitial metalloproteinases neprilysin and IDE. Alternatively, elevated metals could promote the sequestration of Aβ by α-2 macroglobulin [15]. In each of these instances, Aβ would be degraded as metal levels increased, consistent with our current findings. These findings support endogenous brain metal metabolism as being a critical factor in determining steady state levels of Aβ. Aluminum is not known to participate in normal neurochemistry, hence it is not surprising that we found no association of Al levels with Aβ. This finding is also consistent with multiple epidemiological studies that have failed to sustain the once-held suspicion that Al intoxication could increase the risk for AD [16]. Similarly, selenium is not normally found in the metal ionic state in tissues, but rather as selenomethionine or selenocysteine and would not be expected to influence Aβ metabolism by the metalloproteinase mechanism envisaged here.
This newly-defined close association of CSF Aβ with biological metal levels in health may explain the vulnerability of Aβ to abnormal interaction with these metals, which could trigger AD [7]. Therefore, we also explored whether the abnormal interaction of metals with Aβ in the brain interstitium in AD is reflected in the CSF. There are very few reports on CSF metal levels in AD. One study reported a significant elevation of copper [3], a second study described decreased zinc levels in living AD subjects [30]. We did not detect such changes, but there were several limitations that may have diminished the power of the study to discriminate CSF metal levels associated with clinical or neuropathological AD. The clinical dementia assessment was performed up to 8.7 years prior to the measurement of Aβ42 and metals in postmortem CSF. Therefore, it is likely that some non-demented individuals developed clinical dementia before death, and that demented participants would have further progressed. Also, the number of AD cases was relatively small. We did not observe an association of CSF metal levels to neuritic (amyloid) plaques, but it has been previously suggested the amyloid plaques do not correlate with total Aβ42 burden in brain tissue [28]. Also, there were no brain Aβ42 values to compare with the CSF values. Future studies that analyze the association of CSF metals and CSF Aβ42 in living subjects are needed to clarify these associations.
Since an intimate biochemical relationship between Aβ and biological metals is revealed by our results, the findings also underscore the potential potency of pharmacological approaches targeting Aβ-metal interactions. One would expect, for example, high-affinity chelators to inhibit Aβ degradation if they could enter the CSF compartment. This could be viewed a favorable since low CSF Aβ levels have predictive value for developing AD [17,41]. However treatment with clioquinol (CQ), a zinc/copper chelator, lowered plasma Aβ in a double-blind phase 2 pilot study in AD subjects [38]. Since plasma Aβ is in a dynamic equilibrium with CSF Aβ [4] it is tempting to assume that the CQ-induced drop in plasma Aβ would have been be associated with a drop in CSF Aβ and therefore might be sinister. However, the levels of Aβ in the CSF/plasma compartments have only been established to be in a dynamic equilibrium over short time scales in healthy humans (eg hours). The nature of the steady-state equilibrium may be quite different on a time scale measured in weeks to months (as in clinical trials or prospective biomarker surveys), or in the presence of a brain amyloid reservoir seen in AD. In attempting to interpret the meaning of the changes in plasma Aβ in the CQ trial, it is important to note that the CQ-treated AD subjects exhibited a significant arrest in cognitive deterioration compared to placebo controls during the epoch after 24 weeks of treatment where the plasma Aβ levels rose. Also, CQ treatment has been reported to stabilize clinical deterioration over 9-14 months in an open-label study of familial AD [21]. Therefore, the rise in plasma Aβ associated with CQ treatment is unlikely to be sinister.
Another important point to consider about the mechanism of action of CQ in this context, is that it is a low-affinity chelator and does not remove metals from tissue. Unlike high-affinity chelators, CQ actually raises brain copper and zinc levels [12]. Animal studies have shown that despite limited blood-brain barrier penetration, CQ concentrates in brain amyloid pathology and is cleared within hours from the brain compartment and plasma [32], so its site of action in amyloid clearance [12] is unlikely to be the CSF. We hypothesize that CQ binds copper and zinc in amyloid aggregates and so promotes Aβ dissociation and clearance, with the metals returning to relatively metal-deficient tissue. It is therefore difficult to predict the consequent change in CSF Aβ levels in the short and long term, as illustrated by a phase 1 study with low doses of CQ showing CSF Aβ levels to rise at day 7 of treatment and then fall at day 21 [36]. This may be due to dissociated Aβ from plaques transiently exiting the brain via the CSF. A second-generation derivative of CQ with much better blood-brain barrier permeability, named PBT2 (http://www.alzforum.org/drg/drc/default.asp?type=drugName&drugName=PBT2), has nearly completed a double-blind phase 2 clinical trial where plasma and CSF Aβ are assayed over a period of 12 weeks of treatment. Data from this study could illuminate the complexities involved in perturbing the equilibria between the brain amyloid compartment, CSF Aβ and plasma Aβ using this pharmacological approach.
The close inverse association that we have identified between CSF Aβ levels and biological metals underscore the importance of brain metal homeostasis as a potential factor in the evolution of AD pathology. These data would also be consistent with a role for APP and its derivatives in participating in metal regulation in brain tissue, as previously considered [25,26].
Acknowledgments
This study was supported by NIH (National Institute on Aging); Grant Numbers 1RO1 AG12686 (to AIB), 1 U01 AG19349-01, 5 R01 AG017155-04, and in part by the Intramural Research Program of the NIH, National Institute on Aging, as well as (to AIB) the Australian Research Council Federation Fellowship, the National health and Medical Research Council of Australia, the Alzheimer Association Zenith Award and the American Health Assistance Foundation. The Victorian Brain Bank Network is supported by The University of Melbourne, The Mental Health Research Institute of Victoria, Victorian Forensic Institute of Medicine and funded by Neurosciences Australia and the National Health & Medical Research Council.
Abbreviations
- Aβ42
β-amyloid ending at residue 42
- AD
Alzheimer's disease
- APP
amyloid protein precursor
- APLP2
amyloid precursor-like protein 2
- CAA
congophilic amyloid angiopathy
- CERAD
consortium to establish a registry for Alzheimer's disease
- CI
confidence interval
- CQ
clioquinol
- CSF
cerebrospinal fluid
- DP
diffuse plaque
- ELISA
enzyme-linked immunosorbent assay
- GLM
general linear regression model
- NFT
neurofibrillary Tangle
- NP
neuritic plaque
- PMI
post-mortem interval
- SOD1
superoxide dismutase 1
- TPEN
N,N,N′,N′-Tetrakis-(2-pyridylmethyl)-Ethylenediamine
- MMP
matrix metalloproteinase
- PMI
post-mortem interval
- SDS
sodium dodecyl sulfate
Footnotes
Conflict of interest: Drs Bush and Cherny are shareholders in and consultants for Prana Biotechnology Ltd. The company had no involvement in the funding, design or interpretation of the studies, and no input in the drafting of the manuscript.
Disclosure statement: Drs Bush, Adlard and Cherny shareholders in and consultants for Prana Biotechnology Ltd. The company had no involvement in the funding, design or interpretation of the studies, and no input in the drafting of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andreasen N, Hesse C, Davidsson P, Wallin A, Minthon L, Winblad B, Vanderstichele H, Vanmechelen E, Blennow K. Cerebrospinal fluid b-amyloid(1-42) in Alzheimer's disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol. 1999;56:673–80. doi: 10.1001/archneur.56.6.673. [DOI] [PubMed] [Google Scholar]
- 2.Atwood CS, Moir RD, Huang X, Bacarra NME, Scarpa RC, Romano DM, Hartshorn MA, Tanzi RE, Bush AI. Dramatic aggregation of Alzheimer Aβ by Cu(II) is induced by conditions representing physiological acidosis. Journal of Biological Chemistry. 1998;273:12817–26. doi: 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- 3.Basun H, Forssell LG, Wetterberg L, Winblad B. Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer's disease. J Neural Transm Park Dis Dement Sect. 1991;3(4):231–58. [PubMed] [Google Scholar]
- 4.Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006;12(7):856–61. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brody DL, Holtzman DM. Morris water maze search strategy analysis in PDAPP mice before and after experimental traumatic brain injury. Exp Neurol. 2006;197(2):330–40. doi: 10.1016/j.expneurol.2005.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown AM, Tummolo DM, Rhodes KJ, Hofmann JR, Jacobsen JS, Sonnenberg-Reines J. Selective aggregation of endogenous β-amyloid peptide and soluble amyloid precursor protein in cerebrospinal fluid by zinc. J Neurochem. 1997;69(3):1204–12. doi: 10.1046/j.1471-4159.1997.69031204.x. [DOI] [PubMed] [Google Scholar]
- 7.Bush AI. The Metallobiology of Alzheimer's disease. TINS. 2003;26(4):207–14. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 8.Bush AI, Moir RD, Rosenkranz KM, Tanzi RE. Zinc and Alzheimer's disease. Science. 1995;268:1921–3. doi: 10.1126/science.268.5219.1921. [DOI] [PubMed] [Google Scholar]
- 9.Bush AI, Pettingell WH, Jr, Paradis MD, Tanzi RE. Modulation of Aβ adhesiveness and secretase site cleavage by zinc. J Biol Chem. 1994;269:12152–8. [PubMed] [Google Scholar]
- 10.Bush AI, Pettingell WH, Multhaup G, Paradis Md, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL, Tanzi RE. Rapid induction of Alzheimer Aβ amyloid formation by zinc. Science. 1994;265:1464–7. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 11.Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273(43):27765–7. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 12.Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim YS, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–76. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 13.Cherny RA, Legg JT, McLean CA, Fairlie D, Huang X, Atwood CS, Beyreuther K, Tanzi RE, Masters CL, Bush AI. Aqueous dissolution of Alzheimer's disease Aβ amyloid deposits by biometal depletion. Journal of Biological Chemistry. 1999;274:23223–8. doi: 10.1074/jbc.274.33.23223. [DOI] [PubMed] [Google Scholar]
- 14.Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR. Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry. 2003;42(10):2768–73. doi: 10.1021/bi0272151. [DOI] [PubMed] [Google Scholar]
- 15.Du Y, Ni B, Glinn M, Dodel RC, Bales KR, Zhang Z, Hyslop PA, Paul SM. alpha2-Macroglobulin as Aβ-amyloid peptide-binding plasma protein. J Neurochem. 1997;69(1):299–305. [PubMed] [Google Scholar]
- 16.Flaten TP. Aluminium as a risk factor in Alzheimer's disease, with emphasis on drinking water. Brain Res Bull. 2001;55(2):187–96. doi: 10.1016/s0361-9230(01)00459-2. [DOI] [PubMed] [Google Scholar]
- 17.Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow-up study. Lancet neurology. 2006;5(3):228–34. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- 18.Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, Tanzi RE, Bush AI. The Aβ peptide of Alzheimer's Disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–16. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 19.Huang X, Atwood CS, Moir RD, Hartshorn MA, Vonsattel JP, Tanzi RE, Bush AI. Zinc-induced Alzheimer's Aβ1-40 aggregation is mediated by conformational factors. J Biol Chem. 1997;272:26464–70. doi: 10.1074/jbc.272.42.26464. [DOI] [PubMed] [Google Scholar]
- 20.Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall J, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI. Cu(II) potentiation of Alzheimer Aβ neurotoxicity: correlation with cell-free hydrogen peroxide production and metal reduction. Journal of Biological Chemistry. 1999;274:37111–6. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 21.Ibach B, Haen E, Marienhagen J, Hajak G. Clioquinol treatment in familiar early onset of Alzheimer's disease: a case report. Pharmacopsychiatry. 2005;38(4):178–9. doi: 10.1055/s-2005-871241. [DOI] [PubMed] [Google Scholar]
- 22.Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci U S A. 2002;99(11):7705–10. doi: 10.1073/pnas.092034699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci. 1998;158(1):47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 24.Maruyama M, Higuchi M, Takaki Y, Matsuba Y, Tanji H, Nemoto M, Tomita N, Matsui T, Iwata N, Mizukami H, Muramatsu S, Ozawa K, Saido TC, Arai H, Sasaki H. Cerebrospinal fluid neprilysin is reduced in prodromal Alzheimer's disease. Ann Neurol. 2005;57(6):832–42. doi: 10.1002/ana.20494. [DOI] [PubMed] [Google Scholar]
- 25.Maynard CJ, Cappai R, Volitakis I, Cherny RA, Masters CL, Li QX, Bush AI. Gender and genetic background effects on brain metal levels in APP transgenic and normal mice: implications for Alzheimer beta-amyloid pathology. J Inorg Biochem. 2006;100(56):952–62. doi: 10.1016/j.jinorgbio.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 26.Maynard CJ, Cappai R, Volitakis I, Cherny RA, White AR, Beyreuther K, Masters CL, Bush AI, Li QX. Overexpression of Alzheimer's disease β-amyloid opposes the age-dependent elevations of brain copper and iron levels. Journal of Biological Chemistry. 2002;277(47):44670–6. doi: 10.1074/jbc.M204379200. [DOI] [PubMed] [Google Scholar]
- 27.McCullagh P, Nelder J. Generalized linear models. London, UK: Chapman and Hall; 1989. [Google Scholar]
- 28.McLean C, Cherny R, Fraser F, Fuller S, Smith M, Beyreuther K, Bush A, Masters C. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's Disease. Annals of Neurology. 1999;46(6):860–6. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 29.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD).; Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 30.Molina JA, Jimenez-Jimenez FJ, Aguilar MV, Meseguer I, Mateos-Vega CJ, Gonzalez-Munoz MJ, de Bustos F, Porta J, Orti-Pareja M, Zurdo M, Barrios E, Martinez-Para MC. Cerebrospinal fluid levels of transition metals in patients with Alzheimer's disease. J Neural Transm. 1998;105(45):479–88. doi: 10.1007/s007020050071. [DOI] [PubMed] [Google Scholar]
- 31.Opazo C, Huang X, Cherny R, Moir R, Roher A, White A, Cappai R, Masters C, Tanzi R, Inestrosa N, Bush A. Metalloenzyme-like activity of Alzheimer's disease β-amyloid: Cu-dependent catalytic conversion of dopamine, cholesterol and biological reducing agents to neurotoxic H2O2. Journal of Biological Chemistry. 2002;277:40302–8. doi: 10.1074/jbc.M206428200. [DOI] [PubMed] [Google Scholar]
- 32.Opazo C, Luza S, Villemagne VL, Volitakis I, Rowe C, Barnham KJ, Strozyk D, Masters CL, Cherny RA, Bush AI. Radioiodinated clioquinol as a biomarker for β-amyloid:Zn2+ complexes in Alzheimer's disease. Aging Cell. 2006;5:69–79. doi: 10.1111/j.1474-9726.2006.00196.x. [DOI] [PubMed] [Google Scholar]
- 33.Petrovitch H, Nelson J, Snowdon D, Davis DG, Ross GW, Li CY, White L. Microscope field size and the neuropathologic criteria for Alzheimer's disease. Neurology. 1997;49(4):1175–6. doi: 10.1212/wnl.49.4.1175. [DOI] [PubMed] [Google Scholar]
- 34.Petrovitch H, White LR, Ross GW, Steinhorn SC, Li CY, Masaki KH, Davis DG, Nelson J, Hardman J, Curb JD, Blanchette PL, Launer LJ, Yano K, Markesbery WR. Accuracy of clinical criteria for AD in the Honolulu-Asia Aging Study, a population-based study. Neurology. 2001;57(2):226–34. doi: 10.1212/wnl.57.2.226. [DOI] [PubMed] [Google Scholar]
- 35.Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ. Insulin-degrading enzyme regulates extracellular levels of amyloid beta- protein by degradation. J Biol Chem. 1998;273(49):32730–8. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 36.Regland B, Lehmann W, Abedini I, Blennow K, Jonsson M, Karlsson I, Sjogren M, Wallin A, Xilinas M, Gottfries CG. Treatment of Alzheimer's disease with clioquinol. Dement Geriatr Cogn Disord. 2001;12(6):408–14. doi: 10.1159/000051288. [DOI] [PubMed] [Google Scholar]
- 37.Religa D, Strozyk D, Cherny RA, Volitakis I, Haroutunian V, Winblad B, Naslund J, Bush AI. Elevated cortical zinc in Alzheimer disease. Neurology. 2006;67(1):69–75. doi: 10.1212/01.wnl.0000223644.08653.b5. [DOI] [PubMed] [Google Scholar]
- 38.Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny RA, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Aβ amyloid deposition and toxicity in Alzheimer's disease: a pilot phase 2 clinical trial. Archives of Neurology. 2003;60:1685–91. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 39.Rottkamp C, Raina A, Zhu X, Gaier E, Bush A, Atwood C, Chevion M, Perry G, Smith M. Redox-active iron mediates amyloid-β toxicity. Free Radical Biology and Medicine. 2001;30:447–50. doi: 10.1016/s0891-5849(00)00494-9. [DOI] [PubMed] [Google Scholar]
- 40.Schlief ML, Craig AM, Gitlin JD. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J Neurosci. 2005;25(1):239–46. doi: 10.1523/JNEUROSCI.3699-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strozyk D, Blennow K, White LR, Launer LJ. CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology. 2003;60(4):652–6. doi: 10.1212/01.wnl.0000046581.81650.d0. [DOI] [PubMed] [Google Scholar]
- 42.White AR, Du T, Laughton KM, Volitakis I, Sharples RA, Xilinas ME, Hoke DE, Holsinger RM, Evin G, Cherny RA, Hill AF, Barnham KJ, Li QX, Bush AI, Masters CL. Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity. J Biol Chem. 2006;281(26):17670–80. doi: 10.1074/jbc.M602487200. [DOI] [PubMed] [Google Scholar]
- 43.White L, Petrovitch H, Ross GW, Masaki KH, Abbott RD, Teng EL, Rodriguez BL, Blanchette PL, Havlik RJ, Wergowske G, Chiu D, Foley DJ, Murdaugh C, Curb JD. Prevalence of dementia in older Japanese-American men in Hawaii: The Honolulu-Asia Aging Study. Jama. 1996;276(12):955–60. [PubMed] [Google Scholar]