

Abstract
P-type ATPases play essential roles in numerous processes, which in humans include nerve impulse propagation, relaxation of muscle fibers, secretion and absorption in the kidney, acidification of the stomach and nutrient absorption in the intestine. Published evidence suggests that uncharacterized families of P-type ATPases with novel specificities exist. In this study, the fully sequenced genomes of 26 eukaryotes, including animals, plants, fungi and unicellular eukaryotes, were analyzed for P-type ATPases. We report the organismal distributions, phylogenetic relationships, probable topologies and conserved motifs of nine functionally characterized families and 13 uncharacterized families of these enzyme transporters. We have classified these proteins according to the conventions of the functional and phylogenetic IUBMB-approved transporter classification system (www.tcdb.org, Saier et al. in Nucleic Acids Res 34:181–186, 2006; Nucleic Acids Res 37:274–278, 2009).
Electronic supplementary material
The online version of this article (doi:10.1007/s00232-009-9176-2) contains supplementary material, which is available to authorized users.
Keywords: P-type ATPase, Eukaryote, Transport, Phylogeny, Topology, Conserved motif
Introduction
Most functionally characterized P-type ATPases drive the uptake and/or efflux of cations against tremendous concentration gradients (Axelsen and Palmgren 1998), although members of one family within this superfamily catalyze the flipping of phospholipids from the outer monolayer of the plasma membrane bilayer to the inner monolayer (Devaux et al. 2006; Ikeda et al. 2006; Pomorski and Menon 2006). Biochemical evidence for chloride and iron P-type ATPases has been presented, but the molecular identities of these enzymes have not yet been revealed (Baranano et al. 2000; Gerencser 1993; Inagaki et al. 1996; Ueno et al. 2000; Zeng et al. 1999). A common ancestry for all P-type ATPases has been established (Fagan and Saier 1994). These observations suggest that additional families of P-type ATPases with novel specificities are likely to be found.
X-ray structures of eukaryotic Ca2+-, Na+,K+- and H+-ATPases have been described (Olesen et al. 2007; Morth et al. 2007; Pedersen et al. 2007; Toyoshima et al. 2000); and mechanistic analyses have been presented (Berman 2001; Gadsby 2007; Pedersen et al. 2007; Hatori et al. 2007; Kuhlbrandt 2004; Møller et al. 2005; Martin 2005), leading to well-substantiated mechanistic postulates. Møller et al. (1996, 2005) have analyzed the three cytoplasmic domains (B [energy transduction actuator between transmembrane segments {TMSs} 2 and 3, where conserved motifs 1–3 occur], C [nucleotide binding and phosphorylation site domain between TMSs 4 and 5, where motifs 5–8 occur] and J [junctional connecting domain between the C domain and the C-terminal transmembrane domain, where motif 9 occurs]).
Many researchers in the P-type ATPase field use the nomenclature of Axelsen and Palmgren (A&P) (1998), which was developed on the basis of earlier nomenclature suggested by Lutsenko and Kaplan (1995) and Møller et al. (1996). According to this nomenclature, group IA ATPases are the Kdp-type homologues of prokaryotes, which are not found in eukaryotes and have their own distinctive phylogenetic group, topology and mechanism of action. Group IB ATPases form two distinct phylogenetic clusters, each of which contains proteins having different but overlapping sizes and substrate specificities even though their membrane topologies are the same. They represent families 5 (copper) and 6 (heavy metal) in the transporter classification (TC) system. There is no basis for grouping IA ATPases with IB ATPases. Most remaining P-type ATPases known in 1998 were named group II P-type ATPases. A&P divided group II ATPases into four subfamilies (IIA–D). These four families are TC families 1 (Na+, K+), 2 (Ca2+) and 9 (Na+ or K+) corresponding to A&P’s groups IIC, IIA/IIB and IID, respectively. They have essentially the same topology.
Calcium ATPases probably comprise at least five phylogenetic groups (unpublished observations), although A&P recognized only two, which they designated IIA and IIB. Although their topologies, cation specificities and polarity of transport are the same, several features distinguish A&P’s IIA and IIB pumps. IIA pumps may generally bind two Ca2+ ions, while IIB ATPases may bind one; IIA pumps often have regulatory subunits (e.g., phospholamban), whereas IIB ATPases sometimes have built-in autoinhibitory terminal domains; and IIA pumps predominate in (but are not exclusively localized to) internal membranes, whereas IIB ATPases predominate in plasma membranes. Group IIIA ATPases of A&P (TC family 3) expel H+ in eukaryotes but heavy metals in one prokaryote. These display type II topologies. Finally, group IV ATPases of A&P correspond to TC family 8. These phospholipid-flipping proteins also exhibit type II topologies.
Since the early studies, several new families have been discovered; and according to the TC system, each of these is categorized as a distinct family on the basis of phylogenetic clustering patterns. None of these systems is functionally characterized. Several of these families are described and characterized for the first time in this report and several exhibit unexpectedly unique properties.
We here follow up on our earlier publication (Fagan and Saier 1994) as well as those of others (e.g., Axelsen and Palmgren 1998; Møller et al. 1996), analyzing these monophyletic ATPases from genomic, topological and phylogenetic standpoints. We subdivide the P-type ATPase superfamily using the established rules set up for the International Union of Biochemistry and Molecular Biology (IUBMB)–approved, functional and phylogenetic TC system (Saier 2000; Saier et al. 2006, 2009).
Methods
Full-length, established P-type ATPase sequences were retrieved from the TCDB database (Busch and Saier 2002; Saier et al. 2006, 2009) and used to screen recognized proteins encoded within the fully sequenced genomes of 26 eukaryotic organisms. Because of the high degree of sequence similarity among all functional P-type ATPases, this could usually be achieved with a single BLAST search without iterations. In order to ensure a complete set of sequences, the P-type ATPase database (http://www.patbase.kvl.dk/) was also used as a resource. Finally, one protein from each organism was used as a query sequence in NCBI BLAST searches (Altschul et al. 2005; Altschul and Koonin 1998) in order to find any remaining sequences. Redundant and truncated sequences were eliminated, leaving 505 proteins for the analyses reported.
Trees were generated with the neighbor-joining algorithm. The ClustalX program (Thompson et al. 1997) was used with default settings for multiple alignment of homologous protein and rRNA sequences and for construction of the phylogenetic trees displayed in this report. However, other programs (ProtPars [parsimony] and the SuperfamilyTree programs SFT1 and SFT2; Yen et al. unpublished) were used to confirm and extend phylogenetic assignments. The TreeView program (Zhai et al. 2002) was used to view phylogenetic trees in all cases. Topological analyses of multiply aligned protein sequences were performed using the AveHAS program (Zhai and Saier 2001b; see below for more details), while topological analyses of single proteins were conducted using the WHAT program (Zhai and Saier 2001a). The latter results were confirmed using the TMHMM v.2.0 program (Møller et al. 2001). Sequence similarity comparisons between proteins and regions of proteins and statistical significance comparisons were conducted using the GAP, BLAST2 (Tatusova and Madden 1999) and IC (Zhai and Saier 2002) programs. Motif analyses were conducted manually using family-specific and organism-specific multiple alignments.
Average hydropathy was calculated as described by Zhai and Saier (2001b) using the conventions of Kyte and Doolittle (1982). These values were averaged for a sliding window of 19 residues (default settings) and plotted as shown (solid lines in Fig. 4a–c), right-hand scale.
Fig. 4.

Average hydropathy (solid lines, top; scale on left y axis) and average similarity (dotted lines, bottom; scale on right y axis) plots for the three novel topologies of ATPases identified in this study. The plots were generated with the AveHas program (Zhai and Saier 2001b) based on the multiple alignments shown in Supplementary Fig. S3a–c, respectively (see “Methods” for details). a Family 10 proteins, b proteins of families 11–21 and c family 22 proteins. The figure illustrates the basis for assigning the topological types depicted schematically in Fig. 5d–f. Relative values are presented on the y axes as detailed by Zhai and Saier (2001b)
Average similarity was calculated as follows: For each alignment position the amino acyl residues occurring at that position were tallied (numbers of each specific amino acid, squared and divided by the total number of proteins in the alignment) according to the following equation:
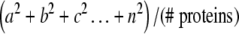 |
a, b, c, … n equals the numbers of each of the represented amino acids at this position. These values were then averaged using a sliding window, which in the present studies was the default setting of the program (19 residues). Relative values were then plotted versus alignment position to generate the “average similarity” plots shown, which the dotted lines in Fig. 4a–c represent. In both plots, relative values are presented.
Results
Objectives and Achievements of this Study
In contrast to all previous studies, we report here an analysis of the occurrence of P-type ATPases encoded within the completely sequenced genomes of the 26 eukaryotic organisms available at the time these studies were initiated, in September 2006. We examine the distribution of the nine functionally characterized families of these enzymes in the different eukaryotic kingdoms, identify 13 new families of unknown specificity (four of which are currently represented by a single protein) and define their organismal distributions and probable topologies. Analyses of functionally recognized conserved motifs reveal that the families of uncharacterized P-type ATPases exhibit more sequence conservation than the characterized families and that all of the essential catalytic residues recognized for these enzymes are conserved, arguing that the uncharacterized proteins are catalytically active. Hydropathy analyses suggest the existence of three novel topological types in addition to the three previously recognized types. Motif analyses reveal conserved familial differences, suggesting distinctive functional roles (see Table 3, Supplementary Table S3).
Table 3.
The nine most conserved motifs characteristic of the 13 functionally uncharacterized (10–22) P-type ATPases
| Consensus | PGD | PAD | TGES | PEGL | DKTGTLT | KGAPE | DPPR | MVTGD | VAVTGDGVND S PALKKADIGVAM |
|---|---|---|---|---|---|---|---|---|---|
| 3.A.3.10 | |||||||||
| Seq. motif | PGD | PCD | TGES | PPEL | DKTGTLT | KGSPE | CPLK | MITGD | TLMCGDGTNDVGALKQAHVGVAL |
| Conservation | –* | –* | :**. | *. :: | **** *:* | ** :** | .–: | : :*** | _: * .*** ***: . ***_: ._* ::: |
| Residue | 850 | 910 | 930 | 1160 | 1200 | 1515 | 1590 | 1615 | 1860 |
| 3.A.3.11 | |||||||||
| Seq. motif | PGN | PCD | TGET | NPAI | DKTGTLT | KGSPE | NKLK | MCTGD | TMFCGDGANDSGALSSADVGLAL |
| Conservation | |||||||||
| Residue | 260 | 270 | 290 | 440 | 480 | 660 | 725 | 745 | 870 |
| 3.A.3.12 | |||||||||
| Seq. motif | PGD | PCD | TGEA | PPAL | DKTGTLT | KGAPE | NRLK | MVTGD | VGMCGDGANDAGALREADVGVGL |
| Conservation | |||||||||
| Residue | 445 | 460 | 480 | 880 | 925 | 1120 | 1235 | 1260 | 1650 |
| 3.A.3.13 | |||||||||
| Seq. motif | PGD | QCD | TGES | PPAL | DKTGTLT | KGSHPE | NRLK | MVTGD | VAMCGDGANDCGALKAAHAGISL |
| Conservation | *_* | _** | **** | **** | ******* | ***** | *_:* | *:*** | *. *** ***** * .***.*..**** |
| Residue | 350 | 365 | 385 | 515 | 555 | 755 | 825 | 850 | 1060 |
| 3.A.3.14 | |||||||||
| Seq. motif | PGD | PCD | TGES | PPAL | DKTGTLT | KGAPE | NKLK | MCTGD | CGFCGDGANDCGALKAADVGISL |
| Conservation | _** | * .* | **** | **** | ******* | ***** | **** | *_*** | _.**** ***** *********: ** |
| Residue | 790 | 810 | 830 | 970 | 1020 | 1200 | 1270 | 1295 | 1430 |
| 3.A.3.15 | |||||||||
| Seq. motif | PGD | PCD | TGES | PPAL | DKTGTLT | KGSHPE | NRIK | MVTGD | VGHMCGDGANDCGALKAAHVGISL |
| Conservation | *** | *** | **** | **** | ******* | ***** | *: :* | ***** | *** *** ***************** |
| Residue | 480 | 495 | 515 | 670 | 715 | 990 | 1060 | 1085 | 1260 |
| 3.A.3.16 | |||||||||
| Seq. motif | PGD | PCD | TGES | PPAL | DKTGTLT | KGSHPE | NKLK | MVTGD | CGMCGDGANDCGALKTADHMGISL |
| Conservation | _** | _** | **** | **** | ******* | ** :** | **** | • :*** | _. **** **** *_*** _** :* :** |
| Residue | 860 | 875 | 895 | 1025 | 1070 | 1260 | 1405 | 1700 | |
| 3.A.3.17 | |||||||||
| Seq. motif | IGD | PVD | TGES | PPAL | DKTGTLT | KGAPE | SPLR | ICSGD | VGFCGDGANDCIALKQADVGVSL |
| Conservation | |||||||||
| Residue | 275 | 290 | 310 | 440 | 485 | 620 | 690 | 710 | 830 |
| 3.A.3.18 | |||||||||
| Seq. motif | PGD | PCD | TGES | PPAL | DKTGTLT | KGSPE | NKLR | MSTGD | VGMCGDGNNDILAIQSANIGIAI |
| Conservation | |||||||||
| Residue | 370 | 380 | 400 | 530 | 575 | 820 | 895 | 915 | 1180 |
| 3.A.3.19 | |||||||||
| Seq. motif | PGD | PCD | TGES | PPSL | DKTGTLT | KGAPE | NPLK | MATGD | __MVGDGANDCGALKQADIGLAL |
| Conservation | |||||||||
| Residue | 290 | 305 | 325 | 440 | 485 | 640 | 725 | 745 | 1045 |
| 3.A.3.20 | |||||||||
| Seq. motif | PGD | PCD | TGES | PPFL | DKTGTLT | KGSPE | NKLK | IISGD | VGMIGDGANDCSAIKQADIGISF |
| Conservation | _** | *_* | ***_ | * .– | ******* | **:** | *. ** | ::*** | :_*: *** :**._: *: :. *:**** |
| Residue | 380 | 395 | 410 | 540 | 580 | 730 | 805 | 830 | 1010 |
| 3.A.3.21 | |||||||||
| Seq. motif | PGD | PVD | TGES | PLDL | DKTGTLT | KGAPT | AAMR | MLTGD | VLVCGDGVNDIAAMREADVSVAM |
| Conservation | |||||||||
| Residue | 345 | 370 | 385 | 395 | 575 | 700 | 770 | 790 | 920 |
| 3.A.3.22 | |||||||||
| Seq. motif | PGD | PCD | TGES | PVWT | DKTGTLT | KGAFE | NQIR | ILTGD | TGMVGDGTNDCGALKISHAGISL |
| Conservation | – | * .* | ***: | *. :_ | ******* | : .– | . _ :: | :: *** | _* _***_**_*. :_:._.:_. |
| Residue | 710 | 720 | 740 | 1000 | 1050 | 1665 | 1745 | 1770 | 2160 |
Format for presentation is as for Supplementary Table S3
The 26 eukaryotes analyzed for P-type ATPases were subdivided into five categories: animals, plants, fungi, unicellular eukaryotes and a ciliate. Full-length homologues from functionally characterized families 1–9 (Fig. 1; Table 1, Supplementary Table S1) and from uncharacterized families 10–22 (Fig. 2; Table 2, Supplementary Table S2) are the focus of this study. Family number is as indicated in the Transporter Classification Database (TCDB http://www.tcdb.org; Saier et al. 2006; 2009). Because the prokaryotic Kdp K+-ATPase complexes (Haupt et al. 2005; Greie and Altendorf 2007) were not detected in any eukaryote, these are not discussed here. Similarly, functionally uncharacterized P-type ATPases (FUPAs) of prokaryotes do not fall into the same families present in eukaryotes. They will be the subject of a separate publication (in preparation). Partial sequences and presumed pseudogenes, of which there are several, were set aside for subsequent analyses (see “Partial Sequences” below).
Fig. 1.

Phylogenetic tree of eukaryotic P-type ATPases within established families of known specificities (TC families 1–9 as indicated). In addition to the proteins encoded within the 26 genomes examined here, at least one established member from each family in TCDB was included. The multiple alignment upon which this tree was based can be seen in Supplementary Fig. S1a, and the dendrogram, which reveals the phylogenetic relationships of all the proteins to each other, is provided in Supplementary Fig. S1b. The CLUSTAL X program (Thompson et al. 1997) with default settings was used to derive the multiple alignment and generate the phylogenetic tree, which was drawn using the TreeView program (Zhai et al. 2002). All sequences used were full-length. Families 1–9 are labeled according to phylogenetic cluster as indicated by number according to the family designations in TCDB (Saier et al. 2006, 2009). Family 7 (Kdp K+-ATPases) is not represented because these proteins have not been found in eukaryotes. These families have been given alternative designations (Axelsen and Palmgren 1998) as follows: family 1, PIIC; family 2, PIIA + PIIB; family 3, PIIIA; family 4, PIIIB; family 5, PIB; family 6, PIB; family 7, PIA; family 8, PIV; family 9, PIID; family 10, PVA; families 13–22, PVB (see Table 4)
Table 1.
Distribution of functionally characterized families of P-type ATPases (families 1–9) encoded within the genomes of 26 eukaryotes
| Organism type | Organism | Family | Full P-ATPases/family (n) | Total/organism or organismal type | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||||
| Animal | Anopheles gambiae | 1 | 3 | 0 | 0 | 1 | 0 | 0 | 5 | 0 | 10 | |
| Animal | Caenorhabditis elegans | 2 | 4 | 0 | 0 | 1 | 0 | 0 | 6 | 0 | 13 | |
| Animal | Drosophila melanogaster | 2 | 3 | 0 | 0 | 1 | 0 | 0 | 6 | 0 | 12 | |
| Animal | Homo sapiens | 6 | 8 | 0 | 0 | 2 | 0 | 0 | 8 | 0 | 24 | |
| Total | 11 | 18 | 0 | 0 | 5 | 0 | 0 | 25 | 0 | 59 | ||
| Plant | Arabidopsis thaliana | 0 | 14 | 12 | 0 | 4 | 4 | 0 | 12 | 0 | 46 | |
| Plant | Oryza sativa | 0 | 14 | 9 | 0 | 6 | 3 | 0 | 9 | 0 | 41 | |
| Total | 0 | 28 | 21 | 0 | 10 | 7 | 0 | 21 | 0 | 87 | ||
| Fungi | Aspergillus fumigatus | 2 | 5 | 3 | 0 | 3 | 0 | 0 | 4 | 3 | 20 | |
| Fungi | Aspergillus nidulans | 0 | 7 | 2 | 0 | 2 | 0 | 0 | 4 | 3 | 18 | |
| Fungi | Aspergillus oryzae | 2 | 6 | 3 | 0 | 4 | 0 | 0 | 4 | 3 | 22 | |
| Fungi | Cryptococcus neoformans | 0 | 3 | 2 | 0 | 1 | 0 | 0 | 4 | 1 | 11 | |
| Fungi | Coccidioides posadasii | 0 | 4 | 1 | 1 | 3 | 0 | 0 | 4 | 3 | 16 | |
| Fungi | Neurospora crassa | 0 | 4 | 1 | 0 | 3 | 0 | 0 | 5 | 3 | 16 | |
| Fungi | Saccharomyces cerevisiae | 0 | 2 | 2 | 0 | 3 | 0 | 0 | 5 | 2 | 14 | |
| Fungi | Schizosaccharomyces pombe | 0 | 2 | 2 | 0 | 1 | 0 | 0 | 5 | 1 | 11 | |
| Total | 4 | 33 | 16 | 1 | 20 | 0 | 0 | 35 | 19 | 128 | ||
| 1-Cell eukaryote | Cyanidioschyzon merolae | 0 | 3 | 1 | 0 | 1 | 0 | 0 | 2 | 0 | 7 | |
| 1-Cell eukaryote | Cryptosporidium parvum | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 5 | |
| 1-Cell eukaryote | Dictyostelium discoideum | 2 | 3 | 1 | 1 | 3 | 0 | 0 | 9 | 0 | 19 | |
| 1-Cell eukaryote | Encephalitozoon cuniculi | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 2 | |
| 1-Cell eukaryote | Entamoeba histolytica | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 9 | 1 | 14 | |
| 1-Cell eukaryote | Leishmania major | 0 | 4 | 1 | 0 | 2 | 0 | 0 | 5 | 1 | 13 | |
| 1-Cell eukaryote | Plasmodium falciparum | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 5 | |
| 1-Cell eukaryote | Trypanosoma brucei | 0 | 5 | 3 | 0 | 0 | 0 | 0 | 4 | 1 | 13 | |
| 1-Cell eukaryote | Trypanosoma cruzi | 0 | 3 | 1 | 0 | 1 | 0 | 0 | 7 | 1 | 13 | |
| 1-Cell eukaryote | Theileria parva | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 4 | |
| 1-Cell eukaryote | Thalassiosira pseudonana | 0 | 4 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 7 | |
| Total | 2 | 32 | 8 | 1 | 7 | 0 | 0 | 47 | 5 | 102 | ||
| Ciliate | Tetrahymena thermophila | 21 | 11 | 0 | 0 | 0 | 0 | 0 | 23 | 0 | 55 | |
| Comprehensive total | 38 | 122 | 45 | 2 | 42 | 7 | 0 | 151 | 24 | 431 | ||
Organisms are grouped according to their kingdom (animals, plants, fungi, unicellular eukaryotes and ciliates). Numbers of enzymes identified in each organism are presented with the familial totals per organismal type as well as the comprehensive totals. Total P-type ATPases per organism are tabulated on the right. For a more complete listing of these enzymes, including fragmentary or incomplete sequences, see Supplementary Table SIB. The abbreviations of the proteins and their individual properties and NCBI gene index numbers are included in Supplementary Table SIA
Fig. 2.

Phylogenetic tree of eukaryotic P-type ATPases belonging to families of unknown specificities (TC FUPA families 10–22). In addition to the proteins identified in this study, representatives of families 1–9 (numbered 1–9) from TCDB are included; these cluster loosely together on the left side of the figure. The proteins in established families 1–9, presented in Fig. 1, are depicted separately from those in novel families for clarity. Format of presentation and methodology are as for Fig. 1. The multiple alignment and dendrogram corresponding to this tree can be found in Supplementary Fig. S2a and b, respectively. Families 10–22, labeled according to phylogenetic cluster, are indicated by number in bold type. These numbers correspond to the family designations in TCDB (Saier et al. 2006, 2009; Møller et al. 2008)
Table 2.
Distribution of functionally uncharacterized families of P-type ATPases (families 10–22) encoded within the genomes of 26 eukaryotes
| Full P-ATPases/family (n) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Organism type | Organism | Family | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | Total/organism |
| Animal | Anopheles gambiae | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| Animal | Caenorhabditis elegans | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | |
| Animal | Drosophila melanogaster | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| Animal | Homo sapiens | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| Total | 4 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | ||
| Plant | Arabidopsis thaliana | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| Plant | Oryza sativa | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| Total | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | ||
| Fungi | Aspergillus fumigatus | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| Fungi | Aspergillus nidulans | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| Fungi | Aspergillus oryzae | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| Fungi | Cryptococcus neoformans | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| Fungi | Coccidioides posadasii | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| Fungi | Neurospora crassa | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| Fungi | Saccharomyces cerevisiae | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| Fungi | Schizosaccharomyces pombe | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 3 | |
| Total | 8 | 0 | 0 | 0 | 6 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 15 | ||
| l-Cell eukaryote | Cyanidioschyzon merolae | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| l-Cell eukaryote | Cryptosporidium parvum | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 4 | 6 | |
| l-Cell eukaryote | Dictyostelium discoideum | 1 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | |
| l-Cell eukaryote | Encephalitozoon cuniculi | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| l-Cell eukaryote | Entamoeba histolytica | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| l-Cell eukaryote | Leishmania major | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| l-Cell eukaryote | Plasmodium falciparum | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | |
| l-Cell eukaryote | Trypanosoma brucei | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| l-Cell eukaryote | Trypanosoma cruzi | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| l-Cell eukaryote | Theileria parva | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | |
| l-Cell eukaryote | Thalassiosira pseudonana | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 2 | |
| Total | 11 | 1 | 1 | 0 | 0 | 3 | 0 | 0 | 1 | 0 | 0 | 1 | 6 | 24 | ||
| Ciliate | Tetrahymena thermophila | 1 | 0 | 0 | 0 | 0 | 0 | 11 | 0 | 0 | 1 | 11 | 0 | 0 | 24 | |
| Comprehensive total | 26 | 1 | 1 | 5 | 6 | 3 | 11 | 1 | 1 | 1 | 11 | 1 | 6 | 74 | ||
Format of presentation is as for Table 1. The abbreviations of the proteins and their properties are presented in Supplementary Table S2A. No incomplete (fragmentary) sequences belonging to these 12 families were identified
The full-length sequences, in various combinations, were aligned; and phylogenetic trees and average hydropathy, amphipathicity and similarity plots were generated. Conserved motifs were analyzed for both position and sequence characteristics. Multiple alignments and other supplementary (S) figures and tables as well as descriptions of the methods used can be found online at http://www.biology.ucsd.edu/~msaier/supmat/eukP-ATPase.
Eukaryotic P-Type ATPases of Known Function
Animal Homologues
Table 1 and Supplementary Table S1 summarize the distributions of the nine families of P-type ATPase homologues of known or postulated function encoded within the 26 fully sequenced genomes examined. These distributions are based on function (when known) as well as phylogenetic clustering patterns (see Fig. 1 and Supplementary Fig. S1a, b on our Web site). Multiple alignments of P-type ATPases according to organismal type are shown in Supplementary Fig. S4a–e on our Web site.
Four animals were analyzed: Anopheles gambiae (mosquito), Caenorhabditis elegans (worm), Drosophila melanogaster (fruit fly), and Homo sapiens (human). Of these animals, each possesses 1–6 Na+,K+- and/or gastric H+,K+-ATPases (family 1; Gadsby 2007), 3–8 Ca2+-ATPases (family 2; Geisler et al. 2000; Olesen et al. 2007), one or two Cu+-ATPases (family 5; Nies 2003) and 5–8 phospholipid flippases (family 8; Axelsen and Palmgren 1998; Devaux et al. 2006; Ikeda et al. 2006). They lack fungal-type H+- (family 3; Scarborough 2000), Mg2+- (family 4; Maguire 2006), heavy metal (family 6; Nies 2003), bacterial-type K+- (family 7; Haupt et al. 2005; Greie and Altendorf 2007) and K+- or Na+- (family 9; Benito et al. 2002) ATPases. Families of unknown function (FUPA families 10–22, Table 2 and Supplementary Table S2) are described below.
Plant Homologues
The two plant species analyzed, Arabidopsis thaliana (thale cress) and Oryza sativa (rice), show homologue distributions very different from those of animals (Axelsen and Palmgren 2001; Baxter et al. 2003). These plants lack Na+,K+-ATPase homologues (family 1) but possess fungal-type H+-translocating ATPases (family 3, 9–12 per organism; Axelsen et al. 1999). They also encode within their genomes a large number of Ca2+-ATPases (family 2, 14 each; Geisler et al. 2000) and phospholipid flippases (family 8, 9–12 each). Like animals, they exhibit Cu+-ATPases (family 5, 4–6 per organism; Lutsenko et al., 2008); but unlike animals, each has three or four heavy metal ATPases (family 6). Mg2+-ATPases and fungal-type Na+ or K+-export ATPases are lacking (Colangelo & Guerinot, 2006).
Fungal Homologues
Of the eight fungal genomes analyzed, all encode 2–7 Ca2+-ATPases (family 2) as well as 1–3 fungal-type H+-ATPases (family 3; Scarborough 2000). Surprisingly, two of the three Aspergillus species also have a pair of homologues most closely resembling the Na+,K+-ATPases (family 1) of animals. In addition, each of them encodes 1–4 paralogues of Cu+-ATPases (family 5) but no heavy metal ATPases (Nies 2003), four or five phospholipid flippases (family 8) and 1–3 fungal-type Na+ or K+-ATPases (family 9). One fungus, Coccidioides posadasii, has a homologue of the bacterial-type Mg2+-ATPases (family 4; Maguire 2006), but bacterial-type K+-ATPases (family 7; Haupt et al. 2005) are lacking, as is true for all eukaryotes examined. In these respects, except for family 9 representation, the distribution of P-type ATPases in fungi more closely resembles that in animals than in plants.
Ciliate Homologues
The ciliate Tetrahymena thermophila has a most unusual distribution of ATPases (Wang et al. 1997; Wang and Takeyasu 1997). It encodes 21 Na+,K+-ATPases (family 1), 11 Ca2+-ATPases (family 2) and 23 phospholipid flippases (family 8). None of the other functionally characterized families is represented, but many functionally uncharacterized homologues, belonging to families 10–22, are encoded (see below). This surprising distribution may reflect the dependence of ciliates on Ca2+, Na+ and K+ for motility and tactic behavior (Plattner and Klauke 2001; Kung and Saimi 1982).
Homologues of Other Unicellular Eukaryotes
The remaining unicellular eukaryotes represent a diverse group of organisms, and hence, their varied homologue representation is not unexpected. Only the slime mold Dictyostelium discoideum has family 1 Na+,K+-ATPases and a family 4 Mg2+-ATPase, with two paralogues of the former and one member of the latter. All of these organisms, except for the protozoan parasite Encephalitozoon cuniculi, possess 2–5 members of family 2 Ca2+-ATPases. Family 3 fungal-type H+-ATPases are found in six of these 11 unicellular eukaryotes, and the parasitic protist Trypanosoma brucei has three such paralogues; all others have just one. All of these organisms possess phospholipid flippases (family 8, 1–9 paralogues per organism), and five have fungal-type Na+ or K+-ATPases (family 1, Table 1).
Partial Sequences
Several “partial” or “fragmentary” ATPases have been identified. These could be the products of pseudogenes or a consequence of inaccurate sequencing or improper exon recognition. Regardless, within each organismal type, whenever an ATPase family in which a partial sequence was identified (belonging to TC families 1–9), corresponding full-length homologues were identified with one exception. The sole exception was a set of two heavy metal ATPase (family 6) fragments in unicellular eukaryotes. These organisms did not display even one full-length heavy metal ATPase.
Family 10 ATPases—A Novel and Distinctive Family of P-Type ATPases
Ca2+ transport function has been proposed for family 10 ATPases (Cronin et al. 2000), but this postulate remains unsubstantiated (Cronin et al. 2002; R. Y. Hampton, personal communication). Surprisingly, each of the 26 organisms studied has one and only one orthologue within this family. These proteins show phylogenetic relationships to each other reflecting the organismal (18S rRNA) phylogenies (compare Figs. 2, 3). This is not a characteristic of any of the other P-type ATPase families.
Fig. 3.

Neighbor joining phylogenetic tree of 18S rRNAs from the 26 eukaryotic organisms included in this study. Organisms are indicated by their three-letter abbreviations (see Tables 1, 2)
The yeast family 10 ATPase forms an oligomeric endomembrane (endoplasmic reticulum [ER] and Golgi) complex and presumably serves a unique and important function, dissimilar from those of all other P-type ATPases. Cronin et al. (2000, 2002) and Vashist et al. (2002) have provided evidence that this yeast enzyme is important in Ca2+ homeostasis and ER function. A role in the outward flipping of phospholipids has been proposed based on high activity of the complex in phospholipid bilayers (R. Y. Hampton, personal communication), but this proposal needs to be substantiated.
A Sub-Superfamily Present in Animals, Fungi, Slime Molds and Ciliates
Four of the 13 identified families of uncharacterized homologues proved to be more similar to each other than to any of the other P-type ATPases. These are families 13–16 (see Fig. 2). Family 13 homologues are found exclusively in animals; members of family 14 are only from fungi; family 15 proteins derive from the slime mold Dictyostelium discoideum; and family 16 proteins are found exclusively in the ciliate Tetrahymena thermophila. Since the proteins of families 13–16 show similar topologies and motifs (see below), they may comprise a “sub-superfamily.” The phylogenetic results reveal that each cluster is derived from a different group of eukaryotes, and this observation strongly supports the conclusion that no horizontal transfer of genes encoding these proteins has occurred between these four eukaryotic kingdoms since the divergence of these organismal types. Horizontal transfer would give rise to families of ATPases where the members of each family are derived from multiple organismal types. These enzymes may serve a single, unified biochemical function in all 26 organisms included in this study.
Parallel Evolution of Ciliate Families 16 and 20
Interestingly, families 16 and 20, both exclusively from Tetrahymena thermophila, consist of 11 paralogues each. In both families, the phylogenetic relationships of the proteins to each other are the same (see Fig. 2). It is unlikely that this happened by chance. We suggest that these two families evolved in parallel for a common set of functions from two distinct ancestral P-type ATPase-encoding genes and that every gene duplication event that occurred in one family occurred in the other family at about the same time.
Other Families of P-Type ATPases
The phylogenetic tree of all eukaryotic P-type ATPases of unknown function (Fig. 2) revealed eight additional branches or clusters with no functionally characterized member. Six of these branches contain only a single “orphan” protein, which does not cluster closely with any other of the 505 proteins identified in the 26 eukaryotic genomes or with any P-type ATPase in TCDB (Saier et al. 2006, 2009). However, three of these families (families 11, 17 and 18) include homologues in the NCBI database and, thus, have multiple members, and family 19 homologues closely resemble those of family 16. Thus, only two of these proteins are true orphans.
As shown in Table 2, each of the six orphans is derived from a different unicellular organism. It is expected that more homologues will be identified as sequences are deposited into the databases. Additionally, family 22 proteins are unique to the Alveolata. A multiple alignment of ATPases belonging to families 10–22 can be seen in Supplementary Fig. S2a, and the corresponding dendrogram is presented in Supplementary Fig. S2b.
Hydropathy Profiles
Average hydropathy and similarity plots for all phylogenetically distinct families of P-type ATPases were generated and analyzed as shown, e.g., in Fig. 4. These plots were generated using the WHAT (Zhai and Saier 2001a) and AveHAS (Zhai and Saier 2001b) programs. Three topological types are currently recognized (Fig. 5, I–III): type I, copper (family 5) and heavy metal (family 6) ATPases; type II, Na+,K+ and gastric H+,K+ (family 1), Ca2+ (family 2), H+ (family 3), Mg2+ (family 4), phospholipid (family 8) and K+ or Na+ (family 9) ATPases; and type III, bacterial Kdp-type K+ (family 7) ATPases. We have identified three additional topological types as detailed below. The hydropathy plots are depicted in Fig. 4a–c (based on the alignments presented in Supplementary Fig. S3a–c). The six topological types are illustrated in Fig. 5, with the three previously recognized topologies (I–III) shown at the top and the three novel putative type IV–VI topologies shown at the bottom. The latter are based on the alignments shown in Supplementary Fig. S3a–c.
Fig. 5.

Six putative topological types of P-type ATPases. Topologies I–III have been defined previously. Types IV–VI were identified in this work (see text). In type IV, A′ and B′ are the two proposed TMSs preceding TMS 1, so designated because they exhibit sequence similarity to TMSs A and B in family 5 and 6 proteins (type I topology). TMS C in type V ATPases and TMSs D–F in type VI ATPases lack significant sequence similarity with A and B and are designated C–F accordingly. The presence of odd numbers of TMSs preceding TMS1 implies that, in contrast to all well-characterized P-type ATPases, the N-terminal sequences of these proteins are extracellular
The average hydropathy profile for family 10 (26 members, one from each organism) is shown in Fig. 4a. Peaks A′ and B′, preceding peak 1, are well conserved and show statistically significant similarity (up to 8 standard deviations, 24% identity, 40% similarity) to peaks A and B in type I ATPases. Peaks 1–4 are present in a 2 (1 & 2) + 2 (3 & 4) arrangement, as expected for both types I and II ATPases. Peaks 5–10 are found in an arrangement typical of P-type ATPases of type II topology (families 1–4, 8 and 9). Family 10 ATPases may thus represent an amalgam of topological types I and II (see Fig. 5, type IV) with a total of 12 predicted TMSs.
Figure 4a reveals six N-terminal peaks of hydrophobicity, found in only one “fusion” protein, Ani7. This region proved to be homologous to many eukaryotic proteins including the Magnaporthe grisea Pth11 fungal protein, a plasma membrane/vacuolar protein that mediates appresorium differentiation in response to inductive substrate cues (DeZwaan et al. 1999). It contains an N-terminal, 8-cysteine, EGF-like, common in several fungal extracellular membrane proteins (CFEM) domain (Kulkarni et al. 2003; Perez et al. 2006). It plays a role in pathogenesis (Kulkarni et al. 2005). Pth11 gene expression is under the control of the CON7 transcription factor that regulates infection-related morphogenesis in Magnaporthe grisea (Odenbach et al. 2007). We include this “fusion” protein within topological type IV.
P-type ATPases of unknown function in families 11–21 belong to a single apparent topological type as shown in Figs. 4b and 5, type V. These proteins appear to share a single hydrophobic peak (peak C) near the N-termini of these proteins, preceding peak 1. In some proteins, this is the most hydrophobic peak. If it proves to be transmembrane, as expected, the N-termini of these proteins must be extracellular. We refer to this topology as type V.
One protein, Tth30, which alone comprises family 19, resembles topological type V ATPases except for a C-terminally fused domain with six TMSs. This putative domain surprisingly shows no significant sequence similarity with other proteins in the NCBI database. We include this putative fusion protein among type V ATPases.
Finally, family 22 members differ from all others in that, preceding peak 1, at the extreme N-termini of these proteins, there are three exceptionally strong peaks of hydrophobicity (Fig. 4c, referred to as peaks D, E and F). The sequences of these N-terminal regions do not resemble those of other P-type ATPases. If all three hydrophobic peaks correspond to transmembrane segments, the N-termini of these proteins must be extracellular (see Fig. 4, type VI). In other respects, the plot resembles those of type II ATPases.
Conserved Motifs
Nine conserved motifs in P-type ATPases have been identified, and the biochemical functions of several of these are known (see Supplementary Table S3, motifs in families 1–9, and Table 3, motifs in families 10–22) (Møller et al. 1996). These motifs, numbered 1–9, from N terminus to C terminus, were analyzed with respect to family and organismal type. Motifs 1–3 occur between TMSs 2 and 3, motif 4 occurs at the end of TMS 4 and motifs 5–9 occur between TMSs 4 and 5. In general, striking similarities are noted for proteins of a single family, although these motifs occasionally exhibit organismal type-specific characteristics. Importantly, some of the motifs for the different families exhibit distinctive features that are likely to be determinants of substrate specificity or mechanistic function. Surprisingly, these nine motifs are usually better conserved in the functionally uncharacterized ATPases than in the characterized families, clearly suggesting that the former proteins are functional. It is worthy of note that a well-conserved motif that is different from the corresponding motif in the well-characterized ATPases implies retention of function, although that function may be altered by the residue substitution(s) (Burbaum et al. 1990; Oestreicher and Scazzocchio 1995; Waxman et al. 2000). These observations are summarized below and in Table 3 and Supplementary Table S3.
Motif 1, PGD, together with motifs 2 and 3, may facilitate the E1 to E2 transition. The P in motif 1 is most variable, while the G is most conserved. The P is often replaced by V but is occasionally replaced by A, T, I, Y or K. V is particularly common in families 8, 1 and 2. In fact, in family 8, V is the dominant residue in all clusters and fully conserved in most. The D is better conserved than the P, but it can be replaced by other residues such as A, T, N, Q and H. Among the uncharacterized family homologues, the PGD motif is better conserved than in the functionally characterized family members; the P is never replaced by V as the dominant residue, and the D is replaced by N only in family 11 (Table 3, Supplementary Table S3).
Like motif 1, motif 2 (PAD) is well conserved among the functionally characterized families. The P and D are nearly fully conserved, but the A can be replaced by C, V, S, T, I or F with decreasing frequencies in that order. Surprisingly, the second residue (A) shows poor conservation among the 13 novel families, but in 10 of these new families a C replaces the A as the dominant, often fully conserved residue; thus, the consensus motif for these proteins is PCD (Supplementary Table S3).
Motif 3 (TGES) is one of the best-conserved motifs in P-type ATPases. The S at position 4 can be conservatively substituted with T, A or N. In the family 8 phospholipid flippases, this motif is well conserved but differs, being DGET, and the T at position 4 in the ciliate proteins can be R, K or I. Among the 13 unknown families, the TGES motif is extremely well conserved, almost without exception.
Motif 4 (PEGL), which may function in energy transduction, shows much greater variation than motif 3. This motif proved to be well conserved in family 2 Ca2+-ATPases and in the animal and fungal family 1 Na+,K+-ATPases. The consensus motif for family 3 H+-ATPases is P(LIVM)(GA)L, but plants exhibit PIAM (largely conserved), unicellular eukaryotes display PIAL (largely but less well conserved) and fungi have PVGL (fully conserved). The two family 4 homologues from the fungus Coccidioides posadasii and the slime mold Dictyostelium discoideum have the same motif: PEML. Families 5 and 6 display the consensus motif 4: PC(GAVS)(LI). Family 8 phospholipid flippases exhibit P(LIVM)S(LIF), where PISL dominates in fungi, plants and unicellular organisms and animals have (PNT)EEL. Family 9 K+- or Na+-ATPases display the motif P(EA)(GSC)L. In the copper and heavy metal ATPases, this motif is (C)PCAL, where the two cysteines are required for metal binding (Liu et al. 2006). Surprisingly, most of the functionally uncharacterized families exhibit the motif PPAL, where the A is most variable. This motif, therefore, uniquely displays both family and organismal type-specific features, suggesting that it may serve as an ion (or other substrate) specificity determinant. This suggestion is in agreement with the conclusion of Liu et al. (2006) and has been documented for Ca2+- and Na+,K+-ATPases (Horisberger et al. 2004).
Motif 5 (DKTGTLT), the phosphorylation site, is nearly fully conserved in all families and organisms, although the L can be substituted by I or V. All members of the nine characterized and 13 uncharacterized families display this motif without exception (see Table 3, Supplementary Table S3).
Motif 6 (KGAPE), which functions in ATP binding, is reasonably well conserved in families 1–9, especially the KGA submotif; but only the G is strongly conserved. However, in the 13 uncharacterized families, the KGAPE motif is almost fully conserved except that A and S occupy position 3 with about equal frequencies.
Motif 7 (DPPR), important for phosphoryl transfer from ATP to the enzyme, is best conserved in homologues of families 1, 2, 3, 4 and 9. In families 5 and 6, this motif is D(MASTP)(LIVC)(RK), while in family 8 it is (EQDK)(GN)(LVKY)(RK). In the uncharacterized families, the dominant motif at this position is N(RK)L(KR), with some variation. This motif is P-type ATPase family- or subtype-specific, suggesting a possible role in interconnecting substrate selectivity with ATP hydrolysis.
Motif 8 (MVTGD), which functions in the catalysis of enzyme phosphorylation, shows appreciable variability, particularly at positions 1 and 2 as follows: (MLIVA) (LIVQ)(TS)GD. This motif is similarly conserved in characterized as well as uncharacterized families (Table 3).
Finally, the 23-residue “hinge” motif, flexibly interconnecting the cytoplasmic enzyme domain C with the C-terminal hydrophobic domain, plays a role in transition state conformational changes. It is moderately well conserved, with the GDG and ND submotifs being fully conserved in all 13 uncharacterized families (Table 3).
Discussion
In this report we present a detailed analysis of P-type ATPases encoded within 26 fully sequenced eukaryotic genomes. We determined the organismal distributions of the previously recognized families (families 1–9) and identified 13 new families (families 10–22). We also identified three novel topological types of P-type ATPases. Several of these families had been noted before (Axelsen and Palmgren 1998; Lutsenko and Kaplan 1995; Møller et al. 1996, 2005, 2008), although fewer sequences were then available to conduct analyses and make family assignments (see Table 4).
Table 4.
Summary of data on 22 families of P-type ATPases found in eukaryotes
| TC family | Axelsen and Palmgren family | Topological typea | Members (n)b | TMSsc | Eukaryotic representationd | Prokaryotic representation | |
|---|---|---|---|---|---|---|---|
| 1 | Na+,K+ | IIC | II | 38 | 1–10 | A,F,U,C | + |
| 2 | Ca2+ | IIA, IIB | II | 122 | 1–10 | A,P,F,U,C | + |
| 3 | H+ | IIIA | II | 45 | 1–10 | P,F,U | + |
| 4 | Mg2+ | IIIB | II | 2 | 1–10 | F | + |
| 5 | Copper | IB | I | 42 | A,B,1–6 | A,P,C | + |
| 6 | Heavy metal | IB | I | 7 | A,B,1–6 | P | + |
| 7 | K+ | IA | III | 0 | 7 | _5 | + |
| 8 | Phospholipid | IV | II | 151 | 1–10 | A,P,F,U,C | − |
| 9 | K+ or Na+ | IID | II | 24 | 1–10 | F,U | + |
| 10 | ? | V | IV | 26 | A’,B’,1–10 | A,P,F,U,C | − |
| 11 | ? | – | V | 1+ | C,1–10 | U | − |
| 12 | ? | – | V | 1 | C,1–10 | U | − |
| 13 | ? | V | V | 5 | C,1–10 | A | − |
| 14 | ? | V | V | 6 | C,1–10 | F | − |
| 15 | ? | V | V | 3 | C,1–10 | U | − |
| 16 | ? | V | V | 11 | C,1–10 | C | − |
| 17 | ? | V | V | 1+ | C,1–10 | F | − |
| 18 | ? | V | V | 1+ | C,1–10 | U | − |
| 19 | ? | V | V | 1 | C,1–10 | C | − |
| 20 | ? | V | V | 11 | C,1–10 | C | − |
| 21 | ? | V | V | 1 | C,1–10 | U | − |
| 22 | ? | V | VI | 6 | D,E,F,1–10 | U | − |
The type V ATPases of Axelsen and Palmgren (1998) are diverse proteins including those belonging to TC families 10 and 13–20. These proteins exhibit topological types IV and V. Among the “others” presented at the bottom of Table 1 in Axelsen and Palmgren (1998) are bacterial proteins. Proteins 158 and 159 are in TC family 24; protein 96 is most closely related to families 1 and 2, while proteins 156 and 157 are in family 2 and protein 19 is in family 23. For descriptions of bacterial TC families 23–32, see TCDB
aAs defined in this study
bNumber of P-type ATPases per family
cNumber of transmembrane α-helical segments
dA animal, P plant, F fungal, U unicellular, C ciliate, + close homologues are found in the NCBI database. The family 19 member most resembles family 16 ATPases
Some families (families 2 [Ca2+], 8 [phospholipids] and 10 [functionally uncharacterized]) are ubiquitous in eukaryotes, being found in each of the five organismal types examined. Surprisingly, although the members of several ATPase families are present in the various organisms in highly variable numbers, one and only one ATPase of family 10 was found in each of the 26 organisms surveyed without exception. The organismal distribution and topological structure are unique characteristics of this family, suggesting a distinctive function. These proteins are not present in 73 prokaryotic genomes surveyed (unpublished observations), providing evidence that their function relates to the uniquely eukaryotic endomembrane system (Cronin et al. 2000, 2002).
Other families, such as the family 1 (Na+/K+) ATPases, are present in most types of eukaryotes, plants being an exception. Family 3 (H+-ATPases) are present in all types except animals and the ciliate. Only two Mg2+-type ATPases (family 4, common in prokaryotes) were identified, and these were found in just one fungus Cyanidioschyzon merolae and the slime mold Dictyostelium discoideum. Surprisingly, family 6 heavy metal ATPases were found only in plants, while family 9 Na+ or K+-ATPases were restricted to the eight fungi examined as well as five unicellular eukaryotes. Family 7 K+-ATPases, prevalent in bacteria, proved to be lacking in eukaryotes. Thus, each family has a unique organismal distribution.
The 13 functionally uncharacterized families are generally less widely distributed, possibly accounting in part for the lack of functional data for them. However, there appear to be two exceptions. First, family 10 is ubiquitous in eukaryotes but seems to be lacking in prokaryotes as noted above. Second, families 13–16 all cluster more closely to each other on the phylogenetic tree than to any of the other homologues (Fig. 2), and each one is from a different organismal type (animals [family 13], fungi [family 14], slime molds [family 15] and ciliates [family 16]). Thus, only plants and some unicellular eukaryotes lack obvious members of this “sub-superfamily.” We suggest that these proteins may serve a single unique function. Their phylogenetic relationships clearly suggest a lack of horizontal transfer between these organismal types. Finally, families 16 and 20, represented only in Tetrahymena thermophila, each have 11 members, and both sets of proteins display the same phylogenetic relationships (Fig. 2). This observation suggests that these two families evolved strictly in parallel with each other, consistent with a functional interdependence, possibly reflective of the evolutionary development of ciliates.
Other uncharacterized ATPase families were “orphan” enzymes, found in single copy in a single organism. No two of these enzymes were found in the same organism. We considered the possibility that some of these putative ATPases might be encoded by nonfunctional “pseudogenes,” but identification of all nine well-conserved motifs and the completeness of their sequences relative to functionally characterized ATPases led us to suggest that these enzyme transporters are functional. Further, three of the families have additional homologues in the current NCBI protein database (10/08). The unique motif sequences and topological features of some of these protein families may provide clues concerning their transport functions. The analyses reported here should therefore serve as guides for future research aimed at answering questions concerning the functions of the novel P-type ATPases and the several surprising findings revealed by this study.
In 1998, Axelsen and Palmgren (A&P) proposed a system of P-type ATPase classification. At about the same time, we developed the functional and phylogenetic TC system for the classification of all transporters found in nature (Saier 1996, 1998, 2000; Paulsen et al. 1998). An advantage of the TC system is that it is applicable to all transporters. It has been adopted by the IUBMB, is in wide use by transport molecular biologists and has been incorporated into numerous databases including the SwissProt/TREMBL (UniProtKB) protein database.
Table 4 compares the two systems of classification cited above (TCDB and the P-type ATPase-specific system of A&P). TC family 1 (A&P’s group IIC) includes the closely related group of Na+, K+ and gastric H+,K+-ATPases characterized primarily in animals but with family members in fungi, unicellular eukaryotes (as reported here) and bacteria (unpublished results), based on our phylogenetic analyses. TC family 2 consists of Ca2+-ATPases corresponding to the ER (SERCA) and plasma membrane (PM) enzymes of animals. Some of the latter are found in endomembranes, particularly in plants (Geisler et al. 2000). These two types correspond to A&P groups IIA and IIB, respectively. Our results show that functionally similar Ca2+ pumps from eukaryotes and prokaryotes can be subdivided into at least five, and probably more, subclusters (unpublished results).
TC family 3 (A&P’s group IIIA) consists of H+-ATPases in plants, fungi and unicellular eukaryotes but also in bacteria, where one such homologue has been shown to transport heavy metals (Mn2+ and Cd2+; Hao et al. 1999). TC family 4 (A&P’s group IIIB) includes Mg2+-ATPases, mostly prokaryotic. Our results show that these four families as well as the eukaryote-specific phospholipid flippases (TC family 8, A&P’s group IV), lacking in prokaryotes, and the K+ or Na+ export ATPases (TC family 9, A&P’s group IID) of fungi and unicellular eukaryotes, but poorly represented in prokaryotes, are all of the same topology (type II). Thus, members of A&P’s groups IIA, IIB, IIC, IIIA, IIIB and IV all exhibit the type II topology.
TC families 5 (copper) and 6 (heavy metals) are of the type I topology and correspond to A&P’s group IB. They are functionally and phylogenetically distinct, and their members exhibit different size ranges. Family 7 bacterial K+ importers are distantly related to other ATPases, form their own coherent phylogenetic groups and function by a mechanism different from all other P-type ATPases. They exhibit a distinctive topology that we call type III. Thus, as revealed in Table 4, A&P’s group IB consists of two different families (TC families 5 and 6), which both exhibit the type I topology. TC families 1–4, 8 and 9 are of type II topology and TC family 7 is of type III topology.
In these assignments of topological types, we have retained the phylogenetic type designations of A&P whenever possible. Thus, A&P’s group IA consists of enzymes of one topology, phylogenetic group and function, while their group IB consists of two distinct phylogenetic families with differing specificities but the same topology that differs from A&P’s group IA. We see no reason to combine groups IA and IB together into a single category.
TC family 10 forms part of A&P’s group V, but it is of our topological type IV, thus appearing to differ in topology from all other type V ATPases. TC families 11–21 (some of which were either unrecognized by A&P or included in their group V) exhibit our type V topology. Although A&P group TC family 10 with all of these proteins, family 10 proteins form a distinct phylogenetic cluster and exhibit the type IV topology, differing from that of families 11–21, which exhibit the type V topology. Thus, there seems to be no basis for clustering these proteins together as suggested by A&P. Only family 22, with six members in the organisms examined, exhibits the type VI topology (see Figs. 4, 5). It should be noted that while some of our families include just one or a few members, more can sometimes be identified by conducting BLAST searches of the NCBI protein database (e.g., families 11, 17 and 18). Further sequencing efforts will undoubtedly reveal the occurrence of additional members of the families that currently include “orphans.”
In summary, A&P’s group I systems include two functionally and phylogenetically distinguishable families (TC families 5 and 6) of similar topology, which A&P group together as IB. TC families 5 and 6 differ in size range, phylogeny and function. A&P’s group IA does not belong with IB as the former differs in all aspects from the latter (phylogeny, topology, organismal distribution, mechanism and cation specificity). Thus, they should not be categorized together in group I.
Following the convention of A&P to the extent possible, we have designated their group IB as topological type I. A&P’s group II, III and IV ATPases include six TC families (1–4, 8 and 9), all of which have the same type II topology but differing specificities and clustering patterns; members of our family 7 (Kdp-type K+-ATPases) belong to our topological type III. A&P’s group V includes TC family 10 of our type IV topology, while A&P’s remaining group V ATPases (TC families 11–21) consist of proteins with a unified topology, our type V topology. Finally, our family 22, which displays a unique topology (type VI), was also grouped as part of group V by A&P (Table 4).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by NIH Grant GM077402 from the National Institute of General Medical Sciences. We thank Dorjee G. Tamang and Dr. Ming Ren Yen for technical assistance and Randy Y. Hampton for useful discussions.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s00232-009-9176-2) contains supplementary material, which is available to authorized users.
Contributor Information
Mark D. Thever, Email: markthever@gmail.com
Milton H. Saier, Jr., Email: msaier@ucsd.edu
References
- Altschul SF, Koonin EV (1998) Iterated profile searches with PSI-BLAST—a tool for discovery in protein databases. Trends Biochem Sci 23:444–447 [DOI] [PubMed]
- Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schaffer AA, Yu YK (2005) Protein database searches using compositionally adjusted substitution matrices. FEBS J 272:5099–5100 [DOI] [PMC free article] [PubMed]
- Axelsen KB, Palmgren MG (1998) Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol 46:84–101 [DOI] [PubMed]
- Axelsen KB, Palmgren MG (2001) Inventory of the superfamily of P-type ion pumps in Arabidopsis. Plant Physiol 126:696–706 [DOI] [PMC free article] [PubMed]
- Axelsen KB, Venema K, Jahn T, Baunsgaard L, Palmgren MG (1999) Molecular dissection of the C-terminal regulatory domain of the plant plasma membrane H+-ATPase AHA2: mapping of residues that when altered give rise to an activated enzyme. Biochemistry 38:7227–7234 [DOI] [PubMed]
- Baranano DE, Wolosker H, Bae BI, Barrow RK, Snyder SH, Ferris CD (2000) A mammalian iron ATPase induced by iron. J Biol Chem 275:15166–15173 [DOI] [PubMed]
- Baxter I, Tchieu J, Sussman MR, Boutry M, Palmgren MG, Gribskov M, Harper JF, Axelsen KB (2003) Genomic comparison of P-type ATPase ion pumps in Arabidopsis and rice. Plant Physiol 132:618–628 [DOI] [PMC free article] [PubMed]
- Benito B, Garciadeblas B, Rodriguez-Navarro A (2002) Potassium- or sodium-efflux ATPase, a key enzyme in the evolution of fungi. Microbiology 148:933–941 [DOI] [PubMed]
- Berman MC (2001) Slippage and uncoupling in P-type cation pumps; implications for energy transduction mechanisms and regulation of metabolism. Biochim Biophys Acta 1513:95–121 [DOI] [PubMed]
- Burbaum JJ, Starzyk RM, Schimmel P (1990) Understanding structural relationships in proteins of unsolved three-dimensional structure. Proteins 7:99–111 [DOI] [PubMed]
- Busch W, Saier MH Jr (2002) The transporter classification (TC) system, 2002. Crit Rev Biochem Mol Biol 37:287–337 [DOI] [PubMed]
- Colangelo EP, Guerinot MT (2006) Put the metal to the petal: metal uptake and transport throughout plants. Curr Opin Plant Biol 9:322–330 [DOI] [PubMed]
- Cronin SR, Khoury A, Ferry DK, Hampton RY (2000) Regulation of HMG-CoA reductase degradation requires the P-type ATPase Cod1p/Spf1p. J Cell Biol 148:915–924 [DOI] [PMC free article] [PubMed]
- Cronin SR, Rao R, Hampton RY (2002) Cod1p/Spf1p is a P-type ATPase involved in ER function and Ca2+ homeostasis. J Cell Biol 157:1017–1028 [DOI] [PMC free article] [PubMed]
- Devaux PF, Lopez-Montero I, Bryde S (2006) Proteins involved in lipid translocation in eukaryotic cells. Chem Phys Lipids 141:119–132 [DOI] [PubMed]
- DeZwaan TM, Carroll AM, Valent B, Sweigard JA (1999) Magnaporthe grisea pth11p is a novel plasma membrane protein that mediates appressorium differentiation in response to inductive substrate cues. Plant Cell 11:2013–2030 [DOI] [PMC free article] [PubMed]
- Fagan MJ, Saier MH Jr (1994) P-type ATPases of eukaryotes and bacteria: sequence analyses and construction of phylogenetic trees. J Mol Evol 38:57–99 [DOI] [PubMed]
- Gadsby DC (2007) Structural biology: ion pumps made crystal clear. Nature 450:957–959 [DOI] [PubMed]
- Geisler M, Axelsen KB, Harper JF, Palmgren MG (2000) Molecular aspects of higher plant P-type Ca2+-ATPases. Biochim Biophys Acta 1465:52–78 [DOI] [PubMed]
- Gerencser GA (1993) A novel P-type Cl−-stimulated ATPase: phosphorylation and specificity. Biochem Biophys Res Commun 196:1188–1194 [DOI] [PubMed]
- Greie JC, Altendorf K (2007) The K+-translocating KdpFABC complex from Escherichia coli: a P-type ATPase with unique features. J Bioenerg Biomembr 39:397–402 [DOI] [PubMed]
- Hao Z, Chen S, Wilson DB (1999) Cloning, expression, and characterization of cadmium and manganese uptake genes from Lactobacillus plantarum. Appl Environ Microbiol 65:4746–4752 [DOI] [PMC free article] [PubMed]
- Hatori Y, Majima E, Tsuda T, Toyoshima C (2007) Domain organization and movements in heavy metal ion pumps: papain digestion of CopA, a Cu+-transporting ATPase. J Biol Chem 282:25213–25221 [DOI] [PubMed]
- Haupt M, Bramkamp M, Coles M, Kessler H, Altendorf K (2005) Prokaryotic Kdp-ATPase: recent insights into the structure and function of KdpB. J Mol Microbiol Biotechnol 10:120–131 [DOI] [PubMed]
- Horisberger JD, Kharoubi-Hess S, Guennoun S, Michielin O (2004) The fourth transmembrane segment of the Na, K-ATPase alpha subunit: a systematic mutagenesis study. J Biol Chem 279:29542–29550 [DOI] [PubMed]
- Ikeda M, Kihara A, Igarashi Y (2006) Lipid asymmetry of the eukaryotic plasma membrane: functions and related enzymes. Biol Bull 29:1542–1546 [DOI] [PubMed]
- Inagaki C, Hara M, Zeng XT (1996) A Cl–pump in rat brain neurons. J Exp Zool 275:262–268 [DOI] [PubMed]
- Kuhlbrandt W (2004) Biology, structure and mechanism of P-type ATPases. Nat Rev Mol Cell Biol 5:282–295 [DOI] [PubMed]
- Kulkarni RD, Kelkar HS, Dean RA (2003) An eight-cysteine-containing CFEM domain unique to a group of fungal membrane proteins. Trends Biochem Sci 28:118–121 [DOI] [PubMed]
- Kulkarni RD, Thon MR, Pan H, Dean RA (2005) Novel G-protein-coupled receptor-like proteins in the plant pathogenic fungus Magnaporthe grisea. Genome Biol 6:R24 [DOI] [PMC free article] [PubMed]
- Kung C, Saimi Y (1982) Are ions involved in the gating of calcium channels? Ann Rev Physiol 44:519–534 [DOI] [PubMed]
- Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132 [DOI] [PubMed]
- Liu J, Dutta SJ, Stemmler AJ, Mitra B (2006) Metal-binding affinity of the transmembrane site in ZntA: implications for metal selectivity. Biochemistry 45:763–772 [DOI] [PubMed]
- Lutsenko S, Kaplan JH (1995) Organization of P-type ATPases: significance of structural diversity. Biochemistry 34:15607–15613 [DOI] [PubMed]
- Lutsenko S, Gupta A, Burkhead JL, Zuzel V (2008) Cellular multitasking: the dual role of human Cu-ATPases in cofactor delivery and intracellular copper balance. Arch Biochem Biophys 476:22–32 [DOI] [PMC free article] [PubMed]
- Maguire ME (2006) Magnesium transporters: properties, regulation and structure. Front Biosci 11:3149–3163 [DOI] [PubMed]
- Martin DW (2005) Structure–function relationships in the Na+, K+ -pump. Semin Nephrol 25:282–291 [DOI] [PubMed]
- Møller JV, Juul B, le Maire M (1996) Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim Biophys Acta 1286:1–51 [DOI] [PubMed]
- Møller S, Croning MD, Apweiler R (2001) Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 17:646–653 [DOI] [PubMed]
- Møller JV, Nissen P, Sorensen TL, le Maire M (2005) Transport mechanism of the sarcoplasmic reticulum Ca2+-ATPase pump. Curr Opin Struct Biol 15:387–393 [DOI] [PubMed]
- Møller AB, Asp T, Holm PB, Palmgren MG (2008) Phylogenetic analysis of P5 P-type ATPases, a eukaryotic lineage of secretory pathway pumps. Mol Phylogenet Evol 46:619–634 [DOI] [PubMed]
- Morth JP, Pedersen BP, Toustrup-Jensen MS, Sorensen TL, Petersen J, Andersen JP, Vilsen B, Nissen P (2007) Crystal structure of the sodium-potassium pump. Nature 450:1043–1049 [DOI] [PubMed]
- Nies DH (2003) Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol Rev 27:313–339 [DOI] [PubMed]
- Odenbach D, Breth B, Thines E, Weber RW, Anke H, Foster AJ (2007) The transcription factor Con7p is a central regulator of infection-related morphogenesis in the rice blast fungus Magnaporthe grisea. Mol Microbiol 64:293–307 [DOI] [PubMed]
- Oestreicher N, Scazzocchio C (1995) A single amino acid change in a pathway-specific transcription factor results in differing degrees of constitutivity, hyperinducibility and derepression of several structural genes. J Mol Biol 249:693–699 [DOI] [PubMed]
- Olesen C, Picard M, Winther AM, Gyrup C, Morth JP, Oxvig C, Møller JV, Nissen P (2007) The structural basis of calcium transport by the calcium pump. Nature 450:1036–1042 [DOI] [PubMed]
- Paulsen IT, Sliwinski MK, Saier MH Jr (1998) Microbial genome analyses: global comparisons of transport capabilities based on phylogenies, bioenergetics and substrate specificities. J Mol Biol 277:573–592 [DOI] [PubMed]
- Pedersen BP, Buch-Pedersen MJ, Morth JP, Palmgren MG, Nissen P (2007) Crystal structure of the plasma membrane proton pump. Nature 450:1111–1114 [DOI] [PubMed]
- Perez A, Pedros B, Murgui A, Casanova M, Lopez-Ribot JL, Martinez JP (2006) Biofilm formation by Candida albicans mutants for genes coding fungal proteins exhibiting the eight-cysteine-containing CFEM domain. FEMS Yeast Res 6:1074–1084 [DOI] [PubMed]
- Plattner H, Klauke N (2001) Calcium in ciliated protozoa: sources, regulation, and calcium-regulated cell functions. Int Rev Cytol 201:115–208 [DOI] [PubMed]
- Pomorski T, Menon AK (2006) Lipid flippases and their biological functions. Cell Mol Life Sci 63:2908–2921 [DOI] [PMC free article] [PubMed]
- Saier MH Jr (1996) Phylogenetic approaches to the identification and characterization of protein families and superfamilies. Microb Comp Genomics 1:129–150 [DOI] [PubMed]
- Saier MH Jr (1998) Molecular phylogeny as a basis for the classification of transport proteins from bacteria, archaea and eukarya. Adv Microb Physiol 40:81–136 [DOI] [PubMed]
- Saier MH Jr (2000) A functional-phylogenetic classification system for transmembrane solute transporters. Microbiol Mol Biol Rev 64:354–411 [DOI] [PMC free article] [PubMed]
- Saier MH Jr, Tran CV, Barabote RD (2006) TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res 34:181–186 [DOI] [PMC free article] [PubMed]
- Saier MH, Yen MR, Noto K, Tamang DG, Elkan C (2009) The transporter classification database (TCDB): recent advances. Nucleic Acids Res 37:274–278. [DOI] [PMC free article] [PubMed]
- Scarborough GA (2000) The plasma membrane proton-translocating ATPase. Cell Mol Life Sci 57:871–883 [DOI] [PMC free article] [PubMed]
- Tatusova TA, Madden TL (1999) Blast 2 sequences—a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Let 174:247–250 [DOI] [PubMed]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882 [DOI] [PMC free article] [PubMed]
- Toyoshima C, Nakasako M, Nomura H, Ogawa H (2000) Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 405:647–655 [DOI] [PubMed]
- Ueno S, Kaieda N, Koyama N (2000) Characterization of a P-type Na+-ATPase of a facultatively anaerobic alkaliphile, Exiguobacterium aurantiacum. J Biol Chem 275:14537–14540 [DOI] [PubMed]
- Vashist S, Frank CG, Jakob CA, Ng DT (2002) Two distinctly localized P-type ATPases collaborate to maintain organelle homeostasis required for glycoprotein processing and quality control. Mol Biol Cell 13:3955–3966 [DOI] [PMC free article] [PubMed]
- Wang S, Gao D, Penny J, Krishna S, Takeyasu K (1997) P-type ATPases in Tetrahymena. Ann N Y Acad Sci 834:158–160 [DOI] [PubMed]
- Wang S, Takeyasu K (1997) Primary structure and evolution of the ATP-binding domains of the P-type ATPases in Tetrahymena thermophila. Am J Physiol 272:715–728 [DOI] [PubMed]
- Waxman SG, Cummins TR, Dib-Hajj SD, Black JA (2000) Voltage-gated sodium channels and the molecular pathogenesis of pain: a review. J Rehabil Res Dev 37:517–595 [PubMed]
- Zeng W, Dohi S, Shimonaka H, Asano T (1999) Spinal antinociceptive action of Na+-K+ pump inhibitor ouabain and its interaction with morphine and lidocaine in rats. Anesthesiology 90:500–508 [DOI] [PubMed]
- Zhai Y, Saier MH Jr (2001a) A Web-based program (WHAT) for the simultaneous prediction of hydropathy, amphipathicity, secondary structure and transmembrane topology for a single protein sequence. J Mol Microbiol Biotechnol 3:501–502 [PubMed]
- Zhai Y, Saier MH Jr (2001b) A Web-based program for the prediction of average hydropathy, average amphipathicity and average similarity of multiply aligned homologous proteins. J Mol Microbiol Biotechnol 3:285–286 [PubMed]
- Zhai Y, Saier MH Jr (2002) A simple sensitive program for detecting internal repeats in sets of multiply aligned homologous proteins. J Mol Microbiol Biotechnol 4:375–377 [PubMed]
- Zhai Y, Tchieu J, Saier MH Jr (2002) A Web-based Tree View (TV) program for the visualization of phylogenetic trees. J Mol Microbiol Biotechnol 4:69–70 [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.