

Abstract
A four-step synthesis of cis-3,5-disubstituted morpholines from enantiomerically pure amino alcohols is described. The key step in the synthesis is a Pd-catalyzed carboamination reaction between a substituted ethanolamine derivative and an aryl or alkenyl bromide. The morpholine products are generated as single stereoisomers in moderate to good yield. This strategy also provides access to fused bicyclic morpholines, as well as 2,3- and 2,5-disubstituted products.
In recent years drug discovery efforts have revealed several interesting biologically active compounds that contain C-substituted morpholine units.1,2 However, despite the medicinal importance of these molecules, the development of new approaches to their synthesis remains relatively unexplored.1,3 For example, few methods allow the preparation of 3,5-disubstituted morpholines,4 and only two approaches to the stereoselective synthesis of cis-3,5-disubstituted derivatives have been described.2a,5 Both of these strategies are limited in scope, as one affords symmetrically disubstituted (meso) products,5 and the other was used only for the generation of one single compound (cis-3-carbomethoxy-5-allylmorpholine).2a
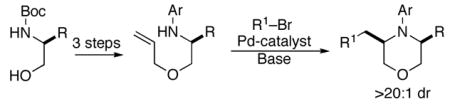
We recently reported a concise asymmetric synthesis of cis-2,6-disubstituted piperazines that involves Pd-catalyzed carboamination reactions of N-allyl ethylenediamine derivatives.6,7 We felt that a similar strategy may be applied to the construction of 3,5-disubstituted morpholines. As shown in Scheme 1, enantiopure N-Boc amino alcohols (1) could be converted to O-allyl ethanolamines 2 using standard methods. These compounds would then be transformed to the desired heterocycles 3 through Pd-catalyzed coupling with an aryl or alkenyl halide.8 This strategy should provide access to a broad array of enantiopure cis-3,5-disubstituted morpholines that are difficult to generate using existing methods.
SCHEME 1. Synthetic Strategy.
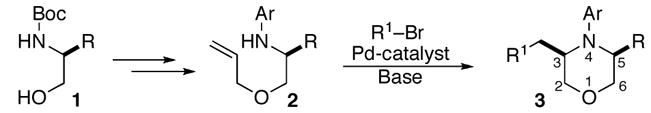
The substrates for the Pd-catalyzed carboamination reactions were synthesized in three steps from commercially available starting materials 1a–e as shown in Scheme 2. Treatment of the N-protected amino alcohols with NaH and allyl bromide afforded allyl ethers 4a–e. Cleavage of the Boc-group followed by Pd-catalyzed N-arylation of the resulting amine trifluoroacetate salts provided 2a–f in moderate to good yield.9
SCHEME 2. Synthesis of Substrates.
a Ligand = (o-biphenyl)PtBu2. b Ligand = P(tBu)3•HBF4. c Ligand = (±)-BINAP.
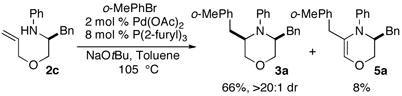
At the beginning of our studies we elected to examine the coupling of 2c with 2-bromotoluene under reaction conditions that had proven optimal in related piperazine-forming carboamination reactions.6 As shown in eq 1, use of a catalyst composed of Pd(OAc)2 and P(2-furyl)3 provided 3a in 66% yield. The main side product observed in this reaction was 5a, although small amounts of several other unidentified side products were also detected. A survey of other ligands (e.g., PPh3, Dpe-phos) did not provide improved results, and our initial choice of solvent (toluene) and base (NaOtBu) also proved optimal. 10
 |
(1) |
The results of our studies on the scope of 3,5-disubstituted morpholine-forming carboamination reactions are illustrated in Table 1. Several different 2-subsituted O-allylethanolamines were effectively converted to the desired heterocycles, including heteroatom-containing substrates derived from methionine (entry 7), serine (entry 8), and tryptophan (entry 9). Although the yields in these reactions were modest (46–66%),11 the diastereoselectivities were uniformly high (> 20:1 dr). The presence of electron-neutral or slightly electron-deficient N-aryl groups on the substrates was tolerated. However, efforts to employ a morpholine precursor bearing an N-(p-methoxyphenyl) moiety led to a poor yield of 3d due to competing N-arylation and Heck arylation of the substrate (entry 3).12 Low yields were also obtained when starting materials with N-p-cyanophenyl groups were used, as competing Heck arylation of the substrate alkene group was again problematic. Similarly, efforts to couple N-Boc-protected substrate 4a with 1-bromo-4-tert-butylbenzene afforded only a Heck arylation product.
TABLE 1.
Synthesis of cis-3,5-Disubstituted Morpholinesa
 | |||
|---|---|---|---|
| entry | substrate | product | yieldb |
| 1 |
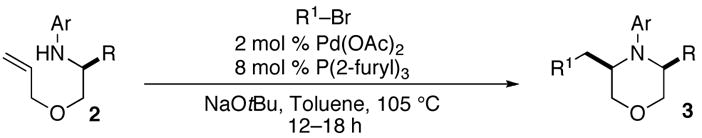
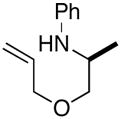 2a |
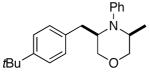 3b |
53% |
| 2 | 2a |
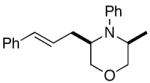 3c |
46%c |
| 3 |
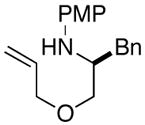 2b |
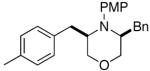 3d |
21% |
| 4 |
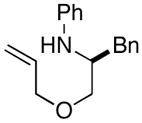 2c |
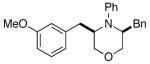 3e |
48% |
| 5 | 2c |
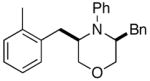 3a |
66% |
| 6 | 2c |
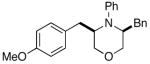 3f |
47% |
| 7 |
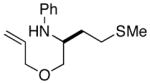 2d |
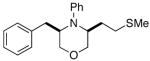 3g |
58% |
| 8 |
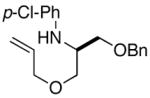 2e |
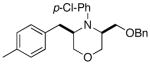 3h |
49% |
| 9 |
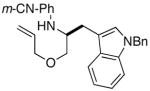 2f |
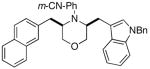 3i |
57% |
Conditions: 1.0 equiv substrate, 2.0 equiv R1Br, 2.0 equiv NaOtBu, 2 mol % Pd(OAc)2, 8 mol % P(2-furyl)3, toluene (0.4 M), 105 °C.
Isolated yield (average of two experiments). All products were formed with >20:1 dr as judged by 1H NMR analysis of crude products prior to purification.
The reaction was conducted using 4.0 equiv of β-bromostyrene, 4.0 equiv of NaOtBu, 4 mol % Pd(OAc)2 and 16 mol % P(2-furyl)3.
In order to further explore the utility of this method for the synthesis of other substituted morpholines, reactions of several N-aryl ethanolamine derivatives with different substitution patterns were examined. As shown in Table 2, substrates 6a–d, which were prepared by O-allylation of 2-(N-phenylamino)cyclohexanol or –cyclopentanol,13 were coupled with aryl bromides using our optimized reaction conditions. These transformations afforded the desired bicyclic morpholines 7a–e in moderate to good yields with excellent diastereoselectivities (>20:1 dr). We also successfully converted 8 and 10 into 2,3-disubstituted morpholine 9 and 2,5-disubstituted morpholine 11 (eq 2–3). However, both 9 and 11 were produced with only modest (2:1) diastereoselectivity.
TABLE 2.
Synthesis of Bicyclic Morpholinesa
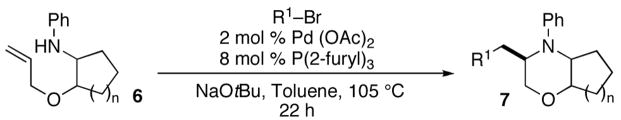 | |||
|---|---|---|---|
| entry | substrate | product | yieldb |
| 1 |
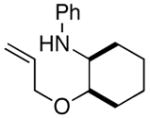 6a |
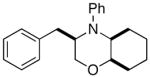 7a |
65% |
| 2 |
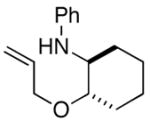 6b |
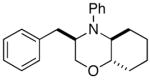 7b |
56% |
| 3 |
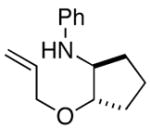 6c |
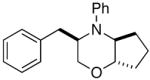 7c |
73% |
| 4 | 6c |
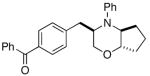 7d |
54% |
| 5 |
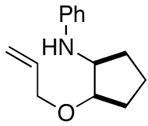 6d |
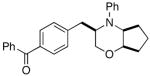 7e |
77% |
Conditions: 1.0 equiv substrate, 2.0 equiv R1Br, 2.0–2.7 equiv NaOtBu, 2 mol %
Isolated yield (average of Pd(OAc)2, 8 mol % P(2-furyl)3, toluene (0.3 M), 105 °C. two or more experiments). All products were formed with >20:1 dr as judged by 1H NMR analysis of crude products prior to purification.
The nature of the aryl halide coupling partner had a significant effect on the yield of the morpholine-forming reactions. Use of electron-rich or electron-neutral derivatives provided acceptable yields of the desired heterocycles. In addition, the coupling of 2a with an alkenyl halide (Table 1, entry 2) was also successful. However, most attempts to employ electron-poor aryl bromides led to complex mixtures of products, although the carboamination reactions of 6c–d with 4-bromobenzophenone (Table 2, entries 4–5) and of 8 with 3-bromobenzonitrile (eq 2) gave useful quantities of desired products. The coupling of 2c with the sterically hindered 2-bromotoluene provided a 66% yield of 3a (Table 1, entry 5), but 1-bromo-2-methylnaphthalene failed to react with 2c under similar conditions.
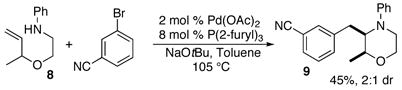 |
(2) |
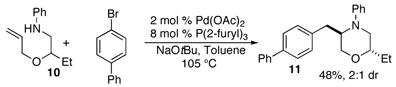 |
(3) |
The mechanism of the morpholine-forming carboamination reactions is likely similar to that of related transformations that generate piperazines, pyrrolidines, and other nitrogen heterocycles.6–8 As shown in Scheme 3, the key intermediate in the conversion of 2 to 3 is palladium(aryl)(amido) complex 12, which is produced by oxidative addition of the aryl bromide to Pd(0) followed by Pd–N bond formation.14 The relative stereochemistry of the substituted morpholine products is most consistent with a pathway involving syn-aminopalladation of 12 through a boat-like transition state (13) to afford 14.15 Reductive elimination from 14 would provide the cis-3,5-disubstituted morpholine products 3. This mechanism also accounts for the conversion of 8 to cis-2,3-disubstituted morpholine 9, and 10 to trans-2,5-disubstituted morpholine 11. These products would arise from transition states 15 and 16, respectively.16
SCHEME 3. Mechanism and Stereochemistry.

As noted above in eq 1, we observed the formation of 3,4-dihydro-2H-1,4-oxazine 5a as a side product in the Pd/P(2-furyl)3 catalyzed coupling of 2c with 2-bromotoluene. This compound is presumably generated via β-hydride elimination from intermediate 14 to provide 17. This complex could then be transformed into unsaturated heterocycle 5a by alkene dissociation and subsequent Heck arylation17 of the resulting product 18 (Scheme 4).
SCHEME 4. Formation of 3,4-dihydro-2H-1,4-oxazine 5a.

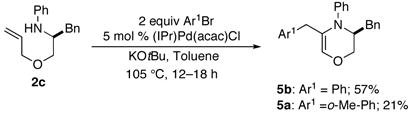
We felt that it may be possible to optimize conditions so that unsaturated compounds such as 5a would be generated as the major products in coupling reactions between 2 and aryl bromides. The mechanism outlined in Scheme 4 suggests that catalysts or ligands that either slow C–C bond-forming reductive elimination, facilitate β–hydride elimination, or both, may favor the conversion of 14 to 17, which in turn leads to generation of 5. Thus, we examined the use of catalysts supported by relatively electron rich monodentate ligands.18 After some experimentation we discovered that use of (IPr)Pd(acac)Cl19 for the coupling of bromobenzene with 2c afforded 5b in 57% yield (eq 4). However, the scope of this reaction is currently limited. For example, use of 2-bromotoluene as the electrophile afforded only 21% yield of 5a. Purification of these products is also difficult due to their hydrolytic lability. Nonetheless, further optimization of conditions or use of this transformation in tandem/sequenced reactions may improve synthetic utility.
 |
(4) |
In conclusion, we have developed a concise asymmetric synthesis of cis-3,5-disubstituted morpholines from readily available enantiopure amino alcohol precursors. The modular nature of this approach permits variation of the morpholine substituents, and also provides access to fused-ring morpholine derivatives. In addition, we have demonstrated that modification of catalyst structure can lead to potentially useful 3,4-dihydro-2H-1,4-oxazine products. The strategies described above significantly expand the range of substituted morpholines that can be prepared in a concise, stereocontrolled manner.
Experimental Section
Representative Procedure for Synthesis of Morpholines via Pd-Catalyzed Carboamination
A Schlenk tube was evacuated, flame dried, and backfilled with nitrogen. The tube was charged with Pd(OAc)2 (2.3 mg, 0.01 mmol), P(2-furyl)3 (9.3 mg, 0.04 mmol), and NaOtBu (96.1 mg, 1.0 mmol). The tube was evacuated and backfilled with nitrogen, then the aryl bromide (1.0 mmol) and a solution of the amine substrate (0.50 mmol) in toluene (1.25 mL) were added to the Schlenk tube (aryl bromides that were solids at room temperature were added as solids following the addition of NaOtBu). The mixture was heated to 105 °C with stirring until the substrate was consumed as judged by GC analysis (12–18 h). The reaction mixture was cooled to rt, quenched with saturated aqueous NH4Cl (3 mL), and extracted with EtOAc (3 × 3 mL). The combined organic layers were concentrated in vacuo and the crude product was purified by flash chromatography on silica gel.
(−)-(3S,5R)-3-Benzyl-5-(2-methylbenzyl)-4-phenylmorpholine (3a)
The representative procedure was employed for the coupling of 2-bromotoluene with 2c. This procedure gave the title compound (121 mg, 68%) as a yellow oil after purification by chromatography with 5% EtOAc/hexanes as the eluant. This material was judged to be of >20:1 dr by 1H NMR analysis before and after purification. [α]23D − 3.1 (c = 1.20, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.43–7.37 (m, 2 H), 7.31–7.14 (m, 7 H), 7.14–7.07 (m, 4 H), 7.07–7.00 (m, 1 H), 3.71 (dd, J = 5.4, 11.5 Hz, 1 H), 3.66–3.46 (m, 5 H), 2.81–2.66 (m, 4 H), 2.23 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 147.5, 138.9, 137.0, 136.5, 130.4, 129.6, 129.5, 129.1, 128.5, 126.3, 126.3, 125.9, 121.7, 119.6, 69.6, 69.6, 57.9, 56.9, 37.0, 33.6, 19.6; IR (film) 1598 cm−1; MS (ESI) 358.2174 (358.2171 calcd for C25H27NO, M + H+).
Supplementary Material
Acknowledgments
The authors thank the NIH-NIGMS (GM071650) for financial support of this work. M.L.L. was supported by the Michigan C.B.I. Training Program (NIH 5T32GM008597-12). Additional support was provided by the Camille and Henry Dreyfus Foundation (Camille Dreyfus Teacher Scholar Award), GlaxoSmithKline, Eli Lilly, Amgen, and 3M.
Footnotes
Supporting Information Available. Experimental procedures, spectroscopic data, and copies of 1H and 13C NMR spectra for all new compounds reported in the text (141 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For a review on the synthesis and biological significance of C-substituted morpholines, see: Wijtmans R, Vink MKS, Schoemaker HE, van Delft FL, Blaauw RH, Rutjes FPJT. Synthesis. 2004:641.
- 2.For selected examples of biologically active cis-3,5-disubstituted morpholines, see: O’Neil SV, Wang Y, Laufersweiler MC, Oppong KA, Soper DL, Wos JA, Ellis CD, Baize MW, Bosch GK, Fancher AN, Lu W, Suchanek MK, Wang RL, De B, Demuth TP., Jr Bioorg Med Chem Lett. 2005;15:5434. doi: 10.1016/j.bmcl.2005.08.109.Allison BD, Phuong VK, McAtee LC, Rosen M, Morton M, Prendergast C, Barrett T, Lagaud G, Freedman J, Li L, Wu X, Venkatesan H, Pippel M, Woods C, Rizzolio MC, Hack M, Hoey K, Deng X, King C, Shankley NP, Rabinowitz MH. J Med Chem. 2006;49:6371. doi: 10.1021/jm060590x.Josien HB, Clader JW, Bara TA, Xu R, Li H, Pissarnitski D, Zhao Z. PCT Int Appl WO 2006004880 A2, January 12, 2006. Chem Abstr. 2006;144:129004.
- 3.For recent approaches to the synthesis of C-substituted morpholines, see: Yar M, McGarrigle EM, Aggarwal VK. Org Lett. 2009;11:257. doi: 10.1021/ol8023727.Penso M, Lupi V, Albanese D, Foschi F, Landini D, Tagliabue A. Synlett. 2008:2451.Wilkinson MC, Bell R, Landon R, Nikiforov PO, Walker AJ. Synlett. 2006:2151.Lanman BA, Myers AG. Org Lett. 2004;6:1045. doi: 10.1021/ol049861t.Tiecco M, Testaferri L, Marini F, Sternativo S, Santi C, Bagnoli L, Temperini A. Tetrahedron: Asymmetry. 2003;14:2651.
- 4.For stereoselective syntheses of trans-3,5-disubstituted morpholines, see: Leijondahl K, Boren L, Braun R, Bäckvall J-E. Org Lett. 2008;10:2027. doi: 10.1021/ol800468h.Dave R, Sasaki NA. Tetrahedron: Asymmetry. 2006;17:388.Dave R, Sasaki NA. Org Lett. 2004;6:15. doi: 10.1021/ol035998s.Takahata H, Takahashi S, Kouno S-i, Momose T. J Org Chem. 1998;63:2224.For non-stereoselective syntheses of 3,5-disubstituted morpholines, see: Revesz L, Blum E, Wicki R. Tetrahedron Lett. 2005;46:5577.Enders D, Meyer O, Raabe G, Runsink J. Synthesis. 1994:66.Barluenga J, Najera C, Yus M. Synthesis. 1978:911.
- 5.D’hooghe MD, Vanlangendonck T, Törnroos KW, De Kimpe N. J Org Chem. 2006;71:4678. doi: 10.1021/jo060313y. [DOI] [PubMed] [Google Scholar]
- 6.(a) Nakhla JS, Wolfe JP. Org Lett. 2007;9:3279. doi: 10.1021/ol071241f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nakhla JS, Schultz DM, Wolfe JP. Tetrahedron. 2009 doi: 10.1016/j.tet.2009.04.017. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For related syntheses of pyrrolidines, imidazolidin-2-ones, isoxazolidines, and pyrazolidines via Pd-catalyzed carboamination reactions, see: Ney JE, Wolfe JP. Angew Chem, Int Ed. 2004;43:3605. doi: 10.1002/anie.200460060.Bertrand MB, Neukom JD, Wolfe JP. J Org Chem. 2008;73:8851. doi: 10.1021/jo801631v.Fritz JA, Wolfe JP. Tetrahedron. 2008;64:6838. doi: 10.1016/j.tet.2008.04.015.Lemen GS, Giampietro NC, Hay MB, Wolfe JP. J Org Chem. 2009;74:2533. doi: 10.1021/jo8027399.Giampietro NC, Wolfe JP. J Am Chem Soc. 2008;130:12907. doi: 10.1021/ja8050487.
- 8.For reviews on Pd-catalyzed carboamination reactions, see: Wolfe JP. Eur J Org Chem. 2007:571.Wolfe JP. Synlett. 2008:2913.
- 9.For a representative reaction sequence, chiral HPLC analysis indicated complete retention of enantiomeric purity (99% ee) during the preparation of substrate 2a from 1a. The Pd-catalyzed carboamination reaction of 2a to 3b also proceeded with no erosion of ee.
- 10.Use of other ligands provided greater amounts of 5a, led to formation of side products resulting from Heck arylation of the starting material, or both. See the Supporting Information for a table of results obtained with other phosphines.
- 11.Side products of general structure 5 were also observed in crude reaction mixtures. NMR analysis indicated these side products were formed as ca. 10–35% of the mixture. See the Supporting Information for further details.
- 12.In some instances side products resulting from sequential N-arylation and Heck arylation of the substrate were also isolated.
- 13.The known trans-2-(N-phenylamino)cycloalkanols were prepared in one step from aniline and cyclohexene oxide or cyclopentene oxide. See: Wang Z, Cui Y-T, Xu Z-B, Qu J. J Org Chem. 2008;73:2270. doi: 10.1021/jo702401t.Arai K, Lucarini S, Salter M, Ohta K, Yamashita Y, Kobayashi S. J Am Chem Soc. 2007;129:8103. doi: 10.1021/ja0708666.The cis-2-(N-phenylamino)cycloalkanols were prepared in three steps from the epoxides. See the Supporting Information for further details.
- 14.(a) Barder TE, Buchwald SL. J Am Chem Soc. 2007;129:12003. doi: 10.1021/ja073747z. [DOI] [PubMed] [Google Scholar]; (b) Yamashita M, Hartwig JF. J Am Chem Soc. 2004;126:5344. doi: 10.1021/ja0315107. [DOI] [PubMed] [Google Scholar]
- 15.Chair-like transition states for intramolecular syn-aminopalladation reactions that generate six-membered rings appear to be less favorable than boat-like transition states due to poor overlap between the alkene Π-system and the Pd–N bond. For additional discussion of boat-like vs. chair-like transition states in Pd-catalyzed carboamination reactions that afford piperazine products, see reference 6b.
- 16.The modest diastereoselectivities observed in the reactions of 8 and 10 are presumably due to relatively small differences in the energies of transition states in which the substrate R-group is oriented in a psueduoaxial vs. pseudoequatorial position. For further discussion, see reference 6b.
- 17.(a) Heck RF. Synlett. 2006:2855. [Google Scholar]; (b) Beller M, Zapf A, Reirmeier TH. In: Transition Metals for Organic Synthesis. 2. Beller M, Bolm C, editors. Wiley-VCH: Weinheim, Germany; 2004. pp. 271–305. [Google Scholar]
- 18.The rate of reductive elimination from Pd(II) decreases as ligand basicity increases and ligand size decreases. However, steric effects can outweigh electronic effects, as electron-rich ligands that are sterically bulky are known to promote reductive elimination. For reviews, see: Christmann U, Vilar R. Angew Chem, Int Ed. 2005;44:366. doi: 10.1002/anie.200461189.Brown JM, Cooley NA. Chem Rev. 1988;88:1031.
- 19.IPr = 1,3-Bis(2,6-diisopropylphenyl)imidazol-2-ylidene. Although this electron-rich ligand is also sterically bulky, it appears that ligand electronic properties play a larger role than steric properties in this particular reaction. For further discussion on the steric and electronic properties of NHC ligands, see: Diez-Gonzalez S, Nolan SP. Coord Chem Rev. 2007;251:874.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.