

Abstract
Vaccination against nicotine is under investigation as a treatment for tobacco dependence. Passive immunization with nicotine-specific antibodies represents a complementary strategy to vaccination. A potential adverse effect of passive immunization in nicotine-dependent individuals is that it may lead to a rapid reduction in brain nicotine levels and trigger withdrawal. The goal of this study was to determine if passive immunization with the nicotine-specific monoclonal antibody Nic311 precipitated withdrawal in nicotine-dependent rats as measured by increases in brain reward thresholds and somatic signs. Another cohort of rats was used to measure brain nicotine levels after Nic311 administration. Nic311 30, 80 or 240 mg/kg reduced brain nicotine concentrations by 45, 83 or 92% compared to controls. None of these Nic311 doses precipitated withdrawal measured at intervals up to 72 hours following antibody administration. Administration of the nicotinic antagonist mecamylamine precipitated a robust nicotine withdrawal syndrome. Therefore, a substantial, but not complete, acute reduction in brain nicotine levels following passive immunization was not sufficient to precipitate nicotine withdrawal in nicotine-dependent rats. The Nic311 doses used have been shown to attenuate the behavioral effects of nicotine, suggesting that the use of passive immunization to treat nicotine addiction is not likely to precipitate withdrawal.
Keywords: Nicotine, passive immunization, monoclonal antibody, withdrawal, mecamylamine
1. Introduction
Nicotine is the primary addictive component in tobacco (Benowitz, 1996). The pharmacokinetics of nicotine, including the extent and rate of nicotine entry into the brain and the rate of nicotine elimination, are key determinants of its rewarding and reinforcing effects (Benowitz, 1990; Henningfield and Keenan, 1993; Hukkanen et al., 2005). Interventions that alter nicotine pharmacokinetics may be useful for attenuating the addictive effects of nicotine and aiding in smoking cessation.
Vaccination against nicotine elicits the production of nicotine-specific antibodies that bind nicotine in serum, reduce or slow its distribution to brain, and slow its elimination (LeSage et al., 2006b). Vaccination of rats against nicotine attenuates a variety of nicotine-related behaviors including the acquisition, maintenance, and reinstatement of nicotine self-administration (de Villiers et al., 2004; LeSage et al., 2006a). Three nicotine vaccines are in clinical trials, and initial reports indicate efficacy in enhancing smoking cessation rates. However, efficacy is limited because it is strongly correlated with the serum nicotine-specific antibody concentrations or titers achieved, which are both modest and characterized by high individual variability (Cornuz et al., 2008; Hatsukami et al., 2005). The development of alternative strategies to reliably produce sufficiently high antibody levels is critical to maximize the efficacy of immunotherapy against nicotine.
Passive immunization against nicotine (the administration of exogenously derived nicotine-specific antibodies, or NicAb) has been studied in animals as a complementary strategy to vaccination. The effects of passive immunization in rats are quite similar to those of vaccination, such as lower brain nicotine concentrations and attenuation of nicotine-induced locomotor sensitization (Carrera et al., 2004; Keyler et al., 2005; Roiko et al., 2008). Passive immunization has a number of potential advantages over vaccination. First, passive immunization provides control over the antibody dose and resulting serum antibody concentration, and suitable doses can produce higher mean serum antibody levels than vaccination. Second, the large individual variability in immune response and resulting serum antibody levels seen with vaccination is circumvented. Third, the effect of passive immunization is immediate as compared to the 1–3 months needed for vaccination to elicit a maximal antibody response. Passive immunization is also being studied as a potential treatment for phencyclidine (Proksch et al., 2000), cocaine (Norman et al., 2007), and methamphetamine addiction or toxicity (Byrnes-Blake et al., 2003). The principal disadvantage of passive immunization with nicotine-specific monoclonal antibodies as a therapeutic strategy for tobacco addiction is its cost (Drucker et al., 2008; McLeod et al., 2007).
A potential adverse effect of both vaccination and passive immunization against nicotine is that, by reducing nicotine levels in brain, they could precipitate a withdrawal syndrome if administered to current smokers. This reduction in brain nicotine would result from antibodies binding nicotine in serum and reducing its free (unbound) fraction, causing a relatively rapid redistribution of nicotine from brain into serum. This is unlikely to be a problem with vaccination because NicAb levels increase gradually over 1–3 months (Lindblom et al., 2005). Passive immunization differs from vaccination in that therapeutic levels of NicAb are achieved immediately, which could more abruptly reduce brain nicotine concentrations.
The goal of this study was to determine whether the acute administration of the monoclonal nicotine-specific antibody Nic311 precipitates a nicotine abstinence syndrome in nicotine dependent rats as assessed by increases in brain reward thresholds (a measure of anhedonia associated with withdrawal) or somatic signs, two well-established measures of nicotine withdrawal (Epping-Jordan et al., 1998; Malin et al., 1992). In a parallel experiment, brain nicotine concentrations were measured to quantitate the extent of reduction in brain nicotine concentrations after Nic311 administration.
2. Materials and Methods
2.1. Animals
Male Holtzman Sprague Dawley rats (Harlan, Indianapolis, IN) weighing 300–325 g at the time of surgery were housed individually in temperature and humidity controlled colony rooms with unlimited access to water under a reversed 12-h light/dark cycle (lights off at 10:00 am). Rats were food-restricted to 18 g/day rat chow to minimize weight gain and limit chronic in-dwelling catheter migration throughout the chronic studies. All protocols were approved by the Minneapolis Medical Research Foundation Animal Care and Use Committee and were in compliance with the National Institutes of Health Guide for Care and Use of Laboratory Animals (Publication No. 85-23, revised 1985).
2.2. Reagents
Nicotine bitartrate or mecamylamine (Sigma Chemical Co., St. Louis, MO) were dissolved in sterile saline. The pH of the nicotine solution was adjusted to 7.4 with dilute NaOH. All nicotine doses and concentrations are expressed as that of the base. The nicotine-specific monoclonal antibody Nic311 is an IgG1κ derived from mice immunized with the immunogen 3′-aminomethylnicotine conjugated to recombinant Pseudomonas exoprotein A, with a Kd for nicotine of 60 nM and < 1% cross-reactivity with nicotine metabolites or a variety of neurotransmitters including acetylcholine (Pentel et al., 2006). Nic311 was purified by protein G chromatography to ≥ 95% of total protein content with endotoxin levels of < 0.2 EU/mg. Nic311 was diluted in phosphate-buffered saline to a concentration of 10 mg/ml for administration of the 30 mg/kg dose, 30 mg/ml for the 80 mg/kg dose, and 50 mg/ml for the 240 mg/kg dose. The Nic311 dose of 80 mg/kg was selected because this is a likely clinical dose; and produces serum NicAb levels similar to vaccination (Roiko et al., 2008). In addition, Nic311 80 mg/kg is equimolar to the steady-state rat body burden of nicotine (during a nicotine infusion of 3.2 mg/kg/day), and the Nic311 doses of 30 and 240 mg/kg bracket this dose (0.4 and 3.5 × nicotine total body burden, respectively). Control IgG was human polyclonal IgG (Sandoglobulin®; Sandoz, Vienna, Austria) that does not bind nicotine or alter nicotine pharmacokinetics or behavior in rats (Keyler et al., 2005).
2.3. Effects of Nic311 on serum and brain nicotine levels
2.3.1. Protocol
Animals were anesthetized with isoflurane and an Alzet 2ML2 osmotic minipump (Durect Cupertino, CA) to deliver nicotine 3.2 mg/kg/day (LeSage et al., 2002). After two days of nicotine exposure (by which time nicotine concentrations had reached steady-state), rats were anesthetized with intramuscular (i.m.) droperidol 2.0 mg/kg and fentanyl 0.04 mg/kg. A temporary femoral cannula was implanted to deliver Nic311 or Control IgG i.v. and then removed. To characterize the effects of Nic311 over the time period corresponding to reward threshold measurement (see below), brain and blood samples were collected 15 or 60 minutes after infusion of Nic311 80 mg/kg (n = 4/time-point) or Control IgG (n = 6/time point). To characterize the dose-response relationship, brain and blood samples were collected 60 minutes after infusion of Nic311 at doses of 30, 80, or 240 mg/kg (n = 4/dose). This time-point was used because the greatest effect on brain nicotine concentrations was observed 60 minutes following infusion of Nic311. Group size was smaller for rats administered Nic311 than for controls due to the limited availability and expense of producing Nic311.
2.3.2. Nicotine assay
Serum and brain nicotine levels were measured by gas chromatography with nitrogen-phosphorous detection (Jacob et al., 1981). Brain nicotine concentrations were corrected for brain blood content (Hieda et al., 1999).
2.3.3. Pharmacokinetic calculations
The total body burden of nicotine during the nicotine infusion at the time treatments were administered was calculated as the product of the steady state concentration of nicotine (52 ± 13 ng/ml as measured on day 2 of the nicotine infusion in the control IgG group) and the steady-state volume of distribution of nicotine as previously reported in this strain of rat (3.5 L/kg) (Keyler et al., 2005). The molar dose of Nic311 was calculated based on a molecular weight of 150 000 kD for Nic311 and 2 nicotine-binding sites per molecule of Nic311. The amount of nicotine in serum after administration of treatments was calculated as the serum nicotine concentration × serum volume, where serum volume is the product of the blood volume (64 ml/kg) and [1- the estimated hematocrit (0.45)] = 35.2 ml/kg.
2.4. Withdrawal assessment
2.4.1. Equipment
Intracranial self-stimulation (ICSS) training and testing occurred in operant conditioning chambers (29 cm × 26 cm × 33 cm high) (Med Associates, St. Albans, VT) placed inside sound-attenuated cubicles. A 5-cm wide metal wheel manipulandum was fixed to the front wall. Brain stimulation was administered with constant-current stimulators (Model #PHM-152, Med-Associates). Rats were connected to the stimulation circuit through bipolar leads (Plastics One, Roanoke, VA) attached to gold-contact swivel commutators (Plastics One). MED-PC IV software was used to control stimulation parameters and for data collection.
2.4.2. Stereotaxic surgery
Animals were anesthetized with i.m. ketamine (75 mg/kg) and xylazine (7.5 mg/kg) and implanted with a bipolar stainless steel electrode (Model MS303/2: Plastics One) in the medial forebrain bundle at the level of the lateral hypothalamus [AP -0.5 and ML ± 1.7 mm from bregma, DV -8.3 mm from dura with the incisor bar set 5 mm above the inter-aural line (Pellegrino, 1979)]. The side of the brain in which the electrode was placed was alternated across subjects. Animals were allowed to recover for at least one week prior to ICSS training. During the first two days of recovery all animals received injections of the antibiotic enrofloxacin, 1.1 mg i.m.
2.4.3. Intracranial self-stimulation (ICSS) procedure
Rats were trained on a modified version of the Kornetsky and Esposito (1979) discrete-trial current-threshold procedure as described previously (see Markou and Koob, 1992). Each trial was initiated with presentation of a non-contingent stimulus (0.1 ms cathodal squarewave pulses at a frequency of 100 Hz for 500 ms) followed by a 7.5-sec window during which a positive response on the wheel manipulandum produced a second, contingent stimulation identical to the first. Lack of responding in the 7.5-second time window was considered a negative response. Each positive or negative response was followed by a variable inter-trial interval averaging 10 sec (range = 7.5 to 12.5 sec), during which time additional responses delayed onset of the subsequent trial by 12.5 sec. Stimulus intensities were presented in four alternating descending and ascending series (step size = 5 μA), with five trials presented at each current intensity step. The current threshold for each series was defined as the midpoint between two consecutive current intensity steps that yielded three or more positive responses and two consecutive current intensity steps that yielded three or more negative responses. The overall threshold for the approximately 45 min session was defined as the mean of the current thresholds from the four alternating series. To assess performance effects (e.g., motor disruption), response latencies (time between onset of the non-contingent stimulus and a positive response) were averaged across all trials in which a positive response was made (Markou and Koob, 1992).
2.4.4. Assessment of somatic signs
Rats were habituated to clear plastic circular chambers for 10 minutes on each of 2 days prior to withdrawal testing. An experimentally blind, trained observer recorded behavior for 10 minutes as described by (Malin et al., 1992). Behavioral categories included gasps/abdominal writhes, teeth chatter/chews, shakes/tremors, ptosis, and other miscellaneous less frequent signs (e.g., scratches, yawns). Multiple successive counts of any sign required a distinct pause between signs. If continuously present, ptosis was only counted once/minute and teeth chattering only once every 15 seconds.
2.4.5 Protocol
Rats were implanted with intra-cranial electrodes and trained for ICSS as described above for at least 15 sessions and until thresholds were stable (i.e., no more than 10% variation over a 5-day period). A chronic in-dwelling jugular cannula was then implanted for antibody delivery as described previously (LeSage et al., 2002). At least 7 days later, rats resumed ICSS threshold assessment, and after ICSS thresholds again stabilized, osmotic minipumps (2ML4) were implanted to deliver a continuous nicotine infusion of 3.2 mg/kg/day. This infusion rate has been shown to reliably induce nicotine dependence as measured using elevated ICSS thresholds and somatic signs following pump removal (spontaneous withdrawal) or administration of a nicotinic antagonist (precipitated withdrawal) (Epping-Jordan et al., 1998). Rats continued to be tested for ICSS during nicotine exposure. After 1–3 weeks of nicotine exposure, rats were assessed for precipitated withdrawal. Duration of nicotine exposure depended on stability of thresholds and was counter-balanced between experimental groups, so that each group had the same average duration of nicotine exposure prior to assessment of precipitated withdrawal.
During assessment of precipitated withdrawal, rats were exposed to one of the following treatments: Control IgG 80 mg/kg i.v. + mecamylamine 1.5 mg/kg s.c. (positive control, n = 6); Control IgG 80 mg/kg i.v. + saline s.c. (negative control, n = 6); Nic311 80 mg/kg i.v. + saline s.c. (n = 6); Nic311 240 mg/kg i.v. + saline s.c. (n = 4). Mecamylamine or saline was administered s.c. immediately after antibody administration. It is well-established that this dose of mecamylamine precipitates robust increases in ICSS thresholds and somatic signs when administered to dependent rats while having no effect on these measures in drug-naïve rats (Watkins et al. 2000; Markou and Paterson 2001; O’Dell et al. 2006; Bruijnzeel et al. 2007). Therefore, the effects of Control IgG+mecamylamine in rats chronically infused with saline were not examined. Group size for rats administered Nic311 240 mg/kg was 4 rather than 6 due to its limited availability and expense.
ICSS thresholds were assessed 0.25, 3, 24, 48 and 72 hours after antibody administration. The s.c. nicotine infusion continued throughout testing. Somatic signs were assessed following the first three ICSS sessions (approximately 1, 4, and 25 hours after infusion).
2.5. Statistical analyses
2.5.1. Effects of Nic311 on serum and brain nicotine concentrations
Mean serum and brain nicotine concentrations following administration of Nic311 (80 mg/kg) or Control IgG were analyzed by two-factor ANOVA with time and treatment as factors, followed by Bonferroni’s post test to compare groups at each time point. Mean serum and brain nicotine concentrations and serum concentrations of the nicotine metabolite cotinine at 60 minutes following administration of Nic311 30, 80, or 240 mg/kg or Control IgG were analyzed by one-way ANOVA followed by Bonferroni’s post-test to compare groups where appropriate.
2.5.2. Withdrawal assessment
Baseline ICSS thresholds were defined as the mean thresholds of the last 5 sessions during nicotine exposure prior to assessment of withdrawal. ICSS thresholds prior to osmotic pump implantation were compared to baseline as described above via t-test to confirm that ICSS thresholds were not altered by nicotine and the defined baseline was appropriate. Baseline thresholds were compared between groups using a one-way ANOVA. To compare differences between groups for the first 24 hours after antibody treatment, ICSS thresholds (expressed as % of baseline) were analyzed by repeated-measures two-factor ANOVA with treatment and session as factors followed by Bonferroni’s post-test for between group comparisons at each test session. ICSS thresholds across sessions for each group were also compared to baseline using a one-way ANOVA followed by Dunnett’s post hoc tests. Further, since the pharmacokinetic data indicated a more substantial reduction in brain nicotine concentrations at 60 minutes compared to 15 minutes, it was possible that averaging across the entire 45 min ICSS session would obscure drug effects. To address this possibility, within-session thresholds of the first ICSS session (15–60 minutes after drug administration) were assessed by t-test to compare the mean threshold from the first two ascending and descending current series (obtained approximately from 0 to 25 minutes of the session) with the mean thresholds from the last two ascending and descending current series (obtained approximately from 25–45 minutes of the session) in each group. Response latencies (before and up to 24 hr after drug infusion) were also analyzed by two-factor ANOVA to check for nonspecific effects. To assess differences in somatic signs between groups, a repeated-measures two-factor ANOVA was used with treatment and session as factors followed by Bonferroni’s post-test.
3. Results
3.1. Effects of Nic311 on serum and brain nicotine concentrations
3.1.1. Time course
There was a a significant effect of treatment (F(1, 12) = 182.2, p < 0.0001) and time (F(1, 12) = 18.4, p = 0.001) on serum nicotine concentration and a significant interaction (F(1, 12) = 17.4, p = 0.001). As shown in Figure 1, Nic311 80 mg/kg significantly increased serum concentrations at both 15 and 60 minutes following antibody infusion (Figure 1, p < 0.001). There was a significant effect of treatment (F(1, 16) = 33.6, p < 0.001) and time (F(1, 16) = 12.94, p < 0.001) on brain nicotine concentrations, but no significant interaction. Nic311 80 mg/kg significantly reduced brain nicotine levels compared to Control IgG at 15 minutes (61% that of controls, p < 0.05) and 1 hour (17% that of controls, p < 0.001) post-infusion.
Fig. 1.

Serum and brain nicotine concentrations (mean ± SD) 15 and 60 min after infusion of Nic311 80 mg/kg. *, ***; p < 0.05, 0.001 compared to control at each time point; (%) indicates brain nicotine levels as percent of control.
3.1.2. Dose response
There was a significant relationship between Nic311 dose and serum nicotine concentrations 60 minutes post-infusion (F(3, 17) = 63.6, p < 0.0001). As shown in Figure 2, Nic311 30 and 80 mg/kg resulted in significantly higher nicotine serum concentrations compared to Control IgG (p < 0.01), and Nic311 240 mg/kg resulted in significantly higher nicotine serum concentrations than all other groups (p < 0.0001). Serum cotinine levels were not significantly different among groups 60 minutes following antibody infusion (control 546 ± 159 ng/ml, Nic311 30 mg/kg 458 ± 134 ng/ml, Nic311 80 mg/kg 451 ± 79 ng/ml, Nic311 240 mg/kg 749 ± 301 ng/ml, p = 0.12), confirming that there was no appreciable binding of cotinine to Nic311 in vivo that could alter nicotine binding. There was a significant relationship between Nic311 dose and brain nicotine concentrations (F(3, 20) = 15.0, p < 0.0001). The Nic311 doses of 30, 80, and 240 mg/kg reduced brain nicotine concentrations to 55%, 17%, and 8% of controls (p < 0.05, p < 0.01, p < 0.001, respectively).
Fig. 2.

Serum and brain nicotine concentrations (mean ± SD) measured 60 minutes post-antibody infusion. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared to Control; ###, p < 0.001 compared to all other groups; (%) indicates brain nicotine levels as percent of control.
3.1.3. Pharmacokinetic calculations
The estimated amount of nicotine in the body at the time treatments were administered was 0.182 mg/kg = 1.12 μmol/kg. The molar doses of Nic311 were 0.4 μmol/kg (for the Nic311 30 mg/kg dose), 1.07 μmol/kg (Nic311 80 mg/kg) and 3.20 μmol/kg (Nic311 240 mg/kg). The molar ratio of Nic311 to amount of nicotine in the body were 0.34 (Nic311 30 mg/kg), 0.96 (Nic311 80 mg/kg) and 2.86 (Nic311 240 mg/kg). The total amount of nicotine in serum after treatment was 0.001 mg/kg (Control), 0.033 mg/kg (Nic311 30 mg/kg), 0.041 mg/kg (Nic311 80 mg/kg) and 0.159 mg/kg (Nic311 240 mg/kg). These amounts of nicotine in serum represented 0.6%, 18%, 23% and 87% of the total estimated amount of nicotine in the body at the time of treatment for the 0 (control) 30, 80 and 240 mg/kg Nic311 doses.
3.2. Withdrawal assessment
3.2.1. Baseline measures
Baseline thresholds during nicotine exposure did not differ from thresholds during the last 5 sessions prior to nicotine pump implantation, indicating that nicotine did not alter ICSS thresholds (data not shown). Baseline thresholds prior to precipitated withdrawal did not differ among groups (Table 1).
Table 1.
Baseline thresholds and response latencies, and response latencies during the first 24 hr post-antibody infusion, for the four experimental groups.
| Group | Baseline Threshold (μA) | Baseline Latency (sec) | Latency (sec) after treatment |
||
|---|---|---|---|---|---|
| 15 min | 3 hour | 24 hour | |||
| Control IgG + Saline | 121 ± 14 | 3.4 ± 0.2 | 3.4 ± 0.1 | 3.5 ± 0.2 | 3.5 ± 0.2 |
| Control IgG + Mecamylamine | 110 ± 16 | 2.7 ± 0.2 | 2.7 ± 0.7 | 3.3 ± 0.4 | 2.8 ± 0.3 |
| Nic311 80 mg/kg + Saline | 113 ± 15 | 2.7 ± 0.2 | 2.8 ± 0.2 | 2.6 ± 0.2 | 2.8 ± 0.2 |
| Nic311 240 mg/kg + Saline | 95 ± 12 | 2.5 ± 0.4 | 2.6 ± 0.3 | 2.5 ± 0.5 | 2.5 ± 0.2 |
3.2.2. ICSS Thresholds
Mecamylamine significantly increased ICSS thresholds whereas Nic311 did not (Figure 3, top). There was a significant overall effect of treatment (F(3, 54) = 4.97, p = 0.011) on ICSS thresholds but no significant effect of time and no significant interaction. ICSS thresholds in rats administered Nic311 80 mg/kg + saline did not differ from those administered Control IgG + saline at any time. Nic311 240 mg/kg + saline resulted in a slight increase in ICSS thresholds (mean 110%, range 109–112%) compared to baseline but this was not significantly different from Control IgG+saline (p = 0.3). Control IgG + mecamylamine resulted in significantly higher ICSS thresholds compared to Control IgG + saline or Nic311 80 mg/kg + saline (p < 0.01) 15 minutes after administration but did not differ from Nic311 240 mg/kg + saline at any time. Drug and antibody administration did not alter response latencies for up to 24 hr post-infusion (Table 1).
Fig. 3.
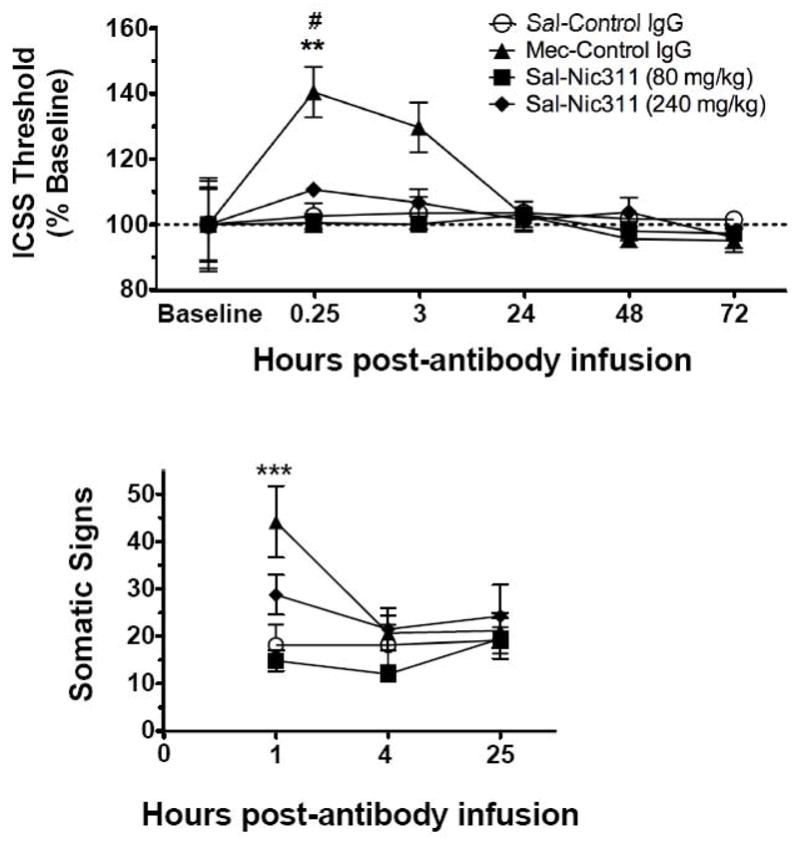
(Top) ICSS thresholds (as percent of baseline, mean ± SE) following administration of either i.v. Control IgG or Nic311, and s.c. mecamylamine or saline. **, p < 0.01 compared to Saline and Nic311 80mg/kg at 0.25 hours; # p < 0.05 compared to baseline. (Bottom) Somatic withdrawal signs observed during the first 25 hr post-antibody infusion. ***, p < 0.001 compared to Control IgG + saline and Nic311 80 mg/kg + saline.
Analysis of within-group data indicated that only Control IgG + mecamylamine at 15 min after drug administration significantly elevated thresholds compared to baseline (F(3, 20) = 4.696, p = 0.012). Analysis of within-session data from the 15 min test showed stability of ICSS thresholds within sessions: there were no differences between the mean thresholds from the first two ascending and descending current series (obtained from approximately 0 to 25 minutes of the session) compared to the mean threshold of the last two ascending and descending current series (obtained from approximately 25–45 minutes of the session) for all groups. This indicates that any potential effects of Nic311 on ICSS thresholds late in the session (i.e., when brain nicotine levels were lowest, see Figure 1) were not obscured by collapsing the data across all four current series.
3.2.3. Somatic signs
Mecamylamine significantly increased somatic signs of withdrawal compared to Nic311 80 mg/kg or Control IgG (Figure 3, bottom). There was a significant overall effect of treatment (F(3, 36) = 4.12, p = 0.022) and time (F(2, 36) = 4.89, p = 0.013) on somatic withdrawal signs, and a significant interaction (F(6, 36) = 3.42, p = 0.009). Control IgG + mecamylamine significantly increased somatic withdrawal signs 1 hour after injection compared to Control IgG + saline or Nic311 80 mg/kg+saline (p < 0.001) but not compared to Nic311 240 mg/kg + saline. Somatic withdrawal signs 1 hr after Nic311 240 mg/kg + saline were slightly elevated but were not significantly different from Control IgG + saline or Nic311 80 mg/kg + saline. There were no differences in somatic withdrawal signs among groups at the remaining time points.
4. Discussion
Previous studies have shown that Nic311 reduces or slows nicotine distribution into brain if administered prior to nicotine administration (Keyler et al., 2005; Pentel et al., 2006). The current study found that Nic311 also rapidly redistributed nicotine out of the brain when administered during an ongoing nicotine infusion. The 80 mg/kg dose of Nic311, which was approximately equimolar to the body burden of nicotine, reduced the brain nicotine concentration by 83%, and the 240 mg/kg dose, a 3-fold molar excess, reduced the brain nicotine concentration by 92% at 60 minutes post-antibody infusion. These large reductions are consistent with previous studies of the effects of passive immunization on brain concentrations of phencyclidine (PCP) (Proksch et al., 2000), methamphetamine (Byrnes-Blake et al., 2003), and desipramine (Pentel et al., 1987) at similar antibody:drug molar ratios. The greater reduction of brain nicotine concentrations at 60 minutes compared to 15 minutes following the 80 mg/kg dose (see Fig 1) was unexpected since maximal effects have occurred sooner with similar dosing of drug-specific antibodies for PCP (Proksch et al., 2000). More detailed study with additional time points would be of interest.
The corresponding serum nicotine concentrations were greatly increased after Nic311 administration. These very high serum nicotine levels are attributable to the high binding capacity of the administered Nic311 and to sampling shortly after Nic311 dosing when a large fraction of the administered antibody would have been present in serum (Keyler et al., 2005). Comparably high serum nicotine concentrations were reported previously when the same Nic311 dose was administered prior to nicotine dosing. The high serum nicotine concentrations resulting from immunization are not toxic because most of the nicotine in serum is bound to antibody and is not pharmacologically active (Keyler et al., 2005). Nicotine serum levels may have been lower at 60 minutes compared to 15 minutes post-antibody infusion (see Fig 1) due to the greater distribution of Nic311 from serum at the later time-point (see Roiko et al., 2008). This would presumably result in the distribution of both antibody-bound and unbound nicotine to other tissues.
This study is the first to evaluate the potential for passive immunization to precipitate nicotine withdrawal, but vaccination has been previously studied in this regard. Vaccination of nicotine-dependent rats with a nicotine conjugate vaccine during continuous nicotine infusion did not increase ICSS thresholds or somatic withdrawal signs (Lindblom et al., 2005). In addition, a Phase I study of a nicotine vaccine did not report any signs or symptoms of nicotine withdrawal in smokers vaccinated against nicotine (Hatsukami et al., 2005). This is most likely attributable to vaccination producing a very gradual increase in serum NicAb concentration over weeks to months so that any resulting changes in brain nicotine concentration also occur gradually. The much more rapid increase in serum NicAb concentration produced by passive immunization would be expected to have a greater potential for producing withdrawal.
In the current study the rapid and substantial redistribution of nicotine out of brain produced by Nic311 did not precipitate nicotine withdrawal whereas the nicotinic acetylcholine receptor (nAChR) antagonist mecamylamine produced robust withdrawal as measured by both brain reward thresholds and somatic signs. The Nic311 dose of 80 mg/kg was studied because it produces substantial effects on nicotine pharmacokinetics and attenuates nicotine-induced locomotor sensitization in rats (Roiko et al., 2008). Nic311 80 mg/kg also produces serum NicAb concentrations comparable to those elicited by vaccination of rats against nicotine and higher than those produced by vaccination in clinical trials (Hatsukami et al., 2005). This Nic311 dose is therefore in the range that might be considered for clinical use. Nic311 80 mg/kg produced no measurable withdrawal, and 240 mg/kg produced only a nonsignificant trend toward increases in ICSS thresholds and somatic signs. It is possible that the small effect of the highest dose would be significant if evaluated using larger groups, but the magnitude of change in ICSS thresholds and somatic signs was quite small compared to that of mecamylamine. These findings imply that the therapeutically desirable effects of Nic311 in rats as modeled by attenuation of locomotor sensitization (Roiko et al., 2008) occur at Nic311 doses that do not precipitate withdrawal. Similarly, vaccination against nicotine blocked the ICSS threshold-reducing (i.e., rewarding) effects of an acute nicotine injection but did not precipitate withdrawal in nicotine-dependent rats (Lindblom et al., 2005). These findings suggest that either active or passive immunization against nicotine should effectively attenuate nicotine’s acute effects in current smokers without precipitating a withdrawal syndrome.
The rate and extent by which nicotine must be removed from the brain to precipitate withdrawal is not well established. Previously, the only means of initiating nicotine withdrawal in nicotine dependent animals were to abruptly discontinue chronic nicotine dosing or to administer an nAChR antagonist. Only limited data are available on the early time course of spontaneous nicotine withdrawal following termination of a nicotine infusion. One study reported that the removal of a nicotine osmotic minipump resulted in the onset of withdrawal in rats 2.5–4 hours and peak withdrawal severity at 6–8 hours, as measured by ICSS and somatic signs (Epping-Jordan et al., 1998). Although serum and brain nicotine concentrations were not measured in this study, the elimination half-life for nicotine in serum or brain in rats is ~1 hour (Ghosheh et al., 1999; Keyler et al., 2005). As an approximation, this suggests that withdrawal after nicotine pump removal was first detected when 82– 94% of nicotine had been eliminated (2.5–4 nicotine half-lives), and peak severity occurred when >99% had been eliminated. Similarly, in humans the onset of tobacco withdrawal has been reported 3–6 hours after cessation of smoking (an estimated 1.5–3 nicotine elimination half-lives in humans) and peak withdrawal at 10–12 hours (5–6 half-lives) (Hendricks et al., 2006; Parrott et al., 1996). Based on these estimates, it is quite surprising that the rapid removal of 83±7% or 92±8% of nicotine from brain after the 80 or 240 mg/kg doses of Nic311, respectively, did not precipitate nicotine withdrawal.
The reasons for this unexpected absence of withdrawal after Nic311 treatment are unclear. Only one nicotine infusion rate was studied to induce dependence, but this nicotine infusion rate is used widely by others and results in robust withdrawal in a variety of strains of rats when terminated (Epping-Jordan et al., 1998; Malin et al., 1992). It is possible that the development of withdrawal requires a delay between the offset of nicotine signaling and the manifestation of behavioral signs. However, consistent with previous reports (Bruijnzeel et al., 2007; Watkins et al., 2000), the administration of mecamylamine produced robust withdrawal within 15 minutes of its administration. It is possible that Nic311 primarily removed nicotine from brain that was not specifically bound to nAChRs, resulting in higher nAChR occupancy than predicted from whole brain levels. Human imaging studies suggest that occupancy of nAChRs can be substantial even under conditions in which total brain levels are very low (Brody et al. 2006; Brody et al. 2008). For example, Brody et al. (2006) reported that only three cigarette puffs produced 75% occupancy of α4β2* nAChRs, a high affinity nAChR subtype implicated in withdrawal (Epping-Jordan et al., 1998; Rahman et al., 2008; Patterson et al., 2009). In the current study, the nicotine remaining in brain after Nic311 administration could have produced sufficient occupancy of α4β2* or other nAChRs to prevent the expression of withdrawal. This would be consistent with high affinity brain nAChRs having a Kd for nicotine of ~ 2 nM (< 1 ng/ml) (Lippiello and Fernandes, 1986).
Another potential contributor to the lack of withdrawal is that the nicotine infusion continued after Nic311 was administered to simulate continued smoking. Pretreatment with Nic311 decreases the early distribution of nicotine to brain but not its chronic accumulation (Pentel et al. 2006). Although not measured in the current study, brain nicotine levels may have increased after the 60 min measurement and mitigated the subsequent manifestation of withdrawal.
A limitation of this study is that it did not include a positive control group assessed for spontaneous withdrawal. Antagonist-precipitated withdrawal was used because it involves an abrupt reduction of nicotine signaling that may better simulate withdrawal induced by the rapid redistribution of nicotine from brain. In addition, it is difficult to predict the precise time point during spontaneous withdrawal that would be associated with reductions in brain nicotine levels similar to those produced by Nic311. Addressing this issue would require a systematic comparison of concurrent changes in ICSS thresholds and brain nicotine levels at numerous time points following cessation of a nicotine infusion or passive immunization, which was beyond the scope of this initial study.
One potential use for passive immunization is in combination with vaccination, to supplement vaccination and ensure that desired antibody levels are achieved, or to provide a more rapid onset of effect. In support of this possibility, combining vaccination with passive immunization results in lower brain nicotine levels in rats and a greater attenuation of nicotine-induced locomotor sensitization than either treatment alone (Roiko et al., 2008). If passive immunization is used concurrently with vaccination, monoclonal antibody might be administered to patients who are still smoking. This study suggests that the precipitation of withdrawal by passive immunization under these circumstances is unlikely.
Acknowledgments
Supported by NIDA grants DA10714, F31-DA021946, F32-DA021935, T32-DA07097, and P50-DA013333 and the Minneapolis Medical Research Foundation Translational Addiction Research Program. No conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Benowitz NL. Pharmacokinetic considerations in understanding nicotine dependence. Ciba Found Symp. 1990;152:186–200. doi: 10.1002/9780470513965.ch11. discussion 200–289. [DOI] [PubMed] [Google Scholar]
- Benowitz NL. Pharmacology of nicotine: addiction and therapeutics. Annu Rev Pharmacol Toxicol. 1996;36:597–613. doi: 10.1146/annurev.pa.36.040196.003121. [DOI] [PubMed] [Google Scholar]
- Brody AL, Mandelkern MA, London ED, Olmstead RE, Farahi J, Scheibal D, Jou J, Allen V, Tiongson E, Chefer SI, Koren AO, Mukhin AG. Cigarette smoking saturates brain alpha 4 beta 2 nicotinic acetylcholine receptors. Arch Gen Psychiatry. 2006;63:907–15. doi: 10.1001/archpsyc.63.8.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody AL, Mandelkern MA, Costello MR, Abrams AL, Scheibal D, Farahi J, London ED, Olmstead RE, Rose JE, Mukhin AG. Brain nicotinic acetylcholine receptor occupancy: effect of smoking a denicotinized cigarette. Int J Neuropsychopharmacol. 2008 Aug 18;:1–12. doi: 10.1017/S146114570800922X. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijnzeel AW, Zislis G, Wilson C, Gold MS. Antagonism of CRF receptors prevents the deficit in brain reward function associated with precipitated nicotine withdrawal in rats. Neuropsychopharmacology. 2007;32:955–963. doi: 10.1038/sj.npp.1301192. [DOI] [PubMed] [Google Scholar]
- Byrnes-Blake KA, Laurenzana EM, Carroll FI, Abraham P, Gentry WB, Landes RD, Owens SM. Pharmacodynamic mechanisms of monoclonal antibody-based antagonism of (+)-methamphetamine in rats. Eur J Pharmacol. 2003;461:119–128. doi: 10.1016/s0014-2999(03)01313-x. [DOI] [PubMed] [Google Scholar]
- Carrera MR, Ashley JA, Hoffman TZ, Isomura S, Wirsching P, Koob GF, Janda KD. Investigations using immunization to attenuate the psychoactive effects of nicotine. Bioorg Med Chem. 2004;12:563–570. doi: 10.1016/j.bmc.2003.11.029. [DOI] [PubMed] [Google Scholar]
- Cornuz J, Zwahlen S, Jungi WF, Osterwalder J, Klingler K, van Melle G, Bangala Y, Guessous I, Muller P, Willers J, Maurer P, Bachmann MF, Cerny T. A vaccine against nicotine for smoking cessation: a randomized controlled trial. PLoS ONE. 2008;3:e2547. doi: 10.1371/journal.pone.0002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villiers SH, Lindblom N, Kalayanov G, Gordon S, Johansson AM, Svensson TH. Active immunization against nicotine alters the distribution of nicotine but not the metabolism to cotinine in the rat. Naunyn Schmiedebergs Arch Pharmacol. 2004;370:299–304. doi: 10.1007/s00210-004-0960-3. [DOI] [PubMed] [Google Scholar]
- Drucker A, Skedgel C, Virik K, Rayson D, Sellon M, Younis T. The cost burden of trastuzumab and bevacizumab therapy for solid tumours in Canada. Current oncology. 2008;15:136–142. doi: 10.3747/co.v15i3.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epping-Jordan MP, Watkins SS, Koob GF, Markou A. Dramatic decreases in brain reward function during nicotine withdrawal. Nature. 1998;393:76–79. doi: 10.1038/30001. [DOI] [PubMed] [Google Scholar]
- Ghosheh O, Dwoskin LP, Li WK, Crooks PA. Residence times and half-lives of nicotine metabolites in rat brain after acute peripheral administration of [2′-(14)C]nicotine. Drug Metab Dispos. 1999;27:1448–1455. [PubMed] [Google Scholar]
- Hatsukami DK, Rennard S, Jorenby D, Fiore M, Koopmeiners J, de Vos A, Horwith G, Pentel PR. Safety and immunogenicity of a nicotine conjugate vaccine in current smokers. Clin Pharmacol Ther. 2005;78:456–467. doi: 10.1016/j.clpt.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Hendricks PS, Ditre JW, Drobes DJ, Brandon TH. The early time course of smoking withdrawal effects. Psychopharmacology (Berl) 2006;187:385–396. doi: 10.1007/s00213-006-0429-9. [DOI] [PubMed] [Google Scholar]
- Henningfield JE, Keenan RM. Nicotine delivery kinetics and abuse liability. J Consult Clin Psychol. 1993;61:743–750. doi: 10.1037//0022-006x.61.5.743. [DOI] [PubMed] [Google Scholar]
- Hieda Y, Keyler DE, VanDeVoort JT, Niedbala RS, Raphael DE, Ross CA, Pentel PR. Immunization of rats reduces nicotine distribution to brain. Psychopharmacology (Berl) 1999;143:150–157. doi: 10.1007/s002130050930. [DOI] [PubMed] [Google Scholar]
- Hukkanen J, Jacob P, 3rd, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57:79–115. doi: 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- Jacob P, 3rd, Wilson M, Benowitz NL. Improved gas chromatographic method for the determination of nicotine and cotinine in biologic fluids. J Chromatogr. 1981;222:61–70. doi: 10.1016/s0378-4347(00)81033-6. [DOI] [PubMed] [Google Scholar]
- Keyler DE, Roiko SA, Benlhabib E, LeSage MG, St Peter JV, Stewart S, Fuller S, Le CT, Pentel PR. Monoclonal nicotine-specific antibodies reduce nicotine distribution to brain in rats: dose- and affinity-response relationships. Drug Metab Dispos. 2005;33:1056–1061. doi: 10.1124/dmd.105.004234. [DOI] [PubMed] [Google Scholar]
- Kornetsky C, Esposito RU. Euphorigenic drugs: effects on the reward pathways of the brain. Fed Proc. 1979;38:2473–6. [PubMed] [Google Scholar]
- LeSage MG, Keyler DE, Hieda Y, Collins G, Burroughs D, Le C, Pentel PR. Effects of a nicotine conjugate vaccine on the acquisition and maintenance of nicotine self-administration in rats. Psychopharmacology (Berl) 2006a;184:409–416. doi: 10.1007/s00213-005-0027-2. [DOI] [PubMed] [Google Scholar]
- LeSage MG, Keyler DE, Pentel PR. Current status of immunologic approaches to treating tobacco dependence: vaccines and nicotine-specific antibodies. Aaps J. 2006b;8:E65–75. doi: 10.1208/aapsj080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeSage MG, Keyler DE, Shoeman D, Raphael D, Collins G, Pentel PR. Continuous nicotine infusion reduces nicotine self-administration in rats with 23-h/day access to nicotine. Pharmacol Biochem Behav. 2002;72:279–289. doi: 10.1016/s0091-3057(01)00775-4. [DOI] [PubMed] [Google Scholar]
- Lindblom N, de Villiers SH, Semenova S, Kalayanov G, Gordon S, Schilstrom B, Johansson AM, Markou A, Svensson TH. Active immunisation against nicotine blocks the reward facilitating effects of nicotine and partially prevents nicotine withdrawal in the rat as measured by dopamine output in the nucleus accumbens, brain reward thresholds and somatic signs. Naunyn Schmiedebergs Arch Pharmacol. 2005;372:182–194. doi: 10.1007/s00210-005-0019-0. [DOI] [PubMed] [Google Scholar]
- Lippiello PM, Fernandes KG. The binding of L-[3H]nicotine to a single class of high affinity sites in rat brain membranes. Mol Pharmacol. 1986;29:448–54. [PubMed] [Google Scholar]
- Malin DH, Lake JR, Newlin-Maultsby P, Roberts LK, Lanier JG, Carter VA, Cunningham JS, Wilson OB. Rodent model of nicotine abstinence syndrome. Pharmacol Biochem Behav. 1992;43:779–784. doi: 10.1016/0091-3057(92)90408-8. [DOI] [PubMed] [Google Scholar]
- Markou A, Koob GF. Construct validity of a self-stimulation threshold paradigm: effects of reward and performance manipulations. Physiol Behav. 1992;51:111–119. doi: 10.1016/0031-9384(92)90211-j. [DOI] [PubMed] [Google Scholar]
- Markou A, Paterson NE. The nicotinic antagonist methyllycaconitine has differential effects on nicotine self-administration and nicotine withdrawal in the rat. Nicotine Tob Res. 2001;3:361–73. doi: 10.1080/14622200110073380. [DOI] [PubMed] [Google Scholar]
- McLeod C, Bagust A, Boland A, Dagenais P, Dickson R, Dundar Y, Hill RA, Jones A, Mujica Mota R, Walley T. Adalimumab, etanercept and infliximab for the treatment of ankylosing spondylitis: a systematic review and economic evaluation. Health technology assessment. 2007;11:1–158. iii–iv. doi: 10.3310/hta11280. [DOI] [PubMed] [Google Scholar]
- Norman AB, Tabet MR, Norman MK, Buesing WR, Pesce AJ, Ball WJ. A chimeric human/murine anticocaine monoclonal antibody inhibits the distribution of cocaine to the brain in mice. J Pharmacol Exp Ther. 2007;320:145–153. doi: 10.1124/jpet.106.111781. [DOI] [PubMed] [Google Scholar]
- O’Dell LE, Bruijnzeel AW, Smith RT, Parsons LH, Merves ML, Goldberger BA, Richardson HN, Koob GF, Markou A. Diminished nicotine withdrawal in adolescent rats: implications for vulnerability to addiction. Psychopharmacology (Berl) 2006;186:612–9. doi: 10.1007/s00213-006-0383-6. [DOI] [PubMed] [Google Scholar]
- Parrott AC, Garnham NJ, Wesnes K, Pincock C. Cigarette smoking and abstinence: comparative effects upon cognitive task performance and mood state over 24 hours. Human Psychopharmacology. 1996;11:391 – 400. [Google Scholar]
- Patterson F, Jepson C, Strasser AA, Loughead J, Perkins KA, Gur RC, Frey JM, Siegel S, Lerman C. Varenicline improves mood and cognition during smoking abstinence. Biol Psychiatry. 2008;65:144–9. doi: 10.1016/j.biopsych.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino LP, Pellegrino AS, Cushman AJ. A stereotaxic atlas of the rat brain. New York: Plenum Press; 1979. [Google Scholar]
- Pentel P, Pond SM, Schoof D. Redistribution into plasma of tracer doses of desipramine by anti-desipramine antiserum in rats. Biochem Pharmacol. 1987;36:293–295. doi: 10.1016/0006-2952(87)90703-9. [DOI] [PubMed] [Google Scholar]
- Pentel PR, Dufek MB, Roiko SA, Lesage MG, Keyler DE. Differential effects of passive immunization with nicotine-specific antibodies on the acute and chronic distribution of nicotine to brain in rats. J Pharmacol Exp Ther. 2006;317:660–666. doi: 10.1124/jpet.105.097873. [DOI] [PubMed] [Google Scholar]
- Proksch JW, Gentry WB, Owens SM. Anti-phencyclidine monoclonal antibodies provide long-term reductions in brain phencyclidine concentrations during chronic phencyclidine administration in rats. J Pharmacol Exp Ther. 2000;292:831–837. [PubMed] [Google Scholar]
- Rahman S, Lopez-Hernandez GY, Corrigall WA, Papke RL. Neuronal nicotinic receptors as brain targets for pharmacotherapy of drug addiction. CNS Neurol Disord Drug Targets. 2008;7:422–41. doi: 10.2174/187152708786927831. [DOI] [PubMed] [Google Scholar]
- Roiko SA, Harris AC, Keyler DE, Lesage MG, Zhang Y, Pentel PR. Combined active and passive immunization enhances the efficacy of immunotherapy against nicotine in rats. J Pharmacol Exp Ther. 2008;325:985–993. doi: 10.1124/jpet.107.135111. [DOI] [PubMed] [Google Scholar]
- Watkins SS, Stinus L, Koob GF, Markou A. Reward and somatic changes during precipitated nicotine withdrawal in rats: centrally and peripherally mediated effects. J Pharmacol Exp Ther. 2000;292:1053–1064. [PubMed] [Google Scholar]