

Preface
This is the report of the 58th of a series of workshops organised by the European Centre for the Validation of Alternative Methods (ECVAM). The main objective of ECVAM, as defined in 1993 by its Scientific Advisory Committee (ESAC), is to promote the scientific and regulatory acceptance of alternative methods which are of importance to the biosciences, and which reduce, refine or replace the use of laboratory animals. One of the first priorities set by ECVAM was the implementation of procedures that would enable it to become well informed about the state of the art of non-animal test development and validation, and of opportunities for the possible incorporation of alternative methods into regulatory procedures. It was decided that this would be achieved through a programme of ECVAM workshops, each addressing a specific topic, and at which selected groups of independent international experts would review the current status of various types of in vitro tests and their potential uses, and make recommendations about the best ways forward (1).
The workshop was organised by Michael Balls and Valérie Zuang, and took place on 5–7 May 2004, at the Hotel Lido, Angera (VA), Italy, with participants from academia, industry, research, and national and international validation authorities. The aim was to discuss and define principles and criteria for validation via weight-of-evidence approaches, and to provide guidance on the performance of this type of validation. The outcome of the discussions and the recommendations agreed upon by the workshop participants are summarised in this report, which also takes into account some subsequent events and publications.
"Weight of Evidence"
Weight of evidence (WoE) is a phrase used to describe the type of consideration made in a situation where there is uncertainty, and which is used to ascertain whether the evidence or information supporting one side of a cause or argument is greater than that supporting the other side. We all frequently make personal WoE decisions in our daily lives, but more-formal WoE approaches are used in many different kinds of circumstance — for example, in commercial, educational, health, legal and scientific contexts.
WoE is a term which is commonly used in the published policy-making and scientific literature, not least in relation to risk assessment. Weed (2) searched the PubMed service of the US National Library of Medicine for papers published between 1994 and 2004, in which “weight of evidence” appeared in the title and/or the abstract. He concluded, from a review of 92 of 272 such papers, that WoE had three characteristic uses:
metaphorical, where WoE refers to a collection of studies or to an unspecified methodological approach;
methodological, where WoE points to established interpretative methodologies (e.g. systematic narrative review, meta-analysis, causal criteria, and/or quality criteria for toxicological studies), or where WoE means that “all” rather than some subset of the evidence is examined, or rarely, where WoE points to methods using quantitative weights for evidence; and
theoretical, where WoE serves as a label for a conceptual framework.
Weed identified several problems with the use of WoE approaches in risk assessment, including the frequent lack of definition of the term, multiple uses of the term and a lack of consensus about its meaning, and the many different kinds of weights, both qualitative and quantitative, which can be used. Given the central role that the WoE concept plays in risk assessment, he recommended that the many stakeholders involved should “be clear about its definition, its uses and its implications”. Thus, “When we read that a ‘weight of evidence’ approach was taken (a common and often undocumented statement in the literature), what exactly does that mean? What interpretative methods were employed? How were they applied to the available scientific evidence?”
These kinds of questions are of great significance in relation to this report, which considers how a WoE validation procedure (of type 2, above) can be used to evaluate and/or establish the scientific validity and usefulness of test methods and testing strategies for their particular purposes.
Validation and its Importance
The validation process sits between test method and test strategy development and their scientific and regulatory acceptance, and is concerned with the independent evaluation of their reliability and relevance for particular purposes (3, 4). The initial focus in validation was on the performance of alternative test methods as evaluated in dedicated, practical multi-laboratory studies, which usually involved the testing of coded chemicals and the independent analysis of the resulting data (5). The criteria for validation were originally developed by the European Centre for the Validation of Alternative Methods (ECVAM) and the European Chemicals Bureau (ECB) of the European Commission (EC; 6). These critera were subsequently endorsed and mirrored in the procedures of the US Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM; 7) and the Organisation for Economic Cooperation and Development (OECD; 8). It is now widely accepted that validation according to these EC, ICCVAM and/or OECD principles and criteria is a prerequisite for the regulatory acceptance and application of test methods and testing strategies. Without departing from these agreed principles and criteria, ECVAM has recently proposed a modular approach to validation (9), the procedures applied by ICCVAM have been streamlined and summarised (10, 11), and detailed guidance on the validation process, including a consideration of mechanisms for peer reviews and regulatory acceptance, has been published by the OECD (12).
The ECVAM and ICCVAM procedures are illustrated in Appendices 1–2, at the end of this report.
The Need for Weight of Evidence Validation Assessments
As experience was gained in the performance of dedicated, practical validation studies, it became clear that this approach would not always be appropriate, necessary, or even possible, and that a WoE approach to validation would be more appropriate in some situations. For example, there could be existing evidence of sufficient quantity and quality to permit an evaluation of the performance of an alternative method for a particular purpose, without the need for additional practical work. In other circumstances, there might not be in vivo benchmark data of sufficient breadth, quantity and quality to serve as acceptable reference standards for the practical evaluation of an in vitro method. In addition, the test methods and testing strategies of the future are less and less likely to be direct replacements for existing procedures, but will be based on advancements in the basic sciences of pharmacology and toxicology, involving in silico and in vitro systems, molecular biology and biotechnology.
For these reasons, there is increasing interest in the performance, not only of practical validation studies (i.e. those involving new and dedicated laboratory work), but also WoE validation assessments (i.e. those involving the collection, analysis and weighing of evidence, without any additional dedicated practical studies). This latter type of validation has been referred to as the retrospective evaluation of validation status by ICCVAM, and retrospective validation by the OECD. However, the value of describing it as weight-of-evidence validation assessment is that the procedure could be used either retrospectively (based on existing data) and/or prospectively (based on new data, e.g. collected without dedicated laboratory work).
Five main types of WoE validation assessment could be envisaged (13):
The re-evaluation of a previous practical validation study (or series of studies).
The analysis of data obtained with the same test protocol in different laboratories, but at different times, in studies that were not intended to be parts of a validation exercise.
The analysis of data obtained in one or more laboratories, by using relatively minor variations of a protocol that was used in an earlier practical validation study.
The assessment of the validation status of a testing strategy comprising the use of data from several test methods, each of which had been previously evaluated either in an multi-laboratory validation study or in a WoE validation assessment, or via a different approach to testing, such as read-across for chemical hazard and risk analysis.
The evaluation of all the existing data generated from all the above situations, with consideration given to data generated from validation studies, as well as data generated when using the test method or testing strategy for routine testing purposes.
An example of the first type of WoE validation assessment procedure would be when a test method was being proposed for a slightly different purpose than that for which it was originally validated (i.e. in support of an extension to the scope of the scientific validity). In the second type, it is likely that the protocols used at different times in the various laboratories involved, as well as other protocol parameters, such as sources and types of test chemicals and other materials, would not have been standardised, but would be clearly defined and sufficiently similar for the data they produced to be taken together and evaluated. The third, fourth and fifth types of WoE validation assessment procedure would require judgements to be made about the performances of the tests, either when they were combined or when there were small alterations to the ways in which they were conducted.
It is imperative that WoE validation assessments are conducted with true independence and transparency, that they are designed and managed according to the highest standards, that those involved have sufficient expertise and experience, that the test methods or testing strategies are ready for evaluation, that there is agreement on: 1) the nature, quantity and quality of the evidence to be considered and its collection; 2) how the resultant data should be weighed; and 3) how the conclusions of the evaluation should be arrived at and reported.
Systematic review
Some guidance as to how WoE validation assessments could be conducted might be gained from the ways in which systematic reviews are employed as a central tool in evidence-based medicine (14). They are used to transparently and objectively evaluate in retrospect, all of the available information on a given and focused question. In contrast to the traditional narrative review, which tends to be biased and express an expert opinion (15), systematic reviews offer a high consistency and explicitness of applied methodology. Further advantages are offered by the increase of statistical power gained when combining information, or the identification of new research areas by the generation of new hypotheses. Indeed, in medicine, such reviews are considered to be the highest level of scientific evidence. According to Horvath and Pewsner (14), the process of systematic review can be divided into six phases:
Preparation of the systematic review
Systematic research of the primary literature
Selection of studies
Assessment of quality
Analysis and synthesis of data
Interpretation of data
Each of these steps has its own challenges. For example, in the first phase, it is usually necessary to convene a balanced and objective group of experts to specify the to-be-analysed problem and to develop a review protocol. An issue of major concern in almost all of these steps is the risk of bias. However, in the third phase, for example, bias can be avoided by defining inclusion and exclusion criteria in advance and in a transparent manner. In the fourth step, the quality is assessed with critical appraisal tools, usually based on quality scales or checklists (16). However, although this topic is widely discussed in the scientific literature today, no internationally-agreed standards are available at present. The analysis of data in step five should involve as little narrative as possible, but should be based on the use of summarising and biostatistical tools. A frequently employed and powerful tool is meta-analysis (17), which Egger et al. (18) see as an “observational study of the evidence”. This statistical method was developed to integrate the findings from individual studies. Basically, meta-analysis methods produce an average of the results from several studies, in which the study sizes are incorporated as weights, i.e. larger studies are given more weight than smaller studies.
Consideration of the ways in which systematic reviews are conducted in the field of diagnostic medicine offers a particularly promising approach, as toxicological tests and diagnostic tests have many similarities. Comparable study designs, the availability of a reference standard test and its performance (i.e. its reliability and relevance, and any limitations which may affect its usefulness in the optimisation of the assessment of the new test), and the methodology (e.g. assessment of test accuracy by using prediction models or thresholds), constitute major commonalities (19). Furthermore, considerations of patient spectrum for diagnostic tests (20), i.e. the population of patients to which the test was applied, might give useful guidance when addressing the applicability domains of toxicological tests. Internationally-agreed guidance is also lacking in this field (21, 22), but an international group of researchers developed the Standards for Reporting of Diagnostic Accuracy (STARD)-initiative in 1999. A checklist of 25 items, comprising items on Title/Abstract/Keywords, Methods, Results and Discussion, was published in several medical journals, to encourage improvements in the quality of reporting and to support the selection of quality criteria (23).
While the principle of meta-analysis also applies to systematic reviews of diagnostic tests, some method adjustment or development was triggered by the particular challenges of this field (24). In particular, the aspect of an imperfect reference standard (test) was given attention, and appropriate tools to account for it in meta-analysis were made available (25).
The potential value of systematic reviews for the field of toxicology is slowly being recognised, and is being discussed in the context of an evidence-based toxicology (26, 27).
Readiness for a WoE Validation Assessment
As with practical validation studies, the decision that a WoE validation assessment with a test method or testing strategy should be undertaken, should rest with a recognised validation authority, such as ECVAM or ICCVAM, or another appropriate body, such as the OECD, a trade association, or a national centre, such as ZEBET.
The rationale for a method or strategy to be considered for WoE validation assessment should include:
a clear definition of the scientific purpose and proposed practical application of the method or strategy;
a clear mechanistic description of its scientific basis;
a convincing case for its relevance to the human or animal in vivo situation, including an explanation of the need for it in relation to other methods or strategies;
an optimised protocol for the test procedure or a detailed indication of how the strategy was constructed and should be applied, and also the provision of each of the protocols used to generate data to be considered in support of the validity of the test method;
an evaluation of any similarities or differences in modes of action between the test method or strategy and the in vivo effects and responses in the species of interest;
a comprehensive statement about any test method or test strategy limitations;
evidence concerning its performance, intra-laboratory reproducibility, and inter-laboratory transferability and reproducibility;
reference to any previous independent reviews of the method or strategy, and the results of such reviews; and
an indication of its potential regulatory role.
Specific information should be made available about the test method(s) involved, which should include all the critical elements, such as:
details of the endpoint(s) measured and how any scoring system used is applied;
how the results are derived, calculated and expressed;
the rationale for the use and details of the application of the prediction model(s) used; and
the nature of any positive, negative and/or vehicle controls (or justification for their absence). ICC-VAM (28) and the OECD (12) have developed lists of information and data that should be provided in support of retrospective validation assessments.
In addition, since the acceptability of the WoE validation assessment itself will also have to be evaluated at a later stage, by peer review and by those with legal and regulatory responsibility for the type of testing concerned, it is vital that the several other criteria are also taken into account when the WoE assessment is being planned by, or on behalf, its sponsors. These include:
clarity of the defined goals;
quality of the overall design;
independence of management;
standards for the relevance, quality and quantity of the evidence to be considered;
independence of collection of evidence;
procedures for weighing of the evidence;
independence of the weighing of evidence procedure;
determination and reporting of the outcome;
plans for the publication in the peer-review literature of a summary report on the study and of its outcome;
plans for the development of publicly-accessible web links, so that the full report and the data involved can be freely accessed;
the transparency of whole process (including the identities, affiliations, and potential conflicts of interest of all the experts involved);
proposals for updating the WoE evaluation, when significant and substantial new information becomes available.
A practical suggestion as to the type of information necessary for evaluating readiness for a WoE assessment by or on behalf of a Sponsor was produced by a sub-group at the workshop (Table 1).
Table 1.
An outline scheme on the type of information necessary for evaluating readiness for a WoE assessment by or on behalf of a sponsor
| Level 1: ideal |
Level 2: provisionally acceptable |
Level 3: questionable/unacceptable |
|
|---|---|---|---|
| Rationale for the proposed test method | |||
| Intended uses/purposes | Clearly specified | Not fully defined | Totally unknown |
| Regulatory rational and applicability | Regulatory use | No clear regulatory application | R&D |
| Scientific (mechanistic) basis for the test | Clear mechanistic understanding of the test system | Mechanism not fully understood | No understanding of mechanism |
| Similarities and differences of modes of action in the test method and the reference species | Test system models the same mechanism/mode of action | Test system partially mimics the same mechanism/mode of action | Purely correlative |
|
| |||
| Test method protocol | |||
| Available for each study, multiple studies, single validated protocol | Multiple studies with single protocol/SOP | Single studies with multiple protocols | Single studies with poorly defined protocols |
| Positive, negative and/or vehicle controls | Positive and negative and vehicle controls | Positive or negative or vehicle control | No controls |
| Prediction model (PM) or data interpretation procedure (DIP) | Well-defined PM | DIP | No PM or DIP |
| Critical elements of protocol | Well known | Poorly defined | Not provided |
| Endpoint measurement | Totally objective | Mixture of objective/subjective | Subjective |
|
| |||
| Test substances | |||
| If chemical: purity, CASRN, concentration | Complete information | Complete information but chemical name/identity | No information |
| If product: % of ingredients, composition | Complete information | Incomplete information | No information |
| Number of chemicals/products evaluated and range of responses/chemical classes covered; response domain of the test (range of toxicity) | Statistically justified numbers of chemicals/products and adequate covering of applicability and response domain | Adequate number of chemicals/products that partially cover the domain | Inadequate number of chemicals/products to cover the domain |
| Coding of chemicals | All coded | Some coded | No coding |
|
| |||
| Reference data used for performance assessment | |||
| Data from species of interest | Available | Partially available or extrapolatable from other species | None available |
| Individual subject data (raw data) | Available | Data summary | No data only classification |
| Data quality (GLP, GCCP, etc) | Full compliance | Non GLP, records available | No records available |
|
| |||
| Test method data and results | |||
| Raw data | Available | No raw data, but appropriate statistical analysis | None available |
| Data quality (GLP, GCCP, etc) | Full compliance | Non GLP, records available | No records available |
|
| |||
| Test method evaluation for accuracy of the predictive performance | |||
| Performance compared to reference and to human situation | Comparable performance across different studies | Inconsistent performance across studies but explainable | No consistency and unexplainable |
|
| |||
| Test method reliability (reproducibility) | |||
| Within laboratory | Assessed from multiple laboratories | Assessed from a single laboratory | No evidence |
| Between laboratories | Assessed with all chemicals; same protocol | Assessed only for a subset chemicals; but which stress the method | No evidence |
|
| |||
| Supporting materials | |||
| All relevant publications, other scientific reports and review (Company in-house WoE) | Peer review publications | Publicly available | Unpublished/unavailable |
Evidence and its Collection
Clearly, the types of evidence to be collected, how it is to be obtained and selected, the extent to which it comprises all of the available material, how its quality is to be checked, and whether it is relevant and reliable, are crucial issues. It must also be clearly established that the collection of evidence is complete, and that it was collected in accordance with the pre-defined criteria and without bias, in order to ensure that it is truly representative of the performance of the test method or strategy. In addition, details concerning how the data are applied and interpreted, e.g. via a prediction model or other decision-making procedure to classify and label chemicals according to a particular type of toxicity, must also be included.
The collection of evidence should be controlled by a group of experts who include information technologists and scientists familiar with the type of method or strategy under evaluation and its intended purpose, but who are independent of both the developers and the proponents of the test procedure or testing strategy, as well as independent of those who will weigh the evidence, once it has been collected and organised. However, developers and proponents can be associated with this part of the process, not least by providing some of the evidence.
All the data for review should initially be classified as provisionally acceptable, until they have been adequately analysed and subjected to a formal set of criteria for accepting and including data in support of, or against, a method or strategy. These criteria (e.g. in the form of inclusion and exclusion criteria as used in systematic reviews) should be defined prior to the commencement of data retrieval and/or transformation and analysis, and clearly indicated in the validation assessment report.
The information on the test method or strategy and results produced with it, as well as the reference data used for assessing the performance of a test, should include:
data relevant to the species of interest (e.g. humans or another target species);
a description of the source and quality of the reference materials used to assess the accuracy of the proposed test method or strategy;
access to the original laboratory records and to all the individual raw data and transformed data;
an assessment of the quality of the data (i.e. whether they were produced according to the principles of Good Laboratory Practice, Good Clinical Practice and/or Good Cell Culture Practice, and with indications of checks and balances through both internal and external quality control audits; see 29); and
an explanation of why any related data were not used.
Ideally, the evidence should be available in the form of peer-reviewed publications. However, company reports might also be acceptable in some circumstances, provided that they are freely available in the public domain or could be made available at the conclusion of the study. Care should be taken to avoid any potential bias in data release (e.g. the publication of only positive findings). If available, useful information concerning human responses and/or effects, including the nature and extent of relevant exposures, should also be taken into account.
Where the evaluation of a test method or testing strategy for its predictive performance has been, or is to be, undertaken in relation to the toxicity of a reference set of chemicals with respect to the known responses of the same set of chemicals in the target species, particular attention should be paid to the choice of reference chemicals and to the quality of the data used to reach decisions about their in vivo effects. The reference chemicals and the reference data should be sufficiently representative of the chemical classes, physical properties, types and mechanisms of toxicity, and degrees and spectrum of effects for which the reliability and relevance of the test method or testing strategy are being evaluated.
The criteria to be used to assess the robustness of a quantitative structure-activity relationship [(Q)SAR] model and its applicability domain, and the (Q)SAR information itself, must be disclosed. Such disclosure would involve providing information on the chemicals used to develop the (Q)SAR model and its associated prediction model. As a minimum, key reference standard chemicals should be identified, and the applicability domain should be clearly defined, to ensure the proper use and interpretation of any (Q)SAR information involved in the assessment. For guidance, see 30.
The information on a test substance used to provide evidence should include, as a minimum:
the chemical purity of a chemical, or of each component of a mixture;
the Chemical Abstract Services Registry Number (CASRN) of a chemical, or of each component of a mixture; or the precise composition of products (mixtures and formulations, where known);
all the concentrations tested and their dosing intervals; and
the level and type of coding of the chemicals/products tested.
It is also essential to know the number and nature of chemicals/products evaluated as the test set, including the nature and concentration of any dilution solvent, with respect to their coverage in relation to the intended applicability domain of the test. In addition, it is essential to know the range of the responses covered by the test set (in order to cover the range from no effect to very high potency).
The Weighing of the Evidence
The performance criteria to be met by a test method or strategy in determining whether it should be judged to be/not to be relevant and reliable for its intended purpose, should be clearly defined in advance of the weighing of the evidence, and should be both reasonable and scientifically-based. Acceptance criteria have been developed by ECVAM, ICCVAM (7) and the OECD (12).
As in the case of those responsible for collecting the evidence, it is vital that those charged with formally assessing the evidence are independent of both the developers and the proponents of the test procedure or testing strategy. Nevertheless, consultation with individuals familiar with the development and use of the test method or testing strategy will also be necessary.
A case-by-case approach will be essential, and different kinds of evidence will have different levels of value in contributing to the overall assessment. This will involve evaluations of the plausibility, relevance, consistency, completeness, breadth and overall strength of the evidence.
The assessment itself cannot be used to improve the evidence, but, in addition to providing a consistent and transparent summary, a case could be made concerning the optimal use of a method or strategy, e.g. for testing only certain classes of chemical.
Conclusions from a WoE Validation Assessment
The WoE validation assessment should lead to a clearly-stated outcome, supported by reasoned and detailed arguments, which must be made publicly available. There are likely to be three main types of conclusions, depending on the degree to which the weighing of the evidence resolves uncertainty about the relevance and reliability of the test method or testing strategy for its proposed purpose (13):
that there is sufficient and consistent evidence that the test method/testing strategy is reliable and relevant for its stated purpose, and that it should be accepted for use for that purpose.
that there is insufficient and/or inconsistent evidence about the relevance and reliability of the test method/testing strategy for its stated purpose, and that additional evidence (of a type, quantity and quality to be specified) should be obtained and a further assessment made.
that there is sufficient evidence that the test procedure/testing strategy is not reliable and relevant for its stated purpose, and that it should not be accepted for use for that purpose.
The outcome of the assessment should be published in a peer-review journal, as well as being submitted to the sponsors of the validation assessment and other relevant bodies for further independent and transparent peer reviews of the assessment as a whole (i.e. design, data collection, weighing of the evidence, and reporting).
The Application of WoE Approaches to Validation
Historically, ECVAM has tended to favour validation via dedicated practical studies, which also characterises the OECD Health Effects Test Guidelines Programme, whereas ICCVAM has favoured the WoE validation assessment approach and independent scientific peer review (Table 2).
Table 2.
Examples of practical validation studies (P) and WoE validation assessments (W) conducted by ECVAM, ICCVAM and the OECD
| Test method | ECVAM | ICCVAM | OECD |
|---|---|---|---|
| The 3T3 NRU test for phototoxic potential | P1 | ||
| The application of the 3T3 NRU phototoxicity test to UV filter chemicals | P2 | ||
| The rat TER skin corrosivity test | P3 | Wa,4,5 | |
| The EPISKIN™ skin corrosivity test | P4 | Wa,4,5 | |
| The EpiDerm™ skin corrosivity test | P5 | Wa,4,5 | |
|
| |||
| The Corrositex™ skin corrosivity test | Wb,8 | W5,9 | |
| The local lymph node assay for skin sensitisation | Wb,10 | W11–14 | W |
| The embryonic stem cell test for embryotoxicity | P15 | ||
| The whole-embryo culture test for embryotoxicity | P16 | ||
| The micromass test for embryotoxicity | P17 | ||
|
| |||
| In vitro methods for predicting acute systemic toxicityc | P18 | P19,20 | |
| Human cell-based (in vitro) assays for the detection of pyrogens | P | W | |
| The colony forming unit granulocyte/macrophage (CFU-GM) assay for predicting acute neutropenia in humans | P | ||
| The upper threshold concentration step-down approach to reduce the number of fish used for acute aquatic toxicity testing | W | ||
| The in vitro micronucleus test | W | W21 | |
|
| |||
| The up-and-down procedure (UDP) for in vivo acute toxicity testing | W22 | ||
| The frog embryo teratogenesis assay — Xenopus (FETAX) | W | ||
| In vitro test methods for detecting ocular corrosives and severe irritants | W5 | ||
| In vitro and in vivo tests for percutaneous absorption | W | ||
| The uterotrophic assay for endocrine disruption | P23 | ||
|
| |||
| The Hershberger assay for endocrine disruption | P24,25 | ||
| The enhanced 28-day assay for endocrine disruption | P26 | ||
| In vitro screening assays for endocrine disruption | W27 | P28 | |
Including a review of the ECVAM study;
including a review of the ICCVAM study;
a joint ECVAM/ICCVAM study.
ECVAM (1997). Statement on the scientific validity of the 3T3 NRU PT test (an in vitro test for photo-toxic potential). ATLA 26, 7–8.
ECVAM (1998). Statement on the application of the 3T3 NRU PT test to UV filter chemicals. ATLA 26, 385–386.
ECVAM (1998). Statement on the scientific validity of the rat skin transcutaneous electrical resistance (TER) test (an in vitro test for skin corrosivity). ATLA 26, 275–277.
ICCVAM (2002). ICCVAM Evaluation of EPISKIN™, EpiDerm™, (EPI-200), and Rat Skin Transcut-aneous Electrical Resistance (TER): In Vitro Test Methods for Assessing Dermal Corrosivity Potential of Chemicals. NIH Publication No. 02–4502, Research Triangle Park, NC, USA: National Institute of Environmental Health Sciences (NIEHS). Website http://iccvam.niehs.nih.gov/methods/epiderm.htm
ICCVAM (2004). Recommended Performance Standards for In vitro Test Methods for Skin Corrosion, NIH Publication No. 04–4510, Research Triangle Park, NC, USA: National Institute of Environmental Health Sciences (NIEHS). Website http://iccvam.niehs.nih.gov/docs/docs.htm#invitro
ECVAM (1998). Statement on the scientific validity of the EPISKIN™ test (an in vitro test for skin corrosivity). ATLA 26, 277–288.
ECVAM (2000). Statement on the application of the EpiDerm™ human skin model for skin corrosivity testing. ATLA 28, 365–366.
ECVAM (2001) Statement on the application of the CORROSITEX® assay for skin corrosivity testing. ATLA 29, 96–97.
ICCVAM (1999). Corrositex®: An In vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. NIH Publication No. 99–4495, Research Triangle Park, NC, USA: National Institute of Environmental Health Sciences (NIEHS). Website http://iccvam.niehs.nih.gov/docs/reports/corprrep.pdf
ECVAM (2000). Statement on the scientific validity of local lymph node assay for skin sensitisation testing. ATLA 28, 366–367.
ICCVAM (1999). The Murine Local Lymph Node Assay: A Test Method for Assessing the Allergic Contact Dermatitis Potential of Chemicals/Compounds. NIH Publication No. 99–4494, Research Triangle Park, NC, USA: National Institute of Environmental Health Sciences (NIEHS). Website http://iccvam.niehs.nih.gov/methods/llnadocs/llnarep.pdf
Sailstad, D., Hattan, D., Hill, R. & Stokes, W. (2001). Evaluation of the murine local lymph node assay (LLNA) I: The ICCVAM review process. Regulatory Toxicology and Pharmacology 34, 258–273.
Dean, J., Twerdok, L., Tice, R., Sailstad. D., Hattan, D. & Stokes, W. (2001). Evaluation of the murine local lymph node assay (LLNA) II: Conclusions and recommendations of an independent scientific peer review panel. Regulatory Toxicology and Pharmacology 34, 274–286.
Haneke, K., Tice, R., Carson, B., Margolin, B. & Stokes, W. (2001). Evaluation of the murine local lymph node assay (LLNA): III. Data analyses completed by the national toxicology program (NTP) interagency center for the evaluation of alternative toxicological methods. Regulatory Toxicology and Pharmacology 34, 287–291.
ECVAM (2002). Statement on the scientific validity of the embryonic stem cell test (EST) — an in vitro test for embryotoxicity. ATLA 30, 265–268.
ECVAM (2002). Statement on the scientific validity of the postimplantation rat whole-embryo culture assay — an in vitro test for embryotoxicity. ATLA 30, 271–273.
ECVAM (2002). Statement on the scientific validity of the micromass test — an in vitro test for embry-otoxicity. ATLA 30, 268–270.
ICCVAM (2006). ICCVAM Test Method Evaluation Report. In vitro Cytotoxicity Test Methods for Estimating Starting Doses for Acute Oral Systemic Toxicity Testing. Website http://iccvam.niehs.nih.gov/methods/BRDU_TMER/AcuteTxTMER_Total.pdf
ICCVAM (2001). Report of the International Workshop on In vitro Methods for Assessing Acute Systemic Toxicity: Results of an International Workshop Organized by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program (NTP) Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM). NIH Publication No. 01–4499, Research Triangle Park, NC, USA: National Institute of Environmental Health Sciences (NIEHS). Website http://iccvam.niehs.nih.gov/methods/invidocs/finalall.pdf
ICCVAM (2001). Guidance Document on Using In Vitro Data to Estimate In Vivo Starting Doses for Acute Toxicity, Based on Recommendations from an International Workshop Organized by the Interagency Coordinating Committee on the Validation of Alternative Methods and the National Toxicology Program (NTP) Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM). NIH Publication No. 01–4500, Research Triangle Park, NC, USA: National Institute of Environmental Health Sciences (NIEHS). Website http://iccvam.niehs.nih.gov/methods/invitro.htm.
OECD (2004). Draft Proposal for a New Guideline 487: In Vitro Micronucleus Test. 13pp. Paris, France: OECD. Website http://www.oecd.org/dataoecd/60/28/32106288.pdf
ICCVAM (2001). The Revised Up-and-Down Procedure: A Test Method for Determining the Acute Oral Toxicity of Chemicals, Volumes I and II. NIH Publication No. 02–4501, Research Triangle Park, NC, USA: National Institute of Environmental Health Sciences (NIEHS). Website http://iccvam.niehs.nih.gov/methods/udpdocs/udpfin/vol_1.pdf and http://iccvam.niehs.nih.gov/methods/udpdocs/udpfin/vol_2.pdf
OECD (Draft). Draft Guideline for the Testing of Chemicals — The Uterotrophic Bioassay in Rodents: A Short-term Screening Test for (Anti) oestrogenic Properties. 22pp. Paris, France: OECD. Website http://www.oecd.org/dataoecd/26/53/37206177.pdf
OECD (2006). Final OECD Report on the Initial Work Towards the Validation of the Rat Hershberger Assay: Phase 1, Androgenic Response to Testosterone propionate, and Anti-androgenic Effects of Flutamide. OECD Environment, Health and Safety Publications. Series on Testing and Assessment No. 62, 209pp. Paris, France: OECD. Website http://www.oecd.org/dataoecd/43/25/37468632.pdf
OECD (Draft). Draft OECD Guideline for the Testing of Chemicals: The Hershberger Bioassay in Rats. 17pp. Paris, France: OECD. Website http://www.oecd.org/dataoecd/49/60/37478355.pdf
OECD (2006). Report of the Validation of the Updated Test Guideline 407 — Repeat Dose 28-day Oral Toxicity Study in Laboratory Rats. OECD Environment, Health and Safety Publications. Series on Testing and Assessment No. 59, 229pp. Paris, France: OECD. Website http://www.oecd.org/dataoecd/56/24/37376909.pdf
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen and Androgen Receptor Binding and Transcriptional Activation Assays. NIH Publication No. 03–4503, Research Triangle Park, NC, USA: National Institute of Environmental Health Sciences (NIEHS). Website http://iccvam.niehs.nih.gov/methods/endodocs/edfinrpt/edfinrpt.pdf
OECD (2004). Draft Guidance Document on Reproductive Testing and Assessment. OECD Environment, Heath and Safety Publications. Series on Testing and Assessment No. 43, 68pp. Website http://www.oecd.org/dataoecd/38/46/34030071.pdf
There is likely to be an accelerating trend toward WoE validation assessments in the future, especially as it is increasingly likely that the non-animal tests of the future will contribute evidence that will be used along with other evidence as components of test batteries and stepwise, decision-tree and integrative testing strategies. WoE approaches to validation will also probably be the predominant approach when existing OECD Test Guidelines are revised and updated in the future. However, it should be noted that, to date, no internationally harmonised comprehensive guidance on the validation of testing strategies or test batteries has been developed at the OECD level (12).
In addition, retrospective validation assessments, based on the ability of tests to give the same predictions as previously obtained, for example, with animal tests, will progressively be replaced by prospective assessments, especially where testing strategies and risk assessment approaches are based on more-modern methods in toxicology, themselves based on a greater understanding of mechanisms of toxicity and the application of emerging biotechnologies such as toxicogenomics and toxicoproteomics (31).
Assuring the Quality of the WoE Validation Assessment Process
It is evident from experience already gained that a number of potentially serious pitfalls can be encountered when planning and conducting a WoE validation assessment, some of which also apply to practical validation studies (13, 32). These include:
implausibility of the test system;
inadequate development of the test method or testing strategy;
lack of evidence and/or poor quality of evidence to support the inclusion of the method or strategy in a validation study;
bias in the selection of the experts to take part in the various phases of the study;
lack of sufficient and relevant experience or expertise among the selected experts;
bias in the availability of, selection and/or presentation of evidence;
failure to establish the relevance of evidence;
lack of a prediction model for applying the outcome of the test or strategy;
lack of clarity and/or precision in the weighing procedure;
inappropriateness of the weighing procedure;
bias in the derivation and/or application of the weighing procedure;
the application of unreasonably demanding, or unreasonably undemanding, performance criteria;
injudicious application of the precautionary principle;
bias in selection of data collection or weighing of evidence panel; and
the politicisation of the whole process.
It is vital that these pitfalls are recognised and, insofar as it is possible, avoided in the planning and management of the study. To meet this need, Balls and Combes (13) suggested that a WoE validation study should involve nine main stages (Figure 1) and a number of independent bodies or specifically-appointed groups, namely:
Figure 1.

An illustration of the main stages in a WoE validation study (13)
a Sponsor or Sponsors (e.g. ECVAM, ICCVAM and/or the OECD);
a Management Team (MT) appointed by the Sponsor(s);
a Data Collection Group appointed by the MT;
an Evidence Assessment Group appointed by the MT; and
a Peer Review Group appointed by the Sponsors, plus further peer review, totally separate from the study, organised by other bodies, such as relevant EC services and/or national and international regulatory authorities.
Central to the success, credibility and acceptability of any WoE validation assessment are the affiliations, independence and integrity of each group of participants, and the quality and transparency of the whole process, as designed, described and managed by the MT.
Given the need for sufficient and specific expertise and experience, some conflicts of interest are unavoidable. This problem should be faced and openly dealt with via documentation and transparency at every stage of the process, so that any bias or conflict of interest is fully declared and appropriate procedures for dealing with them are explained.
Detailed explanations should be given concerning the evidence and its acquisition, and of the procedures for weighing the evidence, and a case should be spelled out to support each conclusion or decision reached, and each recommendation made.
The MT should be accountable to the Sponsor(s) for:
ensuring the quality of the whole WoE process from its initiation until its completion;
avoiding failures due to logistical inconsistencies and avoidable problems, by ensuring that all the stages of the process are conducted according to agreed and acceptable criteria; and
striving to maximise third-party confidence and credibility in the procedures used to review, assess and, where appropriate, endorse the test method or testing strategy concerned as relevant and reliable for its stated purpose.
To avoid wasted effort as a result of problems identified late in a study, it is important that the Sponsor ensures that there is appropriate oversight over the whole course of the WoE evaluation process. This oversight could be conducted directly by the Sponsor or assigned to the Peer Review Panel (PRP) appointed by the Sponsor to evaluate the results of the WoE evaluation assessment. Oversight could involve reviewing the proposals of the MT for membership of the groups responsible for the collection of the evidence and for the weighing of the evidence, as well as ensuring that the criteria and procedures for evidence acquisition and review are: 1) appropriate and made available at every stage; 2) defined and conducted with minimal or no conflicts of interest, and with a lack of bias or a balancing of any unavoidable bias; and 3) defined and conducted by individuals independent of the original study. It would also involve ensuring that the outcome of the review process was made available in a publishable form, and that there was a clear and unambiguous statement of endorsement or rejection of the validity for its intended purposes of the test method or testing strategy under review. The Sponsor (or, if so designated, the PRP) should be consulted by the MT, should a situation arise during the evaluation process, which might require modifications to any of the criteria and procedures defined and agreed at the beginning of the evaluation process.
The PRP should consider the comprehensive final report produced by the MT, which should cover all the essential elements and consider all the essential questions involved in the study. If deemed necessary, the PRP could request, through the Sponsor, additional information, data, and/or analyses. While initially addressed to the Sponsor of the study, the final report should be communicated to the appropriate regulatory agencies and other interested parties, together with all other documentation relating to the study, as well as being made available in the public domain.
Any communication between the PRP and the MT should be conducted in ways which do not compromise their own independence, with regard to criteria and procedures for the acquisition of the evidence, criteria and procedures for the weighing of the evidence, and criteria for judging the performance of the test method or testing strategy in relation to its relevance and reliability for its intended purpose. The PRP must not become some kind of steering group for the evaluation — it must be sufficiently distant and detached to ensure that a critical and truly independent review is provided for the Sponsor and made publicly available.
Conclusions and Recommendations
Conclusions
The performance of dedicated practical validation studies is not always appropriate, necessary, or even possible, and, in such circumstances, a WoE validation assessment is more appropriate, where there is likely to be existing evidence of sufficient quantity and quality to permit an evaluation of fitness for purpose to be made without additional practical work, or where there is a lack of in vivo benchmark data of sufficient breadth, quantity and quality to serve as acceptable reference standards for the practical evaluation of an in vitro method.
WoE validation assessments will be increasingly necessary, to support better risk assessment approaches since the test methods and testing strategies of the future are less and less likely to be direct replacements for existing in vivo animal-based test procedures, but instead, will more likely be focused on effects and responses in humans, and will be based on advancements in the basic sciences of pharmacology and toxicology, involving the integration of mechanistic and other types of information from in silico and in vitro systems, molecular biology and biotechnology.
WoE validation assessment involves making the maximum use of available information by undertaking a structured, systematic, independent and transparent review, without a dedicated multi-laboratory practical study, to establish whether it can be concluded that a test method or testing strategy is reliable and relevant for its intended purpose.
Useful experience can be gained from the previous application of WoE validation assessments, including those concerned with skin penetration, the local lymph node assay (LLNA) and the frog embryo teratogenesis assay — Xenopus (FETAX), the Up-and-Down Procedure for acute oral toxicity, and in vitro tests for endocrine disruption. [Further information is available at http://iccvam.niehs.nih.gov]
Guidance on the conduct of WoE validation assessments might also be gained from a consideration of the ways in which systematic reviews are employed as a central tool in evidence-based medicine, including meta-analysis (defined as “an observational study of the evidence” from several different studies).
WoE validation assessments must be conducted with true independence and transparency, and must be designed and managed according to the highest standards. It is essential that those involved have sufficient expertise and experience, that the test methods or testing strategies are ready for evaluation, and that there is agreement on the nature, quantity and quality of the evidence to be considered and its collection, how the resultant data should be weighed, and how the conclusions of the evaluation should be determined and reported.
As with dedicated practical validation studies, the decision that a WoE validation assessment should be undertaken with a test method or testing strategy, should rest with a recognised validation authority, such as ECVAM or ICC-VAM, or another appropriate body, such as the OECD, a trade association, or a national centre, such as ZEBET. The criteria for readiness for a WoE validation assessment should be clearly defined.
The information used in a WoE validation assessment can be obtained and generated in a variety of ways, including prospectively, retrospectively, concurrently and/or compiled from diverse sources, generated for unrelated purposes.
The types of evidence to be collected, how it is to be obtained and selected, the extent to which it comprises all the available material, how its quality is to be checked, and whether it is relevant and reliable, are crucial issues. It must also be clearly established that the evidence is truly representative of the performance of the procedure or strategy, and that its collection is without bias.
The collection of evidence should be overseen by a group of experts (the Data Collection Group in Figure 1), who are sufficiently familiar with the type of method or strategy under evaluation and its intended purpose, but who are independent of both the developers and the proponents of the test procedure or testing strategy, as well as independent of those who will weight the evidence, once it has been collected and organised. However, developers and proponents can be associated with this part of the process, not least by providing some of the evidence.
The sources of the data for review should be disclosed, and all the data should initially be classified as provisionally acceptable, until they have been adequately evaluated and subjected to a formal set of criteria for accepting and including data in support of, or against, a method or strategy. These criteria should be defined prior to the commencement of data retrieval and review.
The performance criteria to be met by a test method or strategy in determining whether it should be judged to be/not to be relevant and reliable for its intended purpose, should be clearly defined in advance of the weighing of the evidence, and should be both reasonable and scientifically-based.
As in the case of those responsible for collecting the evidence, those charged with formally assessing the evidence (the Evidence Assessment Group in Figure 1) should be independent of both the developers and the proponents of the test procedure or testing strategy. Nevertheless, consultation with individuals familiar with the development and use of the test method or testing strategy will also be necessary.
A case-by-case approach will be essential, and different kinds of evidence will have different levels of value in contributing to the overall assessment. This will involve evaluations of the plausibility, relevance, consistency, volume and overall strength of the evidence.
A WoE validation assessment should lead to a clearly-stated outcome, supported by reasoned and detailed arguments, which must be made publicly available. The outcome of the assessment should be published in a peer-review journal, as well as being submitted to the sponsors of the validation assessment and other relevant bodies for further independent and transparent peer reviews of the study as a whole (design, data collection, WoE assessment, and reporting).
It is clear that a number of potentially serious pitfalls can be encountered when planning and conducting a WoE validation assessment. It is vital that these pitfalls are recognised and, insofar as it is possible, avoided in the planning and management of the study.
In view of the need for sufficient and specific expertise and experience, some conflicts of interest may be unavoidable. When this is the case, it should be dealt with via documentation and transparency at every stage of the WoE validation assessment, so that any bias or conflict of interest is fully declared and appropriate procedures for dealing with them are explained.
A comprehensive final report should be produced, which incorporates the MT and PRP reports and addresses all the essential elements involved in the validation assessment. While initially addressed to the Sponsor(s) of the study, this report should be communicated to the appropriate regulatory agencies and other interested parties, together with all other documentation relating to study, as well as being made publicly available.
Recommendations
ECVAM, ICCVAM, the OECD, and others actively involved in the validation process should take steps to further consider whether their practices and procedures are consistent with the principles elaborated in this report.
ECVAM and ICCVAM should jointly develop a guidance document (GD) on WoE validation assessments, based on the principles outlined in this report, as well as on their own validation principles and experience. Such a GD should be proposed for adoption by appropriate international organisations, such as the EU and the OECD, in order to gain international consensus on the recommended principles and processes.
The GD should allow for sufficient flexibility, so that its provisions are widely applicable on a case-by-case basis, but should be sufficiently rigorous to ensure that the core principles of validation are not violated.
ECVAM and ICCVAM should organise a workshop to discuss the use of meta-analysis and other systematic review tools for weighing data from single studies and combined studies, to explore and understand how and when such tools could be used in WoE validation assessments.
ECVAM and ICCVAM should then develop a set of criteria for weighing the evidence from different types of tests and strategies, ranging from purely correlative methods (based on a prediction model that is not related in anyway [either phylogenetically or mechanistically] to the species of interest), as opposed to mechanistically-based methods (i.e. methods based on mechanisms that also occur in the species of interest).
ECVAM should develop a system that would permit appropriate third parties (independent Peer Review Groups) to investigate whether the agreed principles and guidelines had been adhered to in WoE validation assessments (and also in dedicated practical validation studies), taking into account the independent peer review processes used by other organisations, such as ICCVAM and the OECD.
Appendix 1
a. The ECVAM Process for the Validation of Test Methods1
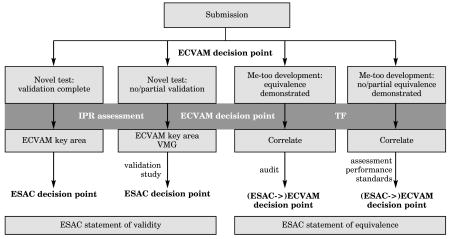
1This further development of the ECVAM validation process is currently under internal discussion at ECVAM.
b. IPR Assessment

c. The ESAC Peer Review Process1,2
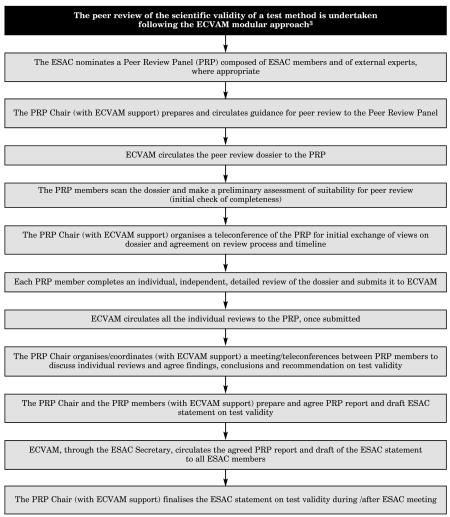
1The ECVAM Scientific Advisory Committee (ESAC) is composed of representatives from the 25 EU Members States, industry, academia and animal welfare, together with representatives of the relevant Commission services (DG Enterprise, DG Research, DG Environment and DG Health and Consumer Protection).
2The ESAC peer review process is currently under discussion at the ESAC.
3Hartung, T., Bremer, S., Casati, S. Coecke, S., Corvi, R., Fortaner, S., Gribaldo, L., Halder, M., Hoffmann, S., Janusch Roi, A., Prieto, P., Sabbioni, E., Scott, L., Worth, A. & Zuang, V. (2004). A modular approach to the ECVAM principles on test validity. ATLA 32, 467–472.
Appendix 2
The ICCVAM Process for Test Method Validation Assessments1
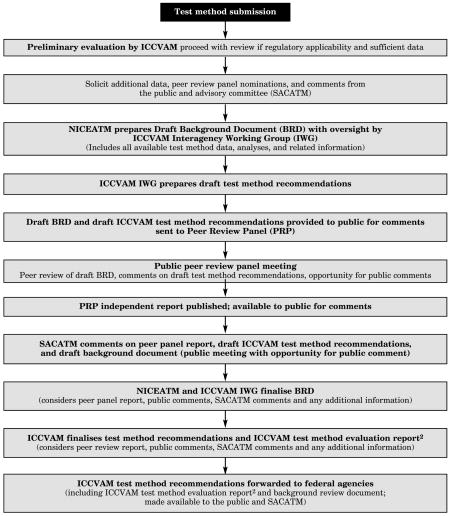
1ICCVAM is composed of representatives from ATSDR, CPSC, DoD, DoE, DoI, DoT, EPA, FDA, NIC, NIH, NIIEHS/NIH, NIOSH/CDC, NLM/NIH, OSHA, USDA.
2The ICCVAM Evaluation Report contains (1) applicable test guidelines, (2) recommended uses, test method protocol, and performance standards, (3) peer-review report as appendix A, and (4) public comments as appendix B.
Footnotes
The authors of this document participated as individuals, and the opinions expressed do not represent the positions of any government agency or other organisation.
References
- 1.ECVAM. ECVAM News & Views. ATLA. 1994;22:7–11. [Google Scholar]
- 2.Weed DL. Weight of evidence: A review of concept and methods. Risk Analysis. 2005;25:1545–1557. doi: 10.1111/j.1539-6924.2005.00699.x. [DOI] [PubMed] [Google Scholar]
- 3.Frazier JM. Scientific Criteria for Validation of In Vitro Toxicity Tests. Paris, France: OECD; 1990. p. 62. OECD Environment Monographs No. 36. [Google Scholar]
- 4.Balls M, Blaauboer B, Brusick D, Frazier J, Lamb D, Pemberton M, Reinhardt C, Roberfroid M, Rosenkranz H, Schmid B, Spielmann H, Stammati A–L, Walum E. The report and recommendations of the CAAT/ERGATT workshop on the validation of toxicity test procedures. ATLA. 1990;18:313–337. [Google Scholar]
- 5.Balls M, Blaauboer BJ, Fentem JH, Bruner L, Combes RD, Ekwall B, Fielder RJ, Guillouzo A, Lewis RW, Lovell DP, Reinhardt CA, Repetto G, Sladowski D, Spielmann H, Zucco F. Practical aspects of the validation of toxicity test procedures. The report and recommendations of ECVAM workshop 5. ATLA. 1995;23:129–147. [Google Scholar]
- 6.Balls M, Karcher W. The validation of alternative test methods. ATLA. 1995;23:884–886. [Google Scholar]
- 7.ICCVAM. Validation and Regulatory Acceptance of Toxicological Test Methods: A Report of the ad hoc Interagency Coordinating Committee on the Validation of Alternative Methods. Research Triangle Park, NC, USA: National Institute of Environmental Health Sciences, (NIEHS); 1997. p. 105. NIH Publication No: 97–3981. Website http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf. [Google Scholar]
- 8.OECD. Report of the OECD Workshop on Harmonisation of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Paris, France: OECD.; 1996 . p. 60. ENV/MC/CHEM(96)9. [Google Scholar]
- 9.Hartung T, Bremer S, Casati S, Coecke S, Corvi R, Fortaner S, Gribaldo L, Halder M, Hoffmann S, Janusch Roi A, Prieto P, Sabbioni E, Scott L, Worth A, Zuang V. A modular approach to the ECVAM principles on test validity. ATLA. 2004;32:467–472. doi: 10.1177/026119290403200503. [DOI] [PubMed] [Google Scholar]
- 10.Stokes W, Schechtman L, Hill R. The Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM): A Review of the ICCVAM Test Method Evaluation Process and International Collaborations with the European Centre for the Validation of Alternative Methods (ECVAM) ATLA. 2002;30(Suppl 2):23–32. doi: 10.1177/026119290203002S04. [DOI] [PubMed] [Google Scholar]
- 11.Schechtman LM, Wind ML, Stokes WS. Streamlining the validation process: The ICCVAM nomination and submission process and guidelines for new, revised and alternative test methods. ALTEX. 2006;22(Special Issue):337–342. [Google Scholar]
- 12.OECD. OECD Series on Testing and Assessment No. 34, ENV/JM/MONO (2005) Vol. 14. Paris, France: OECD; 2005. Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment; p. 96. Website http://appli1.oecd.org/olis/2005doc.nsf/linkto/env-jm-mono(2005)14. [Google Scholar]
- 13.Balls M, Combes R. Validation via weight-of-evidence approaches. ALTEX. 2006;22(Special Issue):288–291. [Google Scholar]
- 14.Horvath AR, Pewsner D. Systematic reviews in laboratory medicine: principles, processes and practical considerations. Clinica Chimica Acta. 2004;342:23–39. doi: 10.1016/j.cccn.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 15.Teagarden JR. Meta-analysis: whither narrative review? Pharmacotherapy. 1989;9:274–284. doi: 10.1002/j.1875-9114.1989.tb04139.x. [DOI] [PubMed] [Google Scholar]
- 16.Katrak P, Bialocerkowski A, Massy-Westropp N, Kumar VS, Grimmer K. A systematic review of the content of critical appraisal tools. BMC Medical Research Methodology. 2004;4:22. doi: 10.1186/1471-2288-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Egger M, Smith DG. Meta-analysis: Potentials and promise. British Medical Journal. 1997;315:1371–1374. doi: 10.1136/bmj.315.7119.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egger M, Smith GD, Phillips AN. Meta-analysis: Principles and procedures. British Medical Journal. 1997;315:1533–1537. doi: 10.1136/bmj.315.7121.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann S, Hartung T. Diagnosis: toxic! — Trying to apply approaches of clinical diagnostics and prevalence in toxicology considerations. Toxicological Sciences. 2005;85:422–428. doi: 10.1093/toxsci/kfi099. [DOI] [PubMed] [Google Scholar]
- 20.Irwig L, Bossuyt PM, Glasziou P, Gatsonis C, Lijmer JG. Designing studies to ensure that estimates of test accuracy are transferable. British Medical Journal. 2002;324:669–671. doi: 10.1136/bmj.324.7338.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whiting P, Rutjes AW, Reitsma J, Bossuyt PM, Kleijnen J. The development of QUADAS: a tool for the quality assessment of studies of diagnostic accuracy included in systematic reviews. BMC Medical Research Methodology. 2003;3:25. doi: 10.1186/1471-2288-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whiting P, Rutjes AW, Dinnes J, Reitsma JB, Bossuyt PM, Kleijnen J. A systematic review finds that diagnostic reviews fail to incorporate quality despite available tools. Journal of Clinical Epidemiology. 2005;58:1–12. doi: 10.1016/j.jclinepi.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 23.Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig LM, Lijmer JG, Moher D, Rennie D, de Vet HC standards for Reporting of Diagnostic Accuracy. Towards complete and accurate reporting of studies of diagnostic accuracy: the STARD initiative. British Medical Journal. 2003;326:41–44. doi: 10.1136/bmj.326.7379.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irwig L, Macaskill P, Glasziou P, Fahey M. Meta-analytic methods for diagnostic test accuracy. Journal of Clinical Epidemiology. 1995;48:119–130. doi: 10.1016/0895-4356(94)00099-c. [DOI] [PubMed] [Google Scholar]
- 25.Walter SD, Irwig L, Glasziou P. Meta-analysis of diagnostic tests with imperfect reference standards. Journal of Clinical Epidemiology. 1999;52:943–951. doi: 10.1016/s0895-4356(99)00086-4. [DOI] [PubMed] [Google Scholar]
- 26.Guzelian PS, Victoroff MS, Halmes NC, Janes RC, Guzelian CP. Evidence-based toxicology: a comprehensive framework for causation. Human and Experimental Toxicology. 2005;24:161–201. doi: 10.1191/0960327105ht517oa. [DOI] [PubMed] [Google Scholar]
- 27.Hoffmann S, Hartung T. Toward an evidence-based toxicology. Human and Experimental Toxicology. 2006;25:497–513. doi: 10.1191/0960327106het648oa. [DOI] [PubMed] [Google Scholar]
- 28.ICCVAM. Research Triangle Park, NC, USA: National Institute of Environmental Health Sciences (NIEHS); 2003. ICCVAM Guidelines for the Nomination and Submission of New, Revised, and Alternative Test Methods; p. 50. NIH Publication No. 03–4508. Website http://iccvam.niehs.nih.gov/docs/guidelines/subguide.htm. [Google Scholar]
- 29.OECD. OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring. Paris, France: OECD; 1998 . Website http://www.oecd.org/document/63/0,2340,en_2649_34381_2346175_1_1_1_1,00.html. [Google Scholar]
- 30.OECD (Draft). . OECD Principles for the Validation, for Regulatory Purposes, of (Quantitative) Structure-Activity Relationship Models. Paris, France: OECD; Website http://www.oecd.org/document/23/0,2340,en_2649_34799_33957015_1_1_1_1,00.html. [Google Scholar]
- 31.Corvi R, Ahr H–J, Albertini A, Blakey DH, Clerici L, Coecke S, Douglas GR, Gribaldo L, Groten JP, Haase B, Hamernik K, Hartung T, Inoue T, Indans I, Maurici D, Orphanides G, Rembges D, Sansone S-A, Snape JR, Toda E, Tong W, van Delft JH, Weis B, Schechtman LM. Meeting Report: Validation of Toxico-genomics-based Test Systems: ECVAM-ICCVAM/NICEATM Considerations for Regulatory Use. Report of the ECVAM-ICCVAM/NICEATM Toxicogenomics Workshop Report. Environmental Health Perspectives. 2006;114:420–429. doi: 10.1289/ehp.8247. Website http://dx.doi.org/ by using the following doi code: 10.1289/ehp.8247. [DOI] [PMC free article] [PubMed]
- 32.Balls M, Combes R. The need for a formal invalidation process for animal and non-animal tests. ATLA. 2005;33:299–308. doi: 10.1177/026119290503300301. [DOI] [PubMed] [Google Scholar]