

Abstract
Survival and maturation of dorsal root ganglia sensory neurons during development depend on target-derived neurotrophins. These target-derived signals must be transmitted across long distances to alter gene expression. Here, we address the possibility that long-range retrograde signals initiated by target-derived neurotrophins activate a specialized transcriptional program. The transcription factor MEF2D is expressed in sensory neurons; we show that expression of this factor is induced in response to target-derived neurotrophins that stimulate the distal axons. We demonstrate that MEF2D regulates expression of an anti-apoptotic bcl-2 family member, bcl-w. Expression of mef2d and bcl-w is stimulated in response to activation of a Trk-dependent ERK5/MEF2 pathway, and our data indicate that this pathway promotes sensory neuron survival. We find that mef2d and bcl-w are members of a larger set of retrograde response genes, which are preferentially induced by neurotrophin stimulation of distal axons. Thus, activation of an ERK5/MEF2D transcriptional program establishes and maintains the cellular constituents of functional sensory circuits.
Introduction
Nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) can elicit a wide variety of cellular responses, including proliferation, migration, survival, axonal outgrowth, synapse formation, and plasticity (Segal, 2003; Chao et al., 2006; Reichardt, 2006). Some of this diversity reflects the two classes of neurotrophin receptors, Trk receptor tyrosine kinases and p75NTR (Casaccia-Bonnefil et al., 1999; Patapoutian and Reichardt, 2001), as Trks and p75NTR induce distinct responses (Yano and Chao, 2000; Lee et al., 2001; Roux and Barker, 2002; Lu et al., 2005; Hempstead, 2006). However, cellular localization of Trk receptor activation could further specify responses (Ginty and Segal, 2002; Segal, 2003).
Neurotrophins were first identified as target-derived trophic factors that bind receptors on axon terminals (Levi-Montalcini and Angeletti, 1968) and elicit both local and long-range effects. Rapid responses initiated at axon terminals by neurotrophins influence direction and extent of axonal growth through kinases, phosphatases, and G proteins that quickly modulate cell morphology or intracellular trafficking (Segal, 2003; Reichardt, 2006). Axon terminal Trk receptors also stimulate long-term changes (Lom and Cohen-Cory, 1999) that depend on transcriptional programs involving factors such as CREB, c-fos, FOXO, NFAT, and MEF2 (Segal et al., 1992; Finkbeiner et al., 1997; Graef et al., 2003; Shalizi et al., 2003; Gan et al., 2005; Zhou and Snider, 2006). These long-term responses to distal axon neurotrophin stimulation require retrograde signaling from the axon terminal to the cell body. Location of neurotrophin stimulation is important for rapid local responses; for example, neurotrophin stimulation reorients axonal growth toward a trophic factor (McCaig et al., 2000; Markus et al., 2002; Zhou and Snider, 2006). However, it is less clear whether location of neurotrophin stimulation also specifies long-term transcriptional responses.
Here, we consider whether transcriptional responses to neurotrophins depend on location of stimulation. NGF in the skin promotes survival, axonal growth, and differentiation of nociceptive neurons of dorsal root ganglia (DRG) (Chen et al., 1999), whereas BDNF promotes survival and differentiation of dorsal root ganglia neurons that innervate specialized touch receptors (Carroll et al., 1998; Watanabe et al., 2000; González-Martínez et al., 2005). Therefore, to identify transcriptional programs that use spatial information, we focus on survival of developing dorsal root ganglion neurons in response to NGF and BDNF stimulation of distal axons. We show the transcription factor MEF2D and the anti-apoptotic Bcl-2 family member Bcl-w (Bcl-2l2) are expressed in sensory neurons and are regulated by target-derived neurotrophins and retrograde signaling pathways. Bcl-w and mef2d are members of a set of retrograde response genes preferentially induced by neurotrophin stimulation of distal axons compared with neurotrophin stimulation of cell bodies. We demonstrate that neurotrophin-induced expression of bcl-w is regulated by ERK5 and MEF2D. Thus, target-derived neurotrophins differentially activate a MEF2 transcriptional program that regulates sensory neuron survival.
Materials and Methods
Cell cultures.
Compartmented chamber cultures (Campenot cultures) were prepared as described previously (Heerssen et al., 2004). Briefly, DRGs from embryonic day 15 (E15) rats were dissected and plated in the center compartment of a Teflon divider (Camp10; Tyler Research) (Campenot, 1982). Cultures were maintained in media consisting of DMEM with 5% horse serum, 1% penicillin-streptomycin, and 0.3 μm cytosine arabinoside (AraC) at 37°C, 7.5% CO2; neurotrophins were added to the cell body compartment at 10 ng/ml BDNF (Peprotech) and 10 ng/ml NGF (Peprotech) and to the axon compartment at a concentration of 100 ng/ml BDNF and 100 ng/ml NGF (100 ng/ml NGF plus BDNF) for 3 d. On day 4, media was replaced and the 0.3 μm AraC omitted. On day 6, neurotrophins were removed from the cell body compartment and reduced to 1 ng/ml in axon compartments for 3–4 d (unless otherwise specified). For each experiment, vehicle control (100 ng/ml BSA in PBS) or neurotrophins (100 ng/ml NGF plus BDNF in vehicle, NGF or BDNF individually) were applied to distal axon or cell body compartments for the indicated time.
Mass cultures consisting of 2.5 × 105 DRG neurons were grown on laminin-coated p35 culture dishes for 2 d in neurotrophin-enriched (100 ng/ml NGF plus BDNF) media with 0.3 μm AraC, followed by 3 d in 10 ng/ml neurotrophins without AraC. Mass cultures were changed to serum and neurotrophin-free media for 2 h and then stimulated for the indicated time with neurotrophins (100 ng/ml NGF plus BDNF) or vehicle control.
Luciferase assays.
African green monkey fibroblast-like (COS-7) cells were maintained in DMEM containing 10% fetal calf serum and 1% penicillin-streptomycin at 37°C, 5% CO2. Firefly luciferase reporter plasmids were transfected into COS cells or DRG neurons using FuGENE 6 (Roche) or by nucleofection (Amaxa Biosystems), respectively, together with TK-pRL, which expresses Renilla luciferase and serves as an internal control. Firefly and Renilla luciferase activity was assessed 72 h after cell transfection. The results shown represent the average of eight independent experiments, with three replicates each.
Plasmids.
3xMRE-Luc, MEF2-VP16 plasmids were described previously (Flavell et al., 2006). plenti-hU6BX RNAi plasmids and TK-pRL were from Cellogenetics and Promega, respectively. The bcl-w plasmid was the generous gift from Susan Cory (The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia). To generate the bcl-w promoter construct, we cloned the 2.5kB upstream of the bcl-w transcription starting site into the pGL3-basic luciferase plasmid. The dominant negative MEK5 (S311A, T315A) and constitutively active MEK5 (S311D, T315D) were obtained from S. Gutkind (National Institutes of Health). The siRNA against MEF2D was designed and inserted in a lentiviral system as described previously (Flavell et al., 2006).
Reagents.
We used the following antibodies: MEF2D (1:2500 for Western blot and 1:500 for immunostaining; BD Transduction Labs), Bcl-w (1:500; Millipore Bioscience Research Reagents), green fluorescent protein (GFP; 1:1000; Roche), pan-actin (1:1000; Cell Signaling), phospho-ERK5 (1:1000; Cell Signaling), ERK5 (1:500; Cell Signaling), phospho-ERK1/2 (1:1000; Cell Signaling), HA (1:1000; Cell Signaling), Tubulin (1:500; Sigma), phospho-Mef2 (1:500; Abcam); secondary antibodies conjugated to HRP (1:10,000; Bio-Rad), Alexa-488 and Alexa-546 (1:1000; Invitrogen). MEF2D 6066 (1:1000 for Western blot and 1:100 for immunostaining) was developed in the Greenberg lab and generated by injecting antigen (a fragment of MEF2D protein, amino acids 292–514) (Han and Prywes, 1995) into rabbit. Characterization and specificity of this antibody are shown in supplemental Figure 2, available at www.jneurosci.org as supplemental material.
Drug treatment.
Distal axons and cell bodies of DRG neurons grown in compartmented cultures were treated either with 200 nm K252A (Calbiochem), 10 μm UO126 (Calbiochem), 50 μm LY294002 (Calbiochem), or DMSO vehicle control for 30 min before neurotrophin stimulation.
Real-time quantitative reverse transcription-PCR.
RNA was harvested from DRG neurons in compartmented chamber cultures or in mass cultures using the RNAqueous-4PCR kit (Ambion). For in vivo analysis, DRG neurons from E18 rats were dissected, and RNA was extracted using TRIzol (Invitrogen). Reverse transcription (RT) was performed using the cDNA archive kit (Applied Biosystems), according to manufacturer's specifications. Quantitative real-time RT-PCR was performed using Taqman Gene expression assays (Applied Biosystems) to assess the expression of c-fos (Rn02105452_s1), bcl-w (Rn00821025_s1), alsin (Mm00511865_m1), igf-1 (Rn00710306_m1), decorin (Rn01503161_m1), enpep (Rn00573861_m1), and mef2d (Rn00578329_m1). Data were normalized by the expression level of gapdh RNA for each sample (Applied Biosystems). Significance was calculated by z-test.
Protein analysis.
Cells were lysed in a nonionic detergent. Protein lysates from 8 to 10 compartmented cultures were pooled, and equal amounts of protein were separated by 7% SDS-PAGE, analyzed by immunoblot, and visualized using the SuperSignal chemiluminescent substrate kit (Pierce).
Immunostaining.
For in vitro analysis, compartmented chamber cultures were fixed in 4% paraformaldehyde (PFA) for 20 min, permeabilized in 0.1% TritonX for 10 min, and blocked in 5% normal goat serum and 0.1% TritonX for 60 min. Cultures were incubated overnight at 4°C in MEF2D (1:500) followed by incubation in goat anti-mouse Alexa 546 (1:1000) for 1 h at room temperature.
For in vivo analysis, MEF2D positive cells were counted from postnatal day 0 (P0) bcl-w+/− or bcl-w−/− animals. Animals were killed and fixed in 4% PFA, cryopreserved in 30% sucrose, and 14 μm slices were prepared. Tissue was subjected to antigen retrieval by 10 mm sodium citrate buffer for 15 min at 95°C, followed by 30 min cooling at room temperature. Tissue sections were permeabilized in 0.5% TritonX for 10 min and blocked in 5% normal goat serum and 0.1% TritonX for 60 min. Sections were incubated overnight at 4°C in MEF2D 6066 (1:100) followed by incubation in goat anti-rabbit Alexa 488 (1:1000) for 1 h at room temperature. Three animals of each genotype were used, and 3–6 dorsal root ganglia from each lumbar and cervical region were counted. Significance was calculated by Student's two-tailed t test.
siRNA knockdown.
For infection of mass cultures or compartmented cultures with siRNA lentivirus constructs, neurons at 3 and 5 d, respectively, were treated with either control Sh6L3–GFP siRNA or Mef2D–GFP siRNA with polybrene (4 μg/ml) for 48 h. After infection, media was changed to DMEM only for 2 h, and then cells were stimulated with 100 ng/ml NGF plus BDNF or vehicle control for 2 h. Neurons were harvested, and protein levels of MEF2D were analyzed by Western blot. RNA was extracted from cell bodies of compartmented chamber cultures using TRIzol (Invitrogen). Quantitative real-time RT-PCR was performed using Taqman Gene expression assays (Applied Biosystems) to assess the expression of c-fos, bcl-w, and mef2d. Data were normalized by the expression level of gapdh RNA for each sample (Applied Biosystems). Significance was calculated by z-test.
Survival assay.
DRG neurons grown in compartmented cultures for 6 d were transferred to serum-free media or to serum-free media supplemented with 100 ng/ml NGF plus BDNF in the distal axon or cell body compartments only. Inhibitors were added to the cell body and distal axon compartments as indicated. We exchanged the media every 12 h to replenish inhibitors and to guard against leakage between compartments. After 72 h, cultures were fixed in 4% paraformaldehyde, and apoptotic cells were visualized using the DeadEnd Fluorometric TdT-mediated dUTP nick end labeling (TUNEL) system kit (Promega), using biotinylated dUTP (5 μm) and Cy3-conjugated avidin (1:1000), and counterstained for 1 min with 4,6-diamidino-2-phenylindole dihydrochloride (DAPI; 1:1000). Each data point represents the mean of five fields per culture from 6 to 12 cultures in 2–6 different experiments scored in a blind manner. Significance was calculated by Student's t test. For in vivo analysis, condensed nuclei using DAPI (1:1000) were counted from P0 bcl-w+/+ or bcl-w−/− animals. Three animals of each genotype were used, and 3–6 dorsal root ganglia from each lumbar and cervical region were counted. Significance was calculated by Student's two-tailed t test.
Animal use.
Timed pregnant rats were purchased from Charles River. Bcl-w−/− mice were a generous gift from Grant MacGregor (University of California, Irvine, Irvine, CA) (Ross et al., 1998). All experimental procedures were done in accordance with the National Institutes of Health guidelines and were approved by the Dana-Farber Cancer Institutional Animal Care and Use Committee.
Results
MEF2D expression is regulated by neurotrophin stimulation of axons and is important for survival of sensory neurons
Neurotrophin stimulation of distal axons in sensory neurons has previously been shown to increase expression levels of the transcription factors c-fos and c-jun (Watson et al., 1999). These transcription factors are rapidly induced by neurotrophin stimulation of either cell bodies or distal axons and so are unlikely to be critical for spatially selective responses to neurotrophin stimulation. MEF2D is a transcription factor that can also be regulated by neurotrophin stimulation (Liu et al., 2003; Shalizi et al., 2003). Therefore, we asked whether this transcription factor is expressed by sensory neurons at the developmental time period when they are sensitive to neurotrophin levels. As shown in Figure 1, A and B, MEF2D is expressed by dorsal root ganglia in late embryonic stages (E18–birth) in vivo. We used quantitative RT-PCR to ask whether mef2d levels are regulated by neurotrophin stimulation of distal axons. Using sensory neurons grown in compartmented cultures, we find that the level of mef2d mRNA is increased within 2 h after selective neurotrophin stimulation of distal axons (Fig. 1C). In contrast, when neurotrophins are applied instead to cell bodies of sensory neurons grown in compartmented cultures, mef2d mRNA levels do not increase. We next asked whether this spatial selectivity is maintained when sensory neurons are stimulated with NGF or BDNF alone rather than a combination of these neurotrophins. Each of the individual neurotrophins can induce mef2d when applied to the distal axons but not when applied to the cell bodies (Fig. 1D). Although either NGF or BDNF alone can induce this response, there is less variability when the two neurotrophins are used in combination. Therefore, in subsequent experiments, we use a combination of the two neurotrophins to stimulate cultured neurons.
Figure 1.

Neurotrophins induce mef2d expression in DRG neurons in response to distal axon stimulation. A, MEF2D protein is detected in vivo in dorsal root ganglia at E18 (lanes 1 and 2 represent 2 separate animals). As a positive control, lysates of neurons over-expressing MEF2VP16 were blotted for MEF2D. B, MEF2D immunostaining of dorsal root ganglia at P0 in vivo. Scale bar, 10 μm. C, Expression of mef2d mRNA in DRG neurons in response to neurotrophin (NT) stimulation. DRG neurons were treated with neurotrophins (100 ng/ml NGF plus BDNF) for 2 h either on distal axons (DA) or on cell bodies (CB). Mef2D mRNA expression is specifically induced by distal axon stimulation and not by cell body stimulation. Expression is compared with DRG neurons treated with vehicle (100 ng/ml BSA). Results represent the mean ± SEM of eight experiments, *p < 0.05. D, Mef2d mRNA expression is upregulated during stimulation of distal axons with NGF alone, BDNF alone, or NGF plus BDNF (NT). Induction of cfos mRNA is shown as a positive control. *p < 0.05. E, MEF2D protein levels were analyzed by Western blotting before and after NT stimulation for 2 h. Normalized relative band density of MEF2D/actin reveal an increase of 21 ± 7% in response to distal axon stimulation.
To determine whether the changes in mef2d mRNA are likely to have functional consequences, we asked whether neurotrophin stimulation also affects the levels of MEF2D protein. As shown in Figure 1E, there is a consistent increase in MEF2D protein in response to neurotrophin stimulation of the distal axons. Furthermore, MEF2D protein is localized to nuclei both before and after neurotrophin stimulation of sensory neurons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Together, these data indicate that mef2d mRNA and MEF2D protein are expressed in developing sensory neurons and regulated by neurotrophin stimulation of distal axons and that the protein is appropriately expressed to participate in responses to target-derived neurotrophins.
MEF2 family members have been implicated in diverse processes in the nervous system, including survival, proliferation, and synaptic regulation (Mao et al., 1999; Liu et al., 2003; Heidenreich and Linseman, 2004; Shalizi and Bonni, 2005; Flavell et al., 2006; Shalizi et al., 2006). To determine whether neurotrophin stimulation of mef2d might participate in neurotrophin-dependent survival of sensory neurons, we reduced the level of MEF2D using mef2d RNAi expressed in lentivirus (Fig. 2A) and then tested the ability of neurotrophins to promote survival in compartmented cultures. After 3 d in neurotrophin-free media, the majority of control sensory neurons undergo apoptosis, as assessed by TUNEL staining. Neurotrophin stimulation of either cell bodies or distal axons maintains neuronal survival (Fig. 2B,C). In cultures where MEF2D has been reduced by lentivirus RNAi, neurotrophin stimulation of the distal axons is unable to prevent apoptosis (Fig. 2B,C). However, neurotrophin stimulation of cell bodies does rescue the survival of some, although not all, sensory neurons. These data indicate that MEF2D is required for neurotrophin-dependent survival of developing sensory neurons, particularly when neuronal survival depends on retrograde signaling mechanisms.
Figure 2.
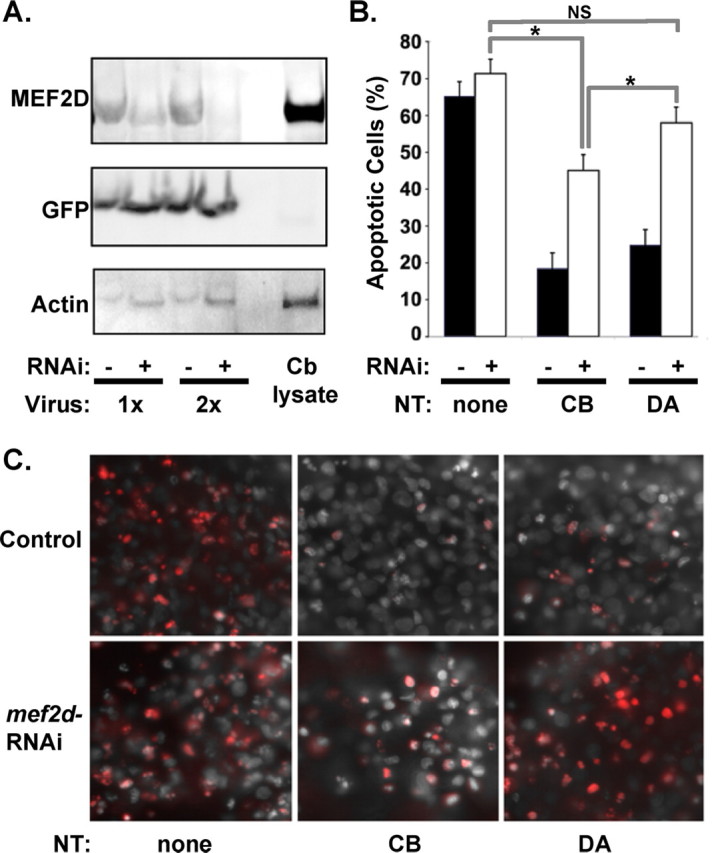
MEF2D is required for survival responses to neurotrophins. A, DRG neurons in culture were infected with lentivirus expressing mef2d–GFP RNAi and cell lysates analyzed by Western blotting with indicated antibodies. Mef2d RNAi (+) results in decreased expression of MEF2D protein levels, whereas a control lentivirus (−) does not. Cerebellar lysate (Cb lysate) was run as a positive control. B, Apoptosis of DRG neurons was analyzed by TUNEL staining. Mef2d RNAi reduces survival of DRG neurons in response to neurotrophins when applied to cell bodies (CB) and blocks survival when applied to distal axons (DA). Results represent the mean ± SEM of five experiments, *p < 0.05. C, Representative images of DRG neuron cell bodies grown in culture and stimulated with neurotrophins or vehicle control on cell bodies or on distal axons. Cells with (mef2d RNAi) or without (control) mef2d reduction are shown. Panels show TUNEL staining (red) and DAPI-stained nuclei in control (top) and in neurons expressing mef2d RNAi (bottom).
The ERK5/MEF2D pathway regulates bcl-w and promotes neuronal survival
We previously found that ERK5 is involved in retrograde signaling and plays an important role in neurotrophin-mediated survival (Watson et al., 2001). Consistent with previous studies, activated ERK5 can phosphorylate, hence activate MEF2 (Fig. 3A) (Kasler et al., 2000; Seyfried et al., 2005). Stimulation of sensory neurons by neurotrophins, applied either to cell bodies (Fig. 3B) or to distal axons (Fig. 3C), increases MEF2 phosphorylation by 17 ± 6% and 20 ± 8%, respectively. To determine whether ERK5, like its substrate MEF2D, has a prominent role in the survival of neurons that depend on retrograde signaling mechanisms, we tested the effects of an ERK5 dominant negative construct (DN–ERK5). As shown in Figure 3D, this construct is able to prevent ERK5 activation. When tested in compartmented cultures, this dominant inhibitory construct decreases sensory neuronal survival as assessed by an increase in apoptosis (Fig. 3E). Furthermore, like mef2d RNAi, DN–ERK5 has a greater effect on neuronal survival when neurons are supported by neurotrophin stimulation of distal axons compared with survival when neurons are supported by neurotrophin stimulation of cell bodies. Thus, ERK5 and MEF2D both contribute to neuronal survival responses initiated by target-derived neurotrophins. These data suggest that an ERK5/MEF2D pathway is needed for retrograde signaling events that promote survival.
Figure 3.

ERK5 regulates MEF2D and promotes survival in response to target derived neurotrophins. A, MEF2D is phosphorylated in response to ERK5 activation. COS cells were transfected with CA–MEK5 and WT–ERK5, or WT–ERK5 alone, and lysates were blotted with an antibody to phospho-MEF2 (p-MEF2). B, MEF2D is phosphorylated in response to cell body stimulation (20 ± 8% increase, p < 0.05) for 20 min. Lysates were blotted with an antibody to phospho-MEF2 and with an antibody to pan-actin as a loading control. C, MEF2D is phosphorylated in response to distal axon stimulation (17 ± 6% increase, p < 0.05) for 2 h. Lysates were blotted with an antibody to phospho-MEF2 and with an antibody to pan-actin as a loading control. D, Neurons were infected with an adenovirus that expresses HA-tagged ERK5-dominant negative form (DN–ERK5), then treated with neurotrophins for 30 min. Compared with uninfected neurons, expression of DN–ERK5 prevents ERK5 phosphorylation without inhibiting ERK1/2 activation. E, ERK5 supports neuronal survival induced by target-derived neurotrophins. Cell apoptosis was measured by TUNEL staining in uninfected neurons and in neurons infected with WT–ERK5 or DN–ERK5 for 2 d. DN–ERK5 blocks survival of neurons that depend on target-derived neurotrophins, whereas DN–ERK5 has a lesser effect on survival supported by cell body stimulation. All data shown are means ± SEM, *p ≤ 0.05.
As MEF2D is a transcription factor, we postulate that the ERK5/MEF2D pathway stimulates expression of anti-apoptotic components, such as members of the bcl-2 family. As shown in Figure 4A, the anti-apoptotic gene bcl-w is upregulated by neurotrophin stimulation of distal axons by NGF plus BDNF, NGF alone, or BDNF alone, and not by cell body stimulation. Similarly, when we analyzed Bcl-w protein levels after cell body or distal axon neurotrophin stimulation, Western blotting revealed a significant increase in Bcl-w protein levels (32 ± 5%) after distal axon stimulation, with no increase observed with cell body stimulation (Fig. 4B). The pattern of induction observed for bcl-w resembles that shown for mef2d, in that expression is selectively induced by neurotrophin stimulation of distal axons. We, therefore, tested induction of mef2d and bcl-w after pharmacologic and genetic interventions that prevent activation of the ERK5 pathway, which is needed for retrograde survival responses. Induction of c-fos serves as a positive control in all experiments, as c-fos is induced in response to neurotrophin stimulation of either cell bodies or of distal axons. Both bcl-w and mef2d induction require Trk activation and so are prevented by K252A, an inhibitor of Trk kinase activity; Trk kinase activity is also required for induction of c-fos (Fig. 4C). Like c-fos induction, induction of bcl-w and mef2d are prevented by UO126, a pharmacologic agent that inhibits MEK1 and MEK5 and thus prevents activation of both ERK1/2 and ERK5 (Fig. 4D). In contrast, DN–MEK5, which inhibits activation of ERK5 but not ERK1/2, prevents induction of bcl-w and mef2d but does not affect c-fos. We postulate that ERK5 acts by regulating MEF2D activity. Therefore, we asked whether inhibition of MEF2D has the same effect on gene expression as DN–MEK5. As shown, mef2d RNAi prevents induction of bcl-w, but it does not affect induction of c-fos (Fig. 4E). Together, these data indicate that distal axon stimulation by neurotrophins regulates expression of bcl-w by a pathway that involves Trk/ERK5 and MEF2D and that the mechanisms regulating bcl-w and mef2d expression diverge from pathways that regulate c-fos.
Figure 4.
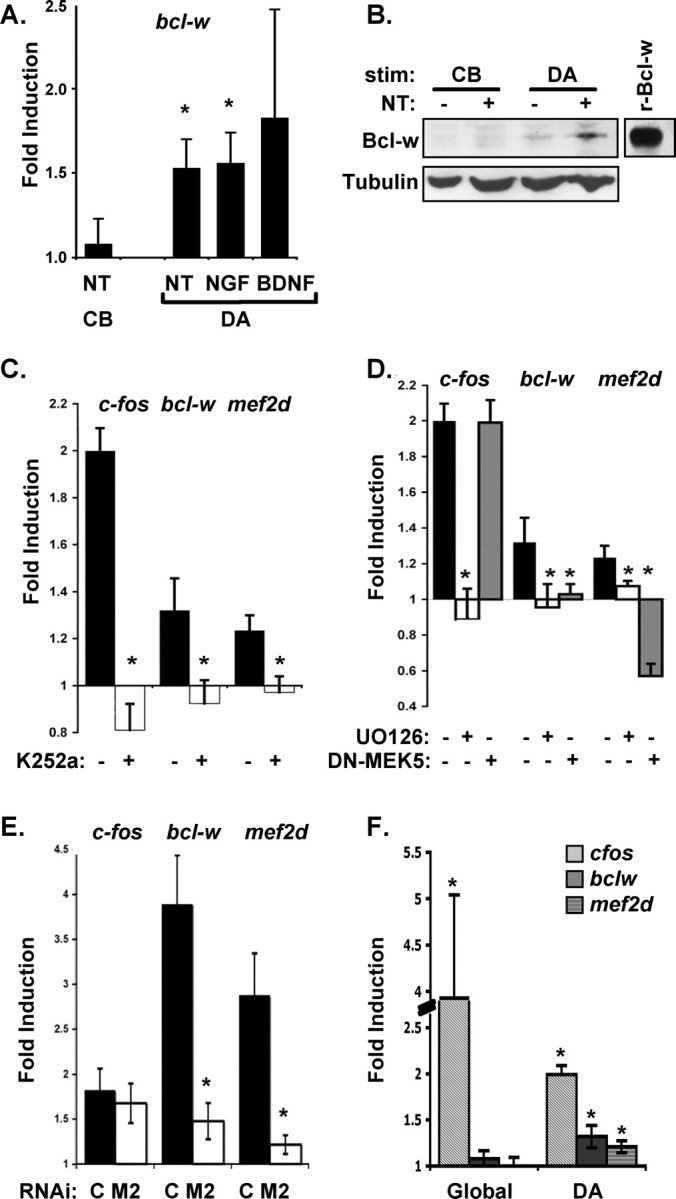
Target-derived neurotrophins induce bcl-w and mef2d expression in compartmented cultures by a Trk-dependent ERK5/MEF2 pathway. A, Expression of bcl-w mRNA in response to neurotrophin stimulation. DRG neurons were treated with neurotrophin (NT = 100 ng/ml NGF plus BDNF) for 2 h either on distal axons (DA) or on cell bodies (CB). Bcl-w mRNA expression is specifically induced by distal axon stimulation and not by cell body stimulation. Bcl-w mRNA is also upregulated during distal axon stimulation with NGF or BDNF alone. All data show mean ± SEM, *p < 0.05. B, Compartmented cultures were stimulated at cell bodies (CB) or distal axons (DA) for 8 h with neurotrophins. Cell body lysates were analyzed by Western blotting for Bcl-w (recombinant-Bclw was the positive control). DA stimulation leads to an increase in Bcl-w protein levels (32 ± 5%). C, The Trk kinase inhibitor K252a or vehicle control was applied to cell bodies and distal axons at 200 nm, a concentration that inhibits phosphorylation of Trk receptors in these neurons. Neurons in compartmented cultures were globally treated or not with K252a, and DA were stimulated with neurotrophins. K252a prevents induction of c-fos, bcl-w and mef2d by target-derived neurotrophins. D, The Erk kinase inhibitor UO126 or vehicle control was applied to cell bodies and distal axons at 50 nm, a concentration that inhibits phosphorylation of ERK kinases. Neurons in compartmented cultures were treated or not with UO126. UO126 prevents bcl-w, mef2d, and c-fos induction by target-derived neurotrophins. Neurons were infected with DN–MEK5 adenovirus for 3 d, then distal axons were stimulated with neurotrophins. DN–MEK5 expression inhibits bcl-w and mef2d mRNA induction but not c-fos induction by target-derived neurotrophins. E, Lentivirus expressing mef2d RNAi (M2) inhibits induction of bcl-w and mef2d, but not induction of c-fos, in response to target-derived neurotrophins (C = control lentivirus). All data show means ± SEM, *p < 0.05. F, Bcl-w and mef2d mRNA expression is specifically induced by distal axon stimulation and not by global stimulation (cultures maintained for 5–7 d).
To explore the relationship between retrograde signals and neurotrophin-dependent responses originating from activated receptors in the plasma membrane of the cell soma, we asked whether simultaneous neurotrophin stimulation of distal axons and cell bodies of dorsal root ganglion neurons grown in mass culture induces these retrograde response genes. Global stimulation leads to dramatic upregulation of c-fos; in contrast, mef2d and bcl-w are only induced by localized stimulation of distal axons (Fig. 4F). We find that neurotrophin stimulation of cell bodies only increases mef2d and bcl-w mRNA levels when combined with a second intervention that can activate ERK5 (Turjanski et al., 2007), such as viral infection or mechanical stress (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), not when combined with neurotrophin stimulation of distal axons (Fig. 4F). Of note, when cultures were maintained for 5–7 d before testing as in Figure 4F, the extent of mef2d and bcl-w induction was lower than that seen in our usual paradigm, wherein cultures are maintained for 8 d before stimulation. These data suggest that induction of mef2d and bclw may be more robust after neurotrophin stimulation of distal axons in neurons with longer processes.
As increases in bcl-w and mef2d in response to neurotrophin stimulation occur at the mRNA level, and induction of bcl-w depends on the transcription factor MEF2D, we postulate that induction of these retrograde response genes is mediated (at least in part) at the transcriptional level. To test this directly, we prepared a bcl-w promoter luciferase construct by fusing the 2.5 kB of upstream sequence from the mouse bcl-w promoter to luciferase. As shown (Fig. 5A), this construct contains one of three conserved consensus MEF2D-binding sites upstream of the start site. When expressed in sensory neurons, the bcl-w-luciferase construct is induced by neurotrophin stimulation of distal axons (Fig. 5B). To determine whether this regulation depends on an ERK5/MEF2D pathway, we analyzed the response of the bcl-w-luciferase construct to constitutive activation of ERK5 or MEF2D in COS cells. Coexpression of constitutively active MEK5 and wild-type ERK5 increases expression of bcl-w-luciferase (Fig. 5C). However, no induction is seen when kinase-inactive MEK5 is expressed together with ERK5. As controls, we assessed the response of a basal promoter and a promoter containing MEF2 response elements (MRE). Although activated MEK5/ERK5 induces expression of bcl-w-luciferase and MRE-luciferase, it does not induce expression of basal constructs. To determine whether bcl-w transcription is also mediated by the ERK5 substrate MEF2, we expressed a constitutively active MEF2 construct (VP16-MEF2) together with bcl-w-luciferase and Renilla luciferase control (Fig. 5D). Constitutively active MEF2 stimulates bcl-w-luciferase and the MRE luciferase but does not induce expression of a basic luciferase construct. Together, these studies indicate that bcl-w transcription can be induced by the ERK5/MEF2D pathway and that relevant response elements are contained in the 2.5 kB region upstream of the bcl-w start site. This may include other promoter elements; for example, two sites that resemble half of a CRE-binding motif are also present in the bcl-w promoter region (−9871 to −9867 and 2048 to 2052, TGACG).
Figure 5.

Bcl-w transcription is ERK5 and MEF2-dependent. A, Schematic of bcl-w 5′ region. Sequence and locations of consensus MEF2-binding sites are indicated. The luciferase construct consisted of the 2.5 kB promoter region, followed by the necleotides ATG and luciferase reporter gene. B, Luciferase reporter assay using bcl-w promoter-Luc cotransfected with pTK–Renilla–Luc for normalization. Two days after transfection by nucleofection, neurons were stimulated with neurotrophins for 2 h. Bcl-w-luciferase expression is neurotrophin dependent. C, D, Bcl-w promoter is ERK5 and MEF2 dependent. Middle and right luciferase reporter assay using bcl-w promoter-Luc, 3xMRE-Luc, or basic Luce cotransfected into COS cells with Renilla luciferase and the indicated expression plasmid(s). Three days after transfection, luciferase activity was measured. Data are from three independent experiments, each of which was conducted in triplicate, and show means ± SEM, *p < 0.05.
Bcl-w, and other retrograde response genes, promote neuronal survival
Bcl-w has not previously been shown to play a role in dorsal root ganglia neuron survival. To determine whether Bcl-w is important for survival of these spinal sensory neurons, we analyzed dorsal root ganglia neurons from bcl-w−/− mice. These mice are viable, but males are infertile because of apoptosis in the developing testes (Print et al., 1998; Ross et al., 1998). Levels of apoptosis were analyzed in vivo in DRGs from P0 bcl-w−/− and wild-type littermates. Cervical and lumbar sections were stained with DAPI and assessed for condensed nuclei as a measure of apoptosis (Fig. 6A). DRGs from either the cervical or lumbar regions of bcl-w−/− mice exhibited significantly higher levels of dying cells compared with wild type (Fig. 6B).
Figure 6.
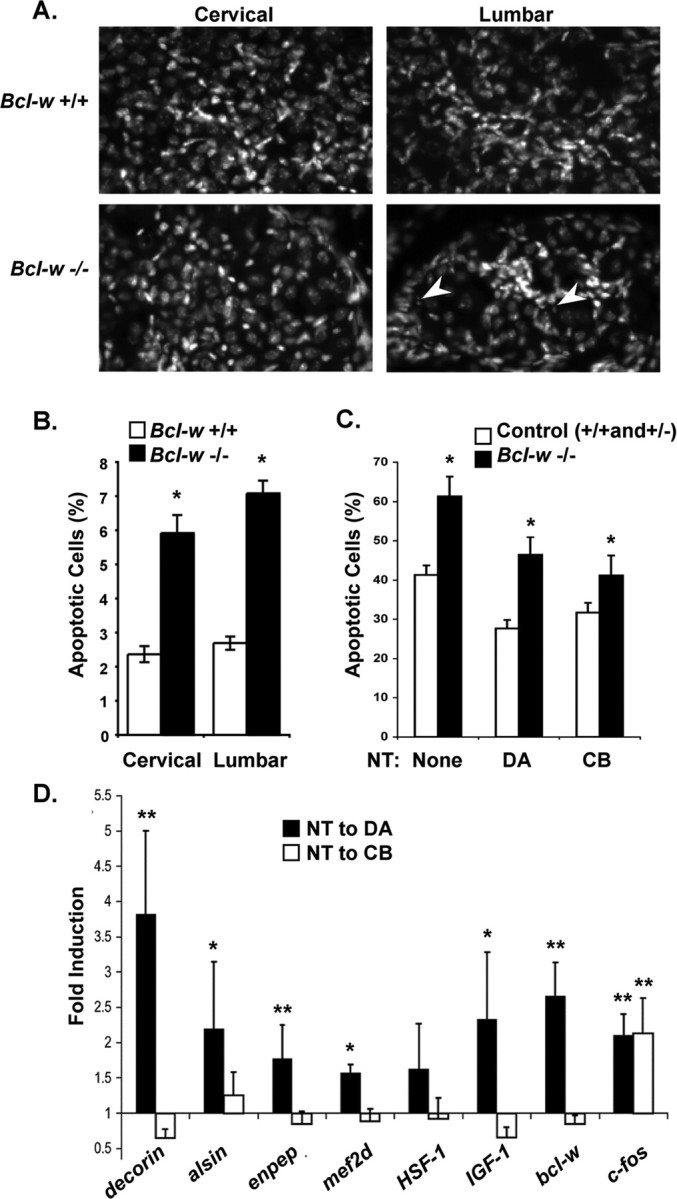
Bcl-w and other retrograde response genes are important for the survival of DRG sensory neurons. A, Representative images of DAPI staining in dorsal root ganglia from cervical and lumbar regions of the spinal cord in bcl-2+/+ and bcl-w−/− mice. Arrows indicate apoptotic cells. Scale bar, 10 μm. B, In vivo analysis of apoptosis. Bcl-w−/− P0 mice show an increase in cell death compared with WT littermates, in both cervical and lumbar DRGs. The percentage of cells in the DRG with condensed nuclei when visualized by DAPI staining are shown. Three animals of each genotype were used, and 3–6 dorsal root ganglia from each lumbar and cervical region were counted. All data show means ± SEM, *p < 0.0001. C, Survival of bclw−/− sensory neurons in compartmented cultures stimulated with neurotrophin applied to the distal axons (DA) or the cell bodies (CB). DRG neurons were dissected at E14 and seeded in compartmented cultures. Bcl-w−/− neurons are more prone to apoptosis in serum free-media compared with bcl-w+/+ and +/− under all conditions tested, *p < 0.05. D, Retrograde response genes: DRG neurons in compartmented cultures were stimulated with NGF and BDNF or vehicle applied either to DA or the cell bodies (CB), for 2 h. RNA was prepared and expression of decorin, alsin, enpeptidase, mef2d, HSF-1, IGF-1, bcl-w, and c-fos were assessed by quantitative RT-PCR and normalized to gapdh in the same sample. Fold induction was measured by comparing normalized level of expression in neurotrophin-treated/vehicle-treated cells for four experiments each involving five cultures for each condition (DA shown first in black). Statistical analysis by z-test, **p ≤ 0.05, *p ≤ 0.10 for a difference from one.
As MEF2D regulates bcl-w and thereby promotes survival, we predict that MEF2D-expressing cells would show decreased survival in the absence of Bcl-w. Therefore, we asked whether the percentage of sensory neurons that express MEF2D is decreased in bcl-w−/− mice. As shown, 30–40% of cervical and lumbar neurons express MEF2D in control neonatal animals. The MEF2D positive cells include neurons that are of various sizes, indicating that this transcription factor is expressed by multiple types of sensory neurons. In bcl-w−/− animals, the pool of MEF2D-expressing neurons is reduced by 23% (percentage of DRG neurons that are MEF2D positive is 37% in control and 28% in bcl-w−/− mice) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Thus, in vivo, bcl-w promotes the survival of DRG neurons, and particularly affects MEF2D-expressing cells.
We then assessed the survival of dorsal root ganglia sensory neurons from bcl-w−/− mice in compartmented culture conditions. These neurons were grown for 1 week with neurotrophins present in the distal axon compartment, at which point Bcl-w is expressed in wild-type cultures. As shown in Figure 6C, Bcl-w is important for survival of dorsal root ganglia neurons. Indeed, survival of bclw−/− sensory cells is compromised (although not eliminated) at 3 d with, or without, neurotrophins. We were surprised to find that bcl-w−/− neurons exhibit reduced survival during neurotrophin deprivation and under conditions where they are sustained by neurotrophins applied to the cell bodies or distal axons. This differs from the results seen when ERK5 or MEF2D activity is inhibited, in that sensory neurons that depend on target-derived neurotrophins are particularly sensitive to inhibition of ERK5 or MEF2D. Therefore, we asked whether ERK5/MEF2D might regulate additional retrograde response genes as well as bcl-w. Other retrograde response genes might contribute to the differential effect of mef2d RNAi and DN–ERK5 on the survival of neurons supported by neurotrophin stimulation of distal axons or cell bodies. We used quantitative RT-PCR to examine a panel of candidate genes that were induced by neurotrophin stimulation of distal axons in Affymetrix gene arrays, and we identified several genes that are preferentially induced by retrograde signaling (Fig. 6D). The genes identified as retrograde response genes include igf-1 and alsin, which encode proteins that have been implicated in promoting neuronal survival (Boillée and Cleveland, 2004; Bondy and Cheng, 2004; Davila et al., 2007). Together, these studies indicate that neurotrophin stimulation of distal axons activates ERK5/MEF2D to initiate a transcriptional program that includes the anti-apoptotic gene bcl-w as well as a set of additional retrograde response genes. Thus, the ERK5/MEF2 pathway, and the resultant gene expression program, is important for neuronal responses to target-derived neurotrophins.
Discussion
The mechanisms by which target-derived neurotrophic factors initiate and propagate survival signals from nerve terminals to a remote neuronal cell body have been the focus of investigation over many years (Hamburger and Levi-Montalcini, 1949; Hendry et al., 1974; Purves, 1986; Korsching, 1993; Campenot, 1994; Ahn et al., 2000; Neet and Campenot, 2001; Heerssen and Segal, 2002; Segal, 2003; Moises et al., 2007). Here, we address the role of transcriptional regulation in mediating responses to target-derived survival signals. We show that selective neurotrophin stimulation of distal axons of sensory neurons results in increased expression of the transcription factor MEF2D. Activation and expression of MEF2D requires the ERK5 MAP kinase. Both ERK5 and MEF2D promote survival of developing sensory neurons; one mechanism for this effect is induction of the anti-apoptotic gene bcl-w. An intriguing feature of this system is that this ERK5/MEF2D survival pathway is predominantly important for the survival of neurons that depend exclusively on neurotrophin support applied to distal axons. Thus, we have identified a transcriptional program that enables neurons to survive once they have contacted a correct target and are incorporated into a functional neural circuit.
Much of the recent work on long-range signals has focused on the mechanisms by which a signal initiated by an activated receptor at the nerve terminal travels retrogradely to a remote cell body to initiate transcriptional changes (Ginty and Segal, 2002). Abundant evidence indicates that neurotrophin binding to Trk receptors initiates both activation and endocytosis of the receptor (Fig. 7A), and the resultant signaling endosomes, containing the ligand-receptor complex, travel by dynein-dependent transport (Fig. 7B) (Grimes et al., 1996; Riccio et al., 1997; Senger and Campenot, 1997; Tsui-Pierchala and Ginty, 1999; Watson et al., 1999; Howe et al., 2001; Neet and Campenot, 2001; Watson et al., 2001; Weible et al., 2001; Delcroix et al., 2003; Heerssen et al., 2004). These specialized signaling endosomes activate a variety of second messenger signaling cascades as they are transported through the axon to the cell soma, including MAP kinases, as well as PI3 kinase and Akt (Kuruvilla et al., 2000; Watson et al., 2001; Wu et al., 2001; Delcroix et al., 2003). The ensuing activation of transcription factors, such as CREB, NFAT, and AP-1, mediate the necessary changes in gene expression in response to target-derived neurotrophins (Riccio et al., 1997, 1999; Watson et al., 1999; Graef et al., 2003). In this way, dynein-dependent transport eliminates the barrier of distance and allows transcriptional changes in response to target-derived neurotrophins.
Figure 7.

Target-derived neurotrophins activate an ERK5/MEF2 transcriptional program to promote survival. A, Neurotrophin binding and Trk activation results in endocytosis of the activated receptor complex. B, The signaling endosome, including downstream messengers, is retrogradely transported by microtubule-dependent dynein motors. C, On reaching the cell body, phosphorylated ERK5 translocates to the nucleus. D, MEF2D is phosphorylated and activated in response to a retrograde ERK5 signal. E, Activated MEF2D initiates transcription of bcl-w, mef2d, and other retrograde response genes (RRGs) to promote survival mediated by target-derived neurotrophins. F, In the case of cell body neurotrophin stimulation, both ERK5 and ERK1/2 are activated; however, expression of bcl-w, mef2d, and other retrograde response genes are not induced. One possible mechanism is through suppression of ERK5 by the ERK1/2 pathway.
Several lines of evidence suggest that transport of signaling endosomes also provides a mechanism by which a neuron can distinguish the location of neurotrophin stimulation and so adjust the nature of its response accordingly. Retinal ganglion cells exhibit increased dendritic arborization in response to BDNF applied at the axon terminals, whereas dendritic arborization is decreased by BDNF stimulation of dendrites within the retina, indicating that the biological response to neurotrophins varies based on location of stimulation (Lom et al., 2002). Here, we provide evidence of additional differential responses and of the underlying mechanisms that account for these spatial distinctions. Our studies have outlined a long-distance signaling pathway that requires activation of ERK5 and MEF2D to initiate a spatially distinct transcriptional program (Fig. 7C,D). This program promotes expression of retrograde response genes, which we define as a set of genes selectively induced by neurotrophin stimulation of distal axons. The retrograde response gene set includes bcl-w and mef2d, as well as igf-1 and alsin (Fig. 7E). These retrograde response genes in aggregate represent an ERK5-regulated expression program that may contribute to distinct biological responses to neurotrophins depending on the location of stimulation.
Gene expression programs regulated by ERK5 or other MAPK cascades are specified by the distinct ERK involved, the kinetics of pathway activation and repression, the subcellular localization of the pathway components, and cross talk among distinct kinase cascades (Raman et al., 2007). Each of these features may contribute to the selective induction of retrograde response genes by distal axon stimulation, with little or no induction by cell body or global stimulation. It is possible that cell body stimulation initiates both the ERK5/MEF2 pathway and a signaling pathway that counteracts ERK5-dependent gene induction (Fig. 7F). As cell body stimulation simultaneously activates the ERK5 and ERK1/2 pathways (Watson et al., 2001), the lack of induction of retrograde response genes may reflect the balance of timing or activity of these distinct MAPKs. Under conditions of heightened ERK1/2 activity, such as cell body stimulation, the effects of ERK5 might be limited. However, in the face of heightened ERK5 activity, as occurs during distal axon stimulation or in response to cell body stimulation after virus infection or other stress signals, activation of ERK5 may override negative effects of ERK1/2 and so lead to retrograde response gene induction. This and other mechanistic explanations for the location selectivity of retrograde response gene induction will require extensive additional investigation and modeling to evaluate how the kinetics and localization of MAPK activation are integrated to produce a specific transcriptional response.
Our studies have focused on pathways that are required for long-range signaling that promote neuronal survival. We found that ERK5 is particularly important for survival in neurons that depend on target-derived neurotrophins. Neuronal survival is compromised by inhibition of ERK5 activity, and this effect is most dramatic when neurons are supported solely by neurotrophin stimulation of axons. Likewise, MEF2D has a greater role in survival of neurons that depend exclusively on neurotrophin stimulation of the axons. Our data indicate that the anti-apoptotic molecule bcl-w, which is regulated by ERK5 and MEF2D, is important for neuronal survival. However, as survival of Bcl-w−/− sensory neurons is compromised regardless of the location of neurotrophin stimulation, we postulate that ERK5 and MEF2D regulate additional anti-apoptotic retrograde response genes. Intriguingly, igf-1 and alsin are among the putative retrograde response genes (Fig. 6E). IGF-1 promotes neuronal survival by binding to IGF–1R and activating PI3 kinase and Akt (Dudek et al., 1997; Bondy and Cheng, 2004), whereas the alsin gene is mutated in ALS2, an early onset motor neuron degenerative disease (Yang et al., 2001; Eymard-Pierre et al., 2002; Rowland, 2005). Alsin protein is a vesicle-associated molecule that has been implicated in the endocytosis and trafficking of neurotrophin receptors (Devon et al., 2006) and so is potentially important for long-range survival pathways. Thus, we suggest that the retrograde response genes in aggregate encode components needed for the prolonged survival and functioning of mature neurons with long axons.
Induction of MEF2D by target-derived neurotrophins promotes survival of sensory neurons appropriately connected in a functional circuit, and this is mediated in part by increased transcription of bcl-w. Although our studies have focused on the roles played by ERK5 and MEF2D in neuronal survival in response to target-derived neurotrophins, it is likely that retrograde signaling and retrograde response genes also function in other spatially distinctive responses to neurotrophins. Interestingly, MEF2 transcription factors have been implicated in regulating synaptic connections. Recent studies have shown that a MEF2-dependent transcription program is critical for establishing synaptic morphology and for regulating synapse number (Shalizi and Bonni, 2005; Flavell et al., 2006; Shalizi et al., 2006). Therefore, increased expression of MEF2D in response to neurotrophin stimulation of distal axons may eliminate supernumerary synapses, while preserving neuronal survival. In this way, target-derived neurotrophins may play a role in maintaining the cellular constituents of newly established functional circuits, while simultaneously refining the synaptic connections.
Footnotes
This work was supported by grants from the National Institutes of Health (NIH) (R.A.S., M.E.G.). We thank Grant MacGregor (University of California, Irvine, Irvine, CA), Susan Corey (The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia), and Steve Gutkind (NIH) for the generous gifts of reagents. We thank Todd Golub for advice. We thank Erin Berry, Kelly Dakin, Parizad Bilimoria, and Athar Malik for technical assistance.
References
- Ahn S, Riccio A, Ginty DD. Spatial considerations for stimulus-dependent transcription in neurons. Annu Rev Physiol. 2000;62:803–823. doi: 10.1146/annurev.physiol.62.1.803. [DOI] [PubMed] [Google Scholar]
- Boillée S, Cleveland DW. Gene therapy for ALS delivers. Trends Neurosci. 2004;27:235–238. doi: 10.1016/j.tins.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Bondy CA, Cheng CM. Signaling by insulin-like growth factor 1 in brain. Eur J Pharmacol. 2004;490:25–31. doi: 10.1016/j.ejphar.2004.02.042. [DOI] [PubMed] [Google Scholar]
- Campenot RB. Development of sympathetic neurons in compartmentalized cultures. II. Local control of neurite survival by nerve growth factor. Dev Biol. 1982;93:13–21. doi: 10.1016/0012-1606(82)90233-0. [DOI] [PubMed] [Google Scholar]
- Campenot RB. NGF and the local control of nerve terminal growth. [Review] J Neurobiol. 1994;25:599–611. doi: 10.1002/neu.480250603. [DOI] [PubMed] [Google Scholar]
- Carroll P, Lewin GR, Koltzenburg M, Toyka KV, Thoenen H. A role for BDNF in mechanosensation. Nat Neurosci. 1998;1:42–46. doi: 10.1038/242. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Gu C, Khursigara G, Chao MV. p75 neurotrophin receptor as a modulator of survival and death decisions. Microsc Res Tech. 1999;45:217–224. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<217::AID-JEMT5>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Chao MV, Rajagopal R, Lee FS. Neurotrophin signalling in health and disease. Clin Sci (Lond) 2006;110:167–173. doi: 10.1042/CS20050163. [DOI] [PubMed] [Google Scholar]
- Chen WP, Chang YC, Hsieh ST. Trophic interactions between sensory nerves and their targets. J Biomed Sci. 1999;6:79–85. doi: 10.1007/BF02256438. [DOI] [PubMed] [Google Scholar]
- Davila D, Piriz J, Trejo JL, Nunez A, Torres-Aleman I. Insulin and insulin-like growth factor I signalling in neurons. Front Biosci. 2007;12:3194–3202. doi: 10.2741/2306. [DOI] [PubMed] [Google Scholar]
- Delcroix JD, Valletta JS, Wu C, Hunt SJ, Kowal AS, Mobley WC. NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron. 2003;39:69–84. doi: 10.1016/s0896-6273(03)00397-0. [DOI] [PubMed] [Google Scholar]
- Devon RS, Orban PC, Gerrow K, Barbieri MA, Schwab C, Cao LP, Helm JR, Bissada N, Cruz-Aguado R, Davidson TL, Witmer J, Metzler M, Lam CK, Tetzlaff W, Simpson EM, McCaffery JM, El-Husseini AE, Leavitt BR, Hayden MR. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc Natl Acad Sci U S A. 2006;103:9595–9600. doi: 10.1073/pnas.0510197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Eymard-Pierre E, Lesca G, Dollet S, Santorelli FM, di Capua M, Bertini E, Boespflug-Tanguy O. Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. Am J Hum Genet. 2002;71:518–527. doi: 10.1086/342359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- Gan L, Zheng W, Chabot JG, Unterman TG, Quirion R. Nuclear/cytoplasmic shuttling of the transcription factor FoxO1 is regulated by neurotrophic factors. J Neurochem. 2005;93:1209–1219. doi: 10.1111/j.1471-4159.2005.03108.x. [DOI] [PubMed] [Google Scholar]
- Ginty DD, Segal RA. Retrograde neurotrophin signaling: Trk-ing along the axon. Curr Opin Neurobiol. 2002;12:268–274. doi: 10.1016/s0959-4388(02)00326-4. [DOI] [PubMed] [Google Scholar]
- González-Martínez T, Fariñas I, Del Valle ME, Feito J, Germanà G, Cobo J, Vega JA. BDNF, but not NT-4, is necessary for normal development of Meissner corpuscles. Neurosci Lett. 2005;377:12–15. doi: 10.1016/j.neulet.2004.11.078. [DOI] [PubMed] [Google Scholar]
- Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, Crabtree GR. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell. 2003;113:657–670. doi: 10.1016/s0092-8674(03)00390-8. [DOI] [PubMed] [Google Scholar]
- Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC. Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J Neurosci. 1996;16:7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V, Levi-Montalcini R. Proliferation, differentiation and degeneration in the spinal ganglia of the chick embryo under normal and experimental conditions. J Exp Zool. 1949;111:457–501. doi: 10.1002/jez.1401110308. [DOI] [PubMed] [Google Scholar]
- Han TH, Prywes R. Regulatory role of MEF2D in serum induction of the c-jun promoter. Mol Cell Biol. 1995;15:2907–2915. doi: 10.1128/mcb.15.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerssen HM, Segal RA. Location, location, location: a spatial view of neurotrophin signal transduction. Trends Neurosci. 2002;25:160–165. doi: 10.1016/s0166-2236(02)02144-6. [DOI] [PubMed] [Google Scholar]
- Heerssen HM, Pazyra MF, Segal RA. Dynein motors transport activated Trks to promote survival of target-dependent neurons. Nat Neurosci. 2004;7:596–604. doi: 10.1038/nn1242. [DOI] [PubMed] [Google Scholar]
- Heidenreich KA, Linseman DA. Myocyte enhancer factor-2 transcription factors in neuronal differentiation and survival. Mol Neurobiol. 2004;29:155–166. doi: 10.1385/MN:29:2:155. [DOI] [PubMed] [Google Scholar]
- Hempstead BL. Dissecting the diverse actions of pro- and mature neurotrophins. Curr Alzheimer Res. 2006;3:19–24. doi: 10.2174/156720506775697061. [DOI] [PubMed] [Google Scholar]
- Hendry IA, Stach R, Herrup K. Characteristics of the retrograde axonal transport system for nerve growth factor in the sympathetic nervous system. Brain Res. 1974;82:117–128. doi: 10.1016/0006-8993(74)90897-x. [DOI] [PubMed] [Google Scholar]
- Howe CL, Valletta JS, Rusnak AS, Mobley WC. NGF signaling from clathrin-coated vesicles. Evidence that signaling endosomes serve as a platform for the Ras-MAPK pathway. Neuron. 2001;32:801–814. doi: 10.1016/s0896-6273(01)00526-8. [DOI] [PubMed] [Google Scholar]
- Kasler HG, Victoria J, Duramad O, Winoto A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol Cell Biol. 2000;20:8382–8389. doi: 10.1128/mcb.20.22.8382-8389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsching S. The neurotrophic factor concept: a reexamination. J Neurosci. 1993;13:2739–2748. doi: 10.1523/JNEUROSCI.13-07-02739.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruvilla R, Ye H, Ginty DD. Spatially and functionally distinct roles of the PI3-K effector pathway during NGF signaling in sympathetic neurons. Neuron. 2000;27:499–512. doi: 10.1016/s0896-6273(00)00061-1. [DOI] [PubMed] [Google Scholar]
- Lee FS, Kim AH, Khursigara G, Chao MV. The uniqueness of being a neurotrophin receptor. Curr Opin Neurobiol. 2001;11:281–286. doi: 10.1016/s0959-4388(00)00209-9. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R, Angeletti PU. Nerve growth factor. Physiol Rev. 1968;48:534–569. doi: 10.1152/physrev.1968.48.3.534. [DOI] [PubMed] [Google Scholar]
- Liu L, Cavanaugh JE, Wang Y, Sakagami H, Mao Z, Xia Z. ERK5 activation of MEF2-mediated gene expression plays a critical role in BDNF-promoted survival of developing but not mature cortical neurons. Proc Natl Acad Sci U S A. 2003;100:8532–8537. doi: 10.1073/pnas.1332804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lom B, Cohen-Cory S. Brain-derived neurotrophic factor differentially regulates retinal ganglion cell dendritic and axonal arborization in vivo. J Neurosci. 1999;19:9928–9938. doi: 10.1523/JNEUROSCI.19-22-09928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lom B, Cogen J, Sanchez AL, Vu T, Cohen-Cory S. Local and target-derived brain-derived neurotrophic factor exert opposing effects on the dendritic arborization of retinal ganglion cells in vivo. J Neurosci. 2002;22:7639–7649. doi: 10.1523/JNEUROSCI.22-17-07639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6:603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- Markus A, Zhong J, Snider WD. Raf and akt mediate distinct aspects of sensory axon growth. Neuron. 2002;35:65–76. doi: 10.1016/s0896-6273(02)00752-3. [DOI] [PubMed] [Google Scholar]
- McCaig CD, Sangster L, Stewart R. Neurotrophins enhance electric field-directed growth cone guidance and directed nerve branching. Dev Dyn. 2000;217:299–308. doi: 10.1002/(SICI)1097-0177(200003)217:3<299::AID-DVDY8>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Moises T, Dreier A, Flohr S, Esser M, Brauers E, Reiss K, Merken D, Weis J, Krüttgen A. Tracking TrkA's trafficking: NGF receptor trafficking controls NGF receptor signaling. Mol Neurobiol. 2007;35:151–159. doi: 10.1007/s12035-007-8000-1. [DOI] [PubMed] [Google Scholar]
- Neet KE, Campenot RB. Receptor binding, internalization, and retrograde transport of neurotrophic factors. Cell Mol Life Sci. 2001;58:1021–1035. doi: 10.1007/PL00000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Print CG, Loveland KL, Gibson L, Meehan T, Stylianou A, Wreford N, de Kretser D, Metcalf D, Köntgen F, Adams JM, Cory S. Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci U S A. 1998;95:12424–12431. doi: 10.1073/pnas.95.21.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves D. The trophic theory of neural connections. Trends Neurosci. 1986;9:486–489. [Google Scholar]
- Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Pierchala BA, Ciarallo CL, Ginty DD. An NGF-Trka-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science. 1997;277:1097–1100. doi: 10.1126/science.277.5329.1097. [DOI] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- Ross AJ, Waymire KG, Moss JE, Parlow AF, Skinner MK, Russell LD, MacGregor GR. Testicular degeneration in Bclw-deficient mice. Nat Genet. 1998;18:251–256. doi: 10.1038/ng0398-251. [DOI] [PubMed] [Google Scholar]
- Roux PP, Barker PA. Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol. 2002;67:203–233. doi: 10.1016/s0301-0082(02)00016-3. [DOI] [PubMed] [Google Scholar]
- Rowland LP. Primary lateral sclerosis, hereditary spastic paraplegia, and mutations in the alsin gene: historical background for the first International Conference. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:67–76. doi: 10.1080/14660820510039032. [DOI] [PubMed] [Google Scholar]
- Segal RA. Selectivity in neurotrophin signaling: theme and variations. Annu Rev Neurosci. 2003;26:299–330. doi: 10.1146/annurev.neuro.26.041002.131421. [DOI] [PubMed] [Google Scholar]
- Segal RA, Takahashi H, McKay RD. Changes in neurotrophin responsiveness during the development of cerebellar granule neurons. Neuron. 1992;9:1041–1052. doi: 10.1016/0896-6273(92)90064-k. [DOI] [PubMed] [Google Scholar]
- Senger DL, Campenot RB. Rapid retrograde tyrosine phosphorylation of TrkA and other proteins in rat sympathetic neurons in compartmented cultures. J Cell Biol. 1997;138:411–421. doi: 10.1083/jcb.138.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfried J, Wang X, Kharebava G, Tournier C. A novel mitogen-activated protein kinase docking site in the N terminus of MEK5alpha organizes the components of the extracellular signal-regulated kinase 5 signaling pathway. Mol Cell Biol. 2005;25:9820–9828. doi: 10.1128/MCB.25.22.9820-9828.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalizi A, Lehtinen M, Gaudilliere B, Donovan N, Han J, Konishi Y, Bonni A. Characterization of a neurotrophin signaling mechanism that mediates neuron survival in a temporally specific pattern. J Neurosci. 2003;23:7326–7336. doi: 10.1523/JNEUROSCI.23-19-07326.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalizi A, Gaudillière B, Yuan Z, Stegmüller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006;311:1012–1017. doi: 10.1126/science.1122513. [DOI] [PubMed] [Google Scholar]
- Shalizi AK, Bonni A. Brawn for brains: the role of MEF2 proteins in the developing nervous system. Curr Top Dev Biol. 2005;69:239–266. doi: 10.1016/S0070-2153(05)69009-6. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Ginty DD. Characterization of an NGF-P-trkA retrograde-signaling complex and age- dependent regulation of TrkA phosphorylation in sympathetic neurons. J Neurosci. 1999;19:8207–8218. doi: 10.1523/JNEUROSCI.19-19-08207.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turjanski AG, Vaqué JP, Gutkind JS. MAP kinases and the control of nuclear events. Oncogene. 2007;26:3240–3253. doi: 10.1038/sj.onc.1210415. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Endo Y, Kimoto K, Katoh-Semba R, Arakawa Y. Functional regulation of tactile sense by brain-derived neurotrophic factor in adult rats during acute inflammation. Neuroscience. 2000;97:171–175. doi: 10.1016/s0306-4522(99)00584-9. [DOI] [PubMed] [Google Scholar]
- Watson FL, Heerssen HM, Moheban DB, Lin MZ, Sauvageot CM, Bhattacharyya A, Pomeroy SL, Segal RA. Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J Neurosci. 1999;19:7889–7900. doi: 10.1523/JNEUROSCI.19-18-07889.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson FL, Heerssen HM, Bhattacharyya A, Klesse L, Lin MZ, Segal RA. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat Neurosci. 2001;4:981–988. doi: 10.1038/nn720. [DOI] [PubMed] [Google Scholar]
- Weible MW, 2nd, Bartlett SE, Reynolds AJ, Hendry IA. Prolonged recycling of internalized neurotrophins in the nerve terminal. Cytometry. 2001;43:182–188. doi: 10.1002/1097-0320(20010301)43:3<182::aid-cyto1047>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Wu C, Lai CF, Mobley WC. Nerve growth factor activates persistent Rap1 signaling in endosomes. J Neurosci. 2001;21:5406–5416. doi: 10.1523/JNEUROSCI.21-15-05406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M, Hung WY, Ouahchi K, Yan J, Azim AC, Cole N, Gascon G, Yagmour A, Ben-Hamida M, Pericak-Vance M, Hentati F, Siddique T. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29:160–165. doi: 10.1038/ng1001-160. [DOI] [PubMed] [Google Scholar]
- Yano H, Chao MV. Neurotrophin receptor structure and interactions. Pharm Acta Helv. 2000;74:253–260. doi: 10.1016/s0031-6865(99)00036-9. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Snider WD. Intracellular control of developmental and regenerative axon growth. Philos Trans R Soc Lond B Biol Sci. 2006;361:1575–1592. doi: 10.1098/rstb.2006.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]