

Abstract
The late stages of biosynthesis of phosphinothricin tripeptide (PTT) involve peptide formation and methylation on phosphorus. The exact timing of these transformations is not known. To provide insight into this question, we developed a heterologous expression system for PhsA, one of three NRPS proteins in PTT biosynthesis. The apparent kcat/Km value for ATP−pyrophosphate exchange activity for d,l-N-acetylphosphinothricin was 3.5 μM−1 min−1, whereas the kcat/Km,app for l-N-acetyldemethylphosphinothricin was 0.5 μM−1 min−1, suggesting the former might be the physiological substrate. Each substrate could be loaded onto the phosphopantetheine arm of the thiolation domain as observed by Fourier transform mass spectrometry (FTMS).
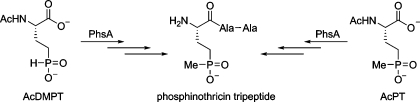
PTT1 (Figure 1A) is produced by Streptomyces viridochromogenes and Streptomyces hygroscopicus(1). The peptide contains the unusual phosphinic acid phosphinothricin (PT), containing two carbon−phosphorus bonds. PTT is widely used as an herbicide under the trade name Herbiace, and the ammonium salt of PT is the active ingredient in BASTA. PT is an inhibitor of glutamine synthetase, resulting in ammonia buildup that is toxic to plants. Transgenic plants engineered to contain the PT resistance gene [pat in S. viridochromogenes(2) or bar in S. hygroscopicus(3)] have found widespread use in agriculture in the form of LibertyLink genetically modified crops such as corn, soy, cotton, and canola. The PTT biosynthetic gene cluster from S. viridochromogenes was recently sequenced (4,5). The final steps in PTT biosynthesis involve the formation of the amide linkages as well as installation of the methyl group on the phosphorus atom (Figure 1A) (6−10). We have been interested in the latter process as it is thought to be an unusual vitamin B12-dependent transformation (11,12) catalyzed by PhpK (13). At present, the exact timing of the methyl transfer step, whether before, during, or after peptide assembly, is not known. A mutant in phsA, encoding one of three nonribosomal peptide synthetase proteins in the PTT gene cluster [PhsA, PhsB, and PhsC (7,8,10)], accumulated N-acetyldemethylphosphinothricin (AcDMPT), suggesting that this phosphinate amino acid is the substrate for PhsA (14,15). PhsA is the initiation module for synthesis of the tripeptide (7) and contains an adenylation (A) domain that activates its substrate amino acid to the corresponding aminoacyl adenylate, a thiolation (T) domain that covalently tethers the amino acid to the phosphopantetheine (PPant) arm of the peptide carrier protein, and a putative N-terminal thioesterase sequence motif of unknown function (10). After the phosphinate amino acid had been charged onto PhsA, the two peptide bond forming events are catalyzed by PhsB and PhsC, each loaded with Ala (7,8).
Figure 1.

(A) Currently accepted biosynthetic pathway for PTT shown with solid arrows. An alternative pathway in which AcDMPT is first methylated and then incorporated into the tripeptide is depicted with dashed arrows. (B) Acylated amino acids tested in this work as substrates for PhsA.
Keller, Wohlleben, and co-workers isolated native PhsA from S. viridochromogenes in small quantities (7). Attempts to express the enzyme in Escherichia coli to obtain larger amounts of protein were not successful. Sufficient native protein was obtained to study its activity by ATP−pyrophosphate exchange assay, demonstrating that AcDMPT was utilized most efficiently under the conditions employed, but that N-acetylphosphinothricin (AcPT) was also a substrate. The observed activity with AcDMPT suggests that the methylation step may be a tailoring reaction that takes place while AcDMPT (or its di- or tripeptide) is tethered to the phosphopantetheine arm of PhsA (or PhsB/PhsC). Given the large quantities of protein that would be required to investigate the molecular details of PTT assembly, we decided to revisit expression of active PhsA in E. coli. The results of this study as well as previous studies by Seto and co-workers (13,16) suggest that an alternative pathway in which AcDMPT is first methylated cannot be ruled out (dashed arrows, Figure 1A).
Initial efforts to express PhsA as an N-terminally His6-tagged protein in E. coli BL21(DE3) produced insoluble protein. To increase solubility, phsA from S. viridochromogenes DSM 40736 was inserted into pMAL-c2X to introduce maltose binding protein (MBP) as a fusion at the N-terminus. Overexpression of the MBP−PhsA fusion protein indeed resulted in greatly improved solubility. However, purification by amylose affinity chromatography did not result in high-purity protein. To improve the purification, the malE-phsA fragment was cloned into pET28b and the MBP−PhsA−His6 protein was heterologously expressed in E. coli. Rapid purification of the fusion protein by Ni-NTA chromatography followed by HiTrap Q HP ion exchange chromatography yielded 7 mg of protein per liter of culture.
As isolated, the recombinantly expressed MBP−PhsA−His6 protein did not contain detectable amounts of the phosphopantetheine arm as monitored by FTMS. Two experiments were performed to examine the in vitro phosphopantetheinylation of the MBP−PhsA−His6 protein by Sfp, a highly promiscuous phosphopantetheinyl transferase from Bacillus subtilis(17). In the first experiment, the CoA analogue BODIPY-FL-N-(2-aminoethyl)maleimidyl-S-CoA was employed to install a fluorescently labeled phosphopantetheine arm on the active site serine residue in the PhsA thiolation domain, which was readily detected spectrofluorometrically after SDS−PAGE (Figure S1). In a second experiment, [3H]acetyl-CoA was utilized as a cosubstrate, resulting in a time-dependent increase in the level of incorporation of tritium into the MBP−PhsA−His6 protein during incubation with Sfp. Maximum incorporation of tritium was near stoichiometric (Figure S2), confirming that Sfp converts the apo MBP−PhsA−His6 protein to its holo form.
The substrate analogues shown in Figure 1B were prepared as described in the . The substrate set was used to probe PhsA activity by the ATP−PPi exchange assay. At 2 mM, l-N-AcAsp and l-N-AcAP4 showed very low activity and were not investigated further. On the other hand, l-N-AcGlu, l-AcDMPT, and d,l-AcPT showed significant activity and the substrate concentration dependence was further investigated. It has been noted for other studies of the kinetics of the ATP−PPi exchange reaction that the unlabeled reactants very rapidly establish an equilibrium and hence that the observed rate of exchange is the equilibrium rate of production of radiolabeled ATP from labeled PPi(18). As such, although the ATP−PPi exchange reaction provides insight into enzyme specificity, it is not straightforward to interpret its kinetics in the framework of the Michaelis−Menten equation (18). However, because of the difficulties associated with measuring direct rates of adenylation (19,20), the ATP−PPi exchange rate has been frequently used to investigate substrate selectivities (e.g., refs (20−22)). Importantly, in cases in which adenylation could be measured directly and the results could be compared with the rates of ATP−PPi exchange, very similar selectivities were observed (20). The initial exchange rates plotted against substrate concentration are provided in Figure S4.
At high concentrations, the observed exchange rate for AcDMPT [125 min−1 (Table 1)] was larger than that for AcPT (84 min−1), consistent with previous results with native PhsA that showed 74% lower activity with AcPT (7), at substrate concentrations of 667 μM (U. Keller, personal communication). The specific activity of the recombinant enzyme for the ATP−pyrophosphate exchange activity under similar conditions was slightly (1.7-fold) higher than that of the native enzyme. However, surprisingly, the apparent Km values of the two potential natural substrates, AcPT and AcDMPT, showed that at low substrate concentrations AcPT is the preferred substrate for amino acid-stimulated ATP−PPi exchange (Table 1). This preference is even more pronounced when considering that AcPT was a mixture of enantiomers, whereas AcDMPT was stereochemically pure. The lower Km,app for AcPT may indicate that this compound is the physiological substrate for PhsA, provided that free AcDMPT and AcPT are both biosynthetic intermediates (i.e., that the P-methyltransferase PhpK acts on AcDMPT in solution). The substrate specificity of the holo MBP−PhsA−His6 protein was further analyzed by competition experiments using FTMS. The protein (12.4 μM) was incubated with a 1:1 mixture of AcDMPT and AcPT with each amino acid at 25, 250, and 450 μM in the presence of 1 mM ATP. After 1 h, the protein was analyzed by FTMS using the phosphopantetheinyl (PPant) ejection assay (23,24). The ions corresponding to PPant loaded with AcPT (466.1778) and AcDMPT (452.1616) were observed (Figure 2) as well as the parent PPant ejection ion (261.1268). In two independent experiments, the ratios of the peak intensities of the AcPT−PPant and AcDMPT−PPant compounds favor the former at all three substrate concentrations. The presence of the PPant ejection ion (Figure S8) and its unknown origin (holo PhsA, AcPT−PhsA, or Ac-DMPT−PhsA) complicate interpretation, but the FTMS experiments confirm that both substrates are not only adenylated but also loaded onto the T-domain.
Table 1. Kinetic Parameters for the N-Acetyl Amino Acid-Stimulated ATP−PPi Exchange Reaction by the MBP−PhsA−His6 Protein in Its Apo and Holo Forms.
| substrate | recombinant PhsA form | kcat (min−1) | Km (μM) | kcat/Km (μM−1 min−1) | relative activity of endogenous enzyme (7) |
|---|---|---|---|---|---|
| (±)-AcPT | apo | 87.9 ± 2.9 | 26.9 ± 2.9 | 3.27 ± 0.37 | 74% |
| holo | 83.5 ± 2.0 | 23.7 ± 1.9 | 3.52 ± 0.29 | ||
| l-AcDMPT | apo | 122.9 ± 2.9 | 198 ± 18 | 0.62 ± 0.06 | 100% |
| holo | 124.7 ± 6.5 | 254 ± 44 | 0.49 ± 0.09 | ||
| N-AcGlu | apo | 25.0 ± 1.1 | 349 ± 55 | 0.0717 ± 0.011 | 11% |
| holo | 28.8 ± 1.5 | 493 ± 82 | 0.0584 ± 0.010 |
Figure 2.

Competition loading results determined by the LC−FTMS PPant ejection assay at the indicated substrate concentrations. Proposed structures of Ppant ejection ions are colored to match corresponding peaks.
The preference for AcPT even at high substrate concentrations where AcDMPT is a better substrate for the PPi exchange reaction could be due to selective substrate loading with a preference for adenylated AcPT over adenylated AcDMPT, a fidelity filter not usually observed in NRPS systems (19,20). Alternatively, the putative thioesterase motif at the N-terminus of PhsA (10) may be involved in proofreading and could catalyze selective hydrolysis of misloaded amino acids in a posttransfer editing step. To test this possibility, the conserved catalytic Ser16 in this motif was mutated to Ala and the loading experiment was repeated. The same selectivity was observed as for the wild-type enzyme, ruling out an editing role for the thioesterase motif (Figure S6).
Collectively, the observations in this study do not clarify the identity of the physiological substrate for PhsA . On one hand, a phsA disruption mutant accumulated AcDMPT, suggesting that this compound is the natural substrate (14,15). On the other hand, the results reported here suggest it may be AcPT. In support of the latter model, Seto and co-workers showed that a mutant producer strain accumulated AcPT and AcPT-Ala-Ala, suggesting free AcDMPT can be methylated by PhpK (16). Similarly, the cell-free extract of a Streptomyces lividans strain carrying a plasmid with a 9.5 kb fragment of the PTT biosynthetic gene cluster from S. hygroscopicus was able to carry out the methylation of both AcDMPT and AcDMPT-Ala-Ala (13). Of note, this DNA fragment carries the phpK and phsA genes but not the genes for PhsB and PhsC. Since AcDMPT-Ala-Ala is unlikely to be loaded onto PhsA, these results suggest the P-methyltransferase can act on free AcDMPT-Ala-Ala (free meaning not bound to an NRPS) and presumably also free AcDMPT. In summary, we have been successful in establishing an overexpression system for PhsA in E. coli that can be used for further studies on this system.
Acknowledgments
We thank Prof. Dr. W. Wohlleben and Prof. Dr. U. Keller for sharing unpublished data and helpful discussions.
Supporting Information Available
Procedures for substrate synthesis (25,26) and molecular biology and supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the National Institutes of Health (Grant PO1 GM077596 to N.L.K. and W.A.v.d.D.) and a Ruth L. Kirschstein National Research Service Award (Grant GM008276 to B.S.E.).
Footnotes
Abbreviations: AcDMPT, N-acetyldemethylphosphinothricin; AcPT, N-acetylphosphinothricin; MBP, maltose binding protein; Ppant, phosphopantetheine; PTT, phosphinothricin tripeptide.
Supplementary Material
References
- Seto H.; Kuzuyama T. (1999) Nat. Prod. Rep. 16, 589–596. [DOI] [PubMed] [Google Scholar]
- Strauch E.; Wohlleben W.; Puhler A. (1988) Gene 63, 65–74. [DOI] [PubMed] [Google Scholar]
- Kumada Y.; Anzai H.; Takano E.; Murakami T.; Hara O.; Itoh R.; Imai S.; Satoh A.; Nagaoka K. (1988) J. Antibiot. 41, 1838–1845. [DOI] [PubMed] [Google Scholar]
- Schwartz D.; Berger S.; Heinzelmann E.; Muschko K.; Welzel K.; Wohlleben W. (2004) Appl. Environ. Microbiol. 70, 7093–7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blodgett J. A.; Zhang J. K.; Metcalf W. W. (2005) Antimicrob. Agents Chemother. 49, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz D.; Alijah R.; Nussbaumer B.; Pelzer S.; Wohlleben W. (1996) Appl. Environ. Microbiol. 62, 570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammel N.; Schwartz D.; Wohlleben W.; Keller U. (1998) Biochemistry 37, 1596–1603. [DOI] [PubMed] [Google Scholar]
- Schwartz D.; Grammel N.; Heinzelmann E.; Keller U.; Wohlleben W. (2005) Antimicrob. Agents Chemother. 49, 4598–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blodgett J. A.; Thomas P. M.; Li G.; Velasquez J. E.; van der Donk W. A.; Kelleher N. L.; Metcalf W. W. (2007) Nat. Chem. Biol. 3, 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eys S.; Schwartz D.; Wohlleben W.; Schinko E. (2008) Antimicrob. Agents Chemother. 52, 1686–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodyer R. D.; Li G.; Zhao H.; van der Donk W. A. (2007) Chem. Commun., 359–361. [DOI] [PubMed] [Google Scholar]
- van der Donk W. A. (2006) J. Org. Chem. 71, 9561–9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamigiri K.; Hidaka T.; Imai S.; Murakami T.; Seto H. (1992) J. Antibiot. 45, 781–787. [DOI] [PubMed] [Google Scholar]
- Alijah R.; Dorendorf J.; Talay S.; Puhler A.; Wohlleben W. (1991) Appl. Microbiol. Biotechnol. 34, 749–755. [DOI] [PubMed] [Google Scholar]
- Wohlleben W.; Alijah R.; Dorendorf J.; Hillemann D.; Nussbaumer B.; Pelzer S. (1992) Gene 115, 127–132. [DOI] [PubMed] [Google Scholar]
- Imai S.; Seto H.; Sasaki T.; Tsuruoka T.; Ogawa H.; Satoh A.; Inouye S.; Niida T.; Otake N. (1985) J. Antibiot. 38, 687–690. [DOI] [PubMed] [Google Scholar]
- Quadri L. E.; Weinreb P. H.; Lei M.; Nakano M. M.; Zuber P.; Walsh C. T. (1998) Biochemistry 37, 1585–1595. [DOI] [PubMed] [Google Scholar]
- Cole F. X.; Schimmel P. R. (1970) Biochemistry 9, 480–489. [DOI] [PubMed] [Google Scholar]
- Keating T. A.; Suo Z.; Ehmann D. E.; Walsh C. T. (2000) Biochemistry 39, 2297–2306. [DOI] [PubMed] [Google Scholar]
- Luo L.; Burkart M. D.; Stachelhaus T.; Walsh C. T. (2001) J. Am. Chem. Soc. 123, 11208–11218. [DOI] [PubMed] [Google Scholar]
- Stachelhaus T.; Marahiel M. A. (1995) J. Biol. Chem. 270, 6163–6169. [DOI] [PubMed] [Google Scholar]
- Gatto G. J. Jr.; McLoughlin S. M.; Kelleher N. L.; Walsh C. T. (2005) Biochemistry 44, 5993–6002. [DOI] [PubMed] [Google Scholar]
- Dorrestein P. C.; Bumpus S. B.; Calderone C. T.; Garneau-Tsodikova S.; Aron Z. D.; Straight P. D.; Kolter R.; Walsh C. T.; Kelleher N. L. (2006) Biochemistry 45, 12756–12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen D. B.; Bumpus S. B.; Aron Z. D.; Kelleher N. L.; Walsh C. T. (2007) J. Am. Chem. Soc. 129, 6366–6367. [DOI] [PubMed] [Google Scholar]
- Selvam C.; Goudet C.; Oueslati N.; Pin J. P.; Acher F. C. (2007) J. Med. Chem. 50, 4656–4664. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Lee K.; Liu P. (2008) Org. Lett. 10, 5521–5524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.