

Abstract
Leukocyte transendothelial migration into inflamed areas is regulated by the integrity of endothelial cell junctions and is stabilized by adhesion molecules including junctional adhesion molecule-A (JAM-A). JAM-A has been shown to participate in homophilic interactions with itself and in heterophilic interactions with leukocyte function-associated antigen-1 (LFA-1) via its first and second immunoglobulin domains, respectively. Using competitive binding assays in conjunction with atomic force microscopy adhesion measurements, we provide compelling evidence that the second domain of JAM-A stabilizes the homophilic interaction because its deletion suppresses the dynamic strength of the JAM-A homophilic interaction. Moreover, binding of the LFA-1 inserted domain to the second domain of JAM-A reduces the dynamic strength of the JAM-A homophilic interaction to the level measured with the JAM-A domain 2 deletion mutant. This finding suggests that LFA-1 binding cancels the stabilizing effects of the second immunoglobulin domain of JAM-A. Finally, our atomic force microscopy measurements reveal that the interaction of JAM-A with LFA-1 is stronger than the JAM-A homophilic interaction. Taken together, these results suggest that LFA-1 binding to JAM-A destabilizes the JAM-A homophilic interaction. In turn, the greater strength of the LFA-1/JAM-A complex permits it to support the tension needed to disrupt the JAM-A homophilic interaction, thus allowing transendothelial migration to proceed.
Introduction
The migration of leukocytes from the blood stream into surrounding tissues is a critical process during immune surveillance as well as inflammatory disease states such as atherosclerosis (1,2). During inflammatory conditions, leukocytes accumulate at the site of injury by first rolling on the endothelium and then undergoing firm adhesion after their activation in response to chemokines (1). These processes are mediated by adhesion molecules. Selectins have been shown to mediate cell rolling. Both integrins and immunoglobulin superfamily members, including the intercellular adhesion molecule-1 (ICAM-1) and the vascular cell adhesion molecule-1 (VCAM-1), mediate firm adhesion of the leukocyte to the endothelium (3). This process is followed by the subsequent migration of the leukocytes across the endothelium.
Transendothelial migration (TEM) of leukocytes into inflamed areas takes place mainly via the paracellular pathway occurring through the junction located between adjacent endothelial cells. Recent reports (4,5) also confirmed the occurrence of migration via the transcellular pathway occurring through the body of the actual cell. The former and more predominant pathway is regulated by the integrity of the endothelial cell junctions, which are stabilized by many molecules (1,6). These molecules include platelet endothelial cellular adhesion molecule-1 (PECAM-1), the junctional adhesion molecule (JAM) family of receptors, and CD99.
This work focuses on JAM-A, a member of the JAM family of receptors that also includes JAM-B, JAM-C, JAM4, and JAML (7). The role of JAM-A was first implicated in transmigration by the finding that both in vitro and in vivo leukocyte transmigration were inhibited by an anti-JAM-A monoclonal antibody (8,9). JAM-A, also known as JAM-1 or F11R, belongs to the immunoglobulin superfamily of receptors. It is expressed as a dimer on the surface of circulating cells but is predominantly present in endothelial and epithelial tight junctions of many different tissues (8,10). JAM-A consists of an intracellular PDZ-domain binding motif, a transmembrane segment, and two extracellular immunoglobulin (Ig) domains. The PDZ-domain binding motif has been shown to associate with the tight junction components occludin, ZO-1, and cingulin and is involved in cell signaling (11,12). The first of these two Ig domains, the membrane-distal Ig domain, is involved in homophilic binding to another JAM-A receptor. This binding can take place across opposing endothelial cells, which comprise the tight junction (in trans), or between adjacent receptors during dimer formation (in cis) (13,14). During leukocyte TEM, the homophilic transendothelial interactions between these receptors must be disrupted to enable a migrating leukocyte to pass through the junction (10).
Previously, it was demonstrated that JAM-A can also interact with leukocyte function-associated antigen-1 (LFA-1) via its second membrane-proximal Ig domain (15,16). Integrin LFA-1 is an αβ heterodimeric transmembrane glycoprotein expressed on the surface of leukocytes (17). The LFA-1/JAM-A interaction plays a key role in the early events of leukocyte TEM. After inflammation, JAM-A is redistributed to the apical portion of the junction, allowing for leukocyte recruitment possibly via a haptotactic gradient (15). However, its role in the underlying mechanism of this process remains ill-defined. It has been postulated that during TEM a trimeric complex forms between LFA-1 on the migrating leukocyte and a junctional JAM-A complex formed in trans (15,18). For TEM to proceed, the JAM-A homophilic interaction must eventually be broken, leading to the loosening of junctional contacts and allowing the leukocyte to migrate.
To our knowledge, the second domain of JAM-A has been implicated only in the heterophilic interaction with LFA-1. Using competitive binding assays in conjunction with atomic force microscopy (AFM), we provide compelling evidence for the role of the second domain of JAM-A in stabilizing the JAM-A homophilic interaction. We postulate that the binding of LFA-1 to the second domain of JAM-A expressed on the endothelium may be the mechanism through which the JAM-A homophilic interactions formed across the endothelial junction are weakened.
Methods
Cells and reagents
The Jurkat and Chinese hamster ovary (CHO) cell lines were maintained in continuous culture in Roswell Park Memorial Institute 1640 and Dulbecco's modified Eagle's F-12 media, respectively. Both cultures were supplemented with 10% heat-inactivated fetal calf serum (Irvine Scientific, Santa Ana, CA), penicillin (50 U/mL; Gibco BRL, Grand Island, NY), and streptomycin (50 μg/mL, Gibco BRL) and were expanded on a 3-day cycle. Recombinant soluble human JAM-A was generated as previously described (16,19). JAM-A contains a cleavage site for tobacco etch virus (TEV) protease between the JAM-A and Fc domains. After digestion of JAM-A with TEV protease, the JAM-A extracellular domain was purified by affinity and anion exchange chromatography. Please note that we refer to JAM-A as JAM-A throughout this article. JAM-A·D1 and JAM-A·D2 were generated accordingly. Rabbit polyclonal antibody against JAM-A was generated by immunizing rabbits with human JAM-A·D1. The isolated extracellular domain of JAM-A was biotinylated using LC-NHS-(+)-biotin (Molecular Biosciences, Boulder, CO) according to manufacturer's instructions. Antibody against LFA-1 (clone TS1/22) was purified from culture supernatant of TS1/22 hybridoma cells (American Type Culture Collection, Manassas, VA) by protein A affinity chromatography.
Solid-phase binding assay
Microtiter wells (Maxisorp; Nunc, Roskilde, Denmark) were coated with JAM-A (10 μg/mL) or bovine serum albumin (BSA; 0 μg/mL) in 10 mM Tris, pH 9.0, and then blocked with 3% BSA. Binding of JAM-A·biotin to immobilized JAM-A was carried out in binding buffer (Tris-buffered saline (TBS), 0.5% BSA, 1 mM Mg2+, 1 mM Ca2+) containing increasing concentrations of JAM-A extracellular domain, JAM-A·D1, JAM-A·D2, or the Fab negative control (AbD Serotec, Düsseldorf, Germany) for 2 h at room temperature. After washing with wash buffer (TBS, 0.05% Tween), bound JAM-A·biotin was detected with peroxidase-conjugated streptavidin and quantified using the tetramethylbenzidine substrate reagent kit (Vector labs, Burlingame, CA). Nonspecific binding to BSA-coated wells was subtracted to calculate specific binding.
CHO cell adhesion assay
CHO cell adhesion assays were carried out as previously described (16) using CHO cells with a stable expression of human JAM-A or transfected with vector control. Microtiter wells (Maxisorp; Nunc) were coated with JAM-A (15 μg/mL) or BSA (15 μg/mL) in 10 mM Tris, pH 9.0, overnight at 4°C and then blocked with 0.5% BSA in phosphate-buffered saline (PBS) for 1 h at room temperature. CHO transfectants were labeled with BCECF-AM and allowed to adhere for 30 min at 37°C in binding buffer (Hank's balanced salt solution, 1 mM Mg2+, 1 mM Ca2+, 10 mM Hepes, pH 7.4). Some wells were preincubated with open inserted (I)-domain, wild-type I-domain, polyclonal antibody (pAb) anti-JAM-A, or soluble JAM-A at 10 μg/mL. Nonadherent cells were removed by washing twice with binding buffer, and the fluorescence of input and adherent cells was determined with a fluorescence plate reader (Tecan, Crailsheim, Germany). Background binding to BSA was negligible and subtracted.
Protein immobilization
A 20 μL aliquot of JAM-A or JAM-A·D2 at 50 μg/mL (single-molecule experiments) or 100 μg/mL (whole-cell experiments) in 0.1 M NaHCO3 (pH 8.6) was adsorbed overnight at 4°C on the center of a 35 mm tissue culture dish (Falcon 353001; Becton Dickinson Labware, Franklin Lakes, NJ). Unbound protein was removed by washing with PBS (10 mM PO43−, 150 mM NaCl, pH 7.3). The exposed surface of the dish was blocked using 0.01% BSA (Sigma, St. Louis, MO) in PBS to eliminate nonspecific binding to the dish surface. One coated dish was used to complete each AFM experiment.
AFM measurements of adhesion forces
AFM force measurements were carried out on an apparatus designed to be operated in the force spectroscopy mode (20–24). Jurkat cells were attached to the AFM cantilever by concanavalin A (Con A)-mediated linkages that were prepared as described previously (24).
A single Jurkat cell was attached to the cantilever by positioning the end of the Con A functionalized cantilever above the center of the cell and carefully lowering it onto the cell for ∼1 s. When attached, the cell was positioned right behind the AFM tip of the cantilever as shown in Fig. 5 A (24,25). To obtain multiple-bond interactions between the Jurkat cell and the immobilized protein, an indentation force of ∼200 pN and a contact time of 2 s were used. The experiments were carried out at a cantilever retraction rate of 5 μm/s at 25°C, as previously described (26). Approximately 50 measurements were acquired in each experiment.
Figure 5.
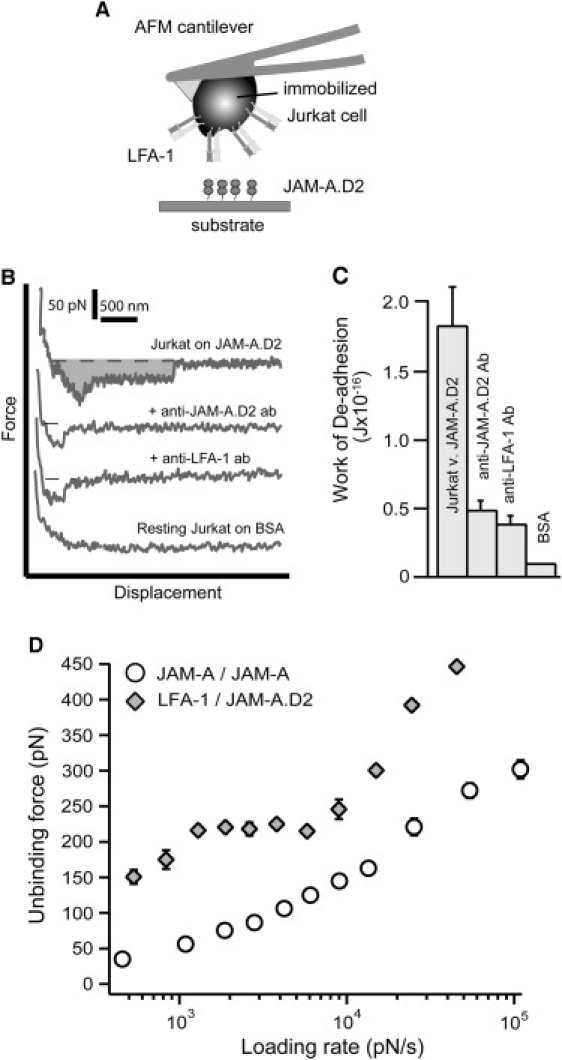
(A) Schematic representation of the experimental setup used to conduct Jurkat AFM cell adhesion studies. The LFA-1-expressing Jurkat cell was attached to the AFM cantilever and allowed to interact with JAM-A·D2 on the sample petri dish. (B) Whole-cell AFM adhesion measurements of Jurkat cells on JAM-A·D2 without treatment, in the presence of antibodies against JAM-A·D2 or LFA-1 (TS1/22), and on BSA. The shaded area represents the work of deadhesion. (C) The average calculated work of deadhesion from AFM whole-cell adhesion measurements of Jurkat cells on JAM-A·D2 without treatment, in the presence of anti-JAM-A·D2 antibody, TS1/22 antibody (anti-LFA-1), and Jurkat cells on BSA. The error is the standard error of three separate experiments. (C) Dynamic force spectra of the LFA-1/JAM-A·D2 (diamond) and JAM-A/JAM-A (circle) unbinding forces were acquired for a loading rate range of ∼500–100,000 pN/s. The error for both loading rate and unbinding force is the standard error of at least three separate experiments.
Alternately, JAM-A receptors were cross-linked to the AFM cantilever. To do so, each cantilever was washed in nanopure water for ∼1 min, irradiated with ultraviolet rays for 5 min, transferred into 2% 3-aminopropyl triethoxysilane in acetone, washed five times in nanopure water, transferred into 0.1% glutaraldehyde in 0.1 M PBS for 30 min at room temperature, washed five times in 0.1 M PBS, and coated with 100 μg/mL of JAM-A, JAM-A·D1, or JAM-A·D2. The cantilevers were incubated overnight at 4°C. Before use, each cantilever was blocked by soaking in 0.01% BSA in TBS for 20 min. Experiments were carried out in TBS buffer. The open I-domain was used at a concentration of 50 μg/mL.
Measurements of unitary LFA-1/JAM-A·D2, JAM-A/JAM-A, and JAM-A/JAM-A·D1 unbinding forces were obtained under conditions that minimized contact between the Jurkat cell or JAM-A receptors on the cantilever tip and the substrate. An adhesion frequency of < 30% in the force measurements ensured that there was a > 85% probability that the adhesion events were mediated by single bonds. Data were corrected for hydrodynamic drag (27,28). The damping coefficient is the slope of the linear fit of the change in hydrodynamic drag force versus cantilever speed and is ∼10 pN·s/μm for protein-coated cantilevers and 9 pN · s/μm for cantilevers with an attached cell. Approximately 3000 measurements were typically acquired over 6 to 8 h for each experiment.
Results
The deletion of the second domain of JAM-A weakens the JAM-A homophilic interaction
Solid-phase binding assays were conducted to compare the effectiveness of JAM-A and JAM-A·D1, which is a deletion mutant missing the second domain of JAM-A, in binding to JAM-A. To our knowledge, only the first domain of JAM-A is required for the JAM-A homophilic interaction, and so we expected the JAM-A·D1 mutant to be equally as effective as JAM-A in competing for JAM-A binding. In these experiments, JAM-A·biotin was allowed to bind to immobilized JAM-A, and specific binding was analyzed in the presence of increasing concentrations of unlabeled inhibitor (Fig. 1). If the JAM-A·biotin was effectively displaced by an inhibitor, a decrease in the percentage of bound JAM-A·biotin ligand was observed. This decrease was very pronounced when JAM-A was used for inhibition but significantly less so when the JAM-A·D1 mutant was used for inhibition (Fig. 1 A). Inhibition was not observed with JAM-A·D2, which is a deletion mutant missing the first domain, or the Fab negative control (Fig. 1 B). The diminished level at which the JAM-A·D1 mutant competed for JAM-A binding was surprising, and indicated that the presence of the second domain may be important for the JAM-A homophilic interaction.
Figure 1.
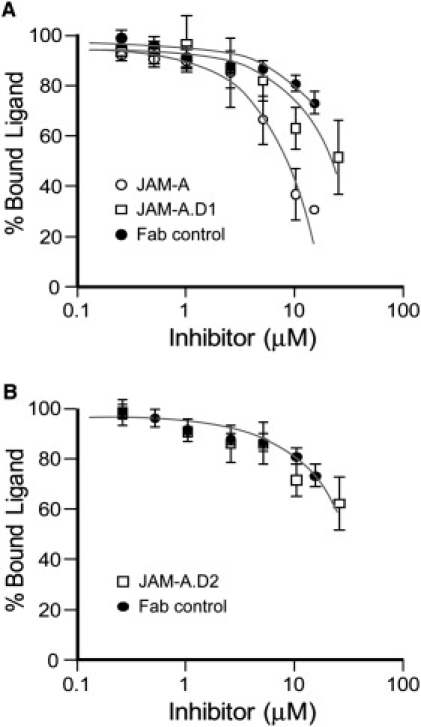
Solid-phase binding assays. (A) Specific binding of JAM-A·biotin to immobilized JAM-A was analyzed in the presence of increasing concentrations of unlabeled JAM-A (circle), JAM-A·D1 (square), and a Fab control (solid circle). (B) Specific binding of JAM-A·biotin to immobilized JAM-A was analyzed in the presence of increasing concentrations of unlabeled JAM-A·D2 (square) and a Fab control (solid circle). The fraction of bound ligand is given as mean ± SE of four separate experiments in each case.
To specifically address the potential impact that the absence of the second domain of JAM-A has on the actual strength of the JAM-A homophilic interaction, we used AFM techniques. Single-molecule AFM adhesion measurements were conducted to confirm that the observed decrease of adhesion strength was the result of domain 2 deletion. In these experiments, JAM-A protein was cross-linked to the AFM cantilever tip and allowed to adhere to either the JAM-A or the JAM-A·D1 substrate. As described in Methods, these measurements were acquired at an adhesion frequency of 30% to ensure that measurements of single JAM-A interactions were obtained. In control experiments, anti-JAM-A antibody further reduced the adhesion frequency by 70% for both interactions and by > 80% when JAM-A adhesion was measured on BSA. The representative AFM force scans of the JAM-A homophilic interaction are shown in Fig. 2 A. The force jump shown in the third and sixth scans of the retract trace represents the unbinding force (fu). The system spring (ks) is determined from the slope of the force-distance curve and is used to calculate the loading rate of each measurement. The loading rate is the product of the system spring and the rate of cantilever retraction. The measurements were conducted at increasing loading rates, which was achieved by varying the rate of cantilever retraction. It is important to study receptor-ligand complexes at a range of loading rates because they might respond differently to pulling forces. Such differences in response to an applied pulling force were observed for a number of receptor-ligand pairs, namely, VLA-4/VCAM-1, selectin/sialyl Lewis X, and LFA-1/ICAM-1 and -2 (29–32). All adhesion forces were sorted according to their respective loading rates and compiled into force histograms. Representative histograms for the JAM-A/JAM-A interaction are shown in Fig. 2 B. The peak unbinding force increased with loading rate (rf). Using the unbinding forces from the peaks of all force histograms, we generated the dynamic force spectra of the JAM-A/JAM-A and JAM-A/JAM-A·D1 interactions. The dynamic force spectra of unbinding forces for a loading rate range of ∼2000–100,000 pN/s are shown in Fig. 2 C. The average unbinding forces acquired for JAM-A·D1 were significantly lower than those acquired with JAM-A (Fig. 2 C). These results confirm that the second domain must play an important role in stabilizing the JAM-A homophilic interaction.
Figure 2.
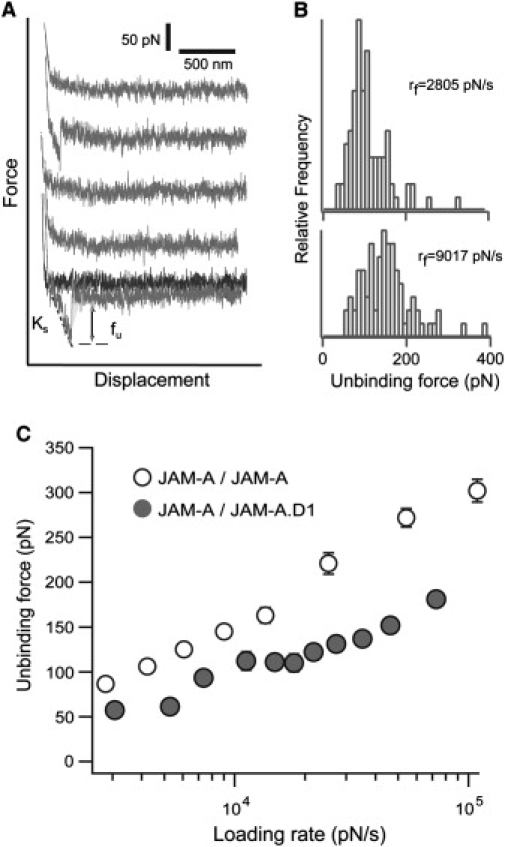
Single-molecule AFM measurements of the JAM-A/JAM-A and JAM-A/JAM-A·D1 interactions. (A) Single-molecule adhesion force scans of the JAM-A homophilic interaction. Adhesion was seen in the second and fifth scans. fu is the unbinding force, and ks represents the system spring. (B) Force histograms of measurements sorted according to loading rate (rf). Representative force histograms for loading rates of 2805 ± 37 pN/s and 9017 ± 102 pN/s are shown. (C) Dynamic force spectrum of the JAM-A/JAM-A (open circle) and JAMA.D1/JAM-A (black circle) interactions. Individual forces were acquired at a loading rate range of ∼2000–100,000 pN/s, sorted according to loading rate, and compiled into force histograms. Each data point in the dynamic force spectrum corresponds to the peak of each force histogram. The error for both loading rate and unbinding force is the standard error of at least three separate experiments.
Additional experiments were performed to determine if domain 2 of JAM-A stabilizes the JAM-A homophilic interaction or if it is involved in actually binding to JAM-A. Single-molecule AFM adhesion studies were conducted for the JAM-A·D2/JAM-A, JAMA·D2/JAM-A·D1, and JAM-A·D2/JAM-A·D2 interactions. Specific interactions were not observed for any of these interactions as confirmed by anti-JAM-A·D1 and anti-JAM-A·D2 antibodies. This finding is a strong indication that the second domain of JAM-A must be involved in stabilizing the JAM-A homophilic interaction, because it does not bind to either itself or domain 1 of JAM-A.
LFA-1 binding weakens the JAM-A homophilic interaction
As discussed previously, LFA-1 has been shown to bind to the second domain of JAM-A (15). We postulated that this binding may interfere with the potential stabilizing effects of the second domain of JAM-A, thus weakening the JAM-A homophilic interaction. To determine the effects of the LFA-1 binding, we measured JAM-A homophilic adhesion in its presence and absence. The I-domain of LFA-1, which has been shown to contain the binding site for JAM-A (16), was used as a surrogate for native LFA-1 in our adhesion assays and our AFM measurements.
We first conducted adhesion assays of CHO transfectants to JAM-A (Fig. 3). Adhesion to JAM-A was assessed using CHO cells transfected with vector only (CHO/vector, white bars) and with JAM-A (CHO/JAM-A, black bars). The vector controls exhibited little binding to JAM-A substrate. The binding of the CHO/JAM-A cells was measured in the presence of both the wild-type and open I-domains. The open I-domain is a soluble recombinant protein that contains disulfide bonds that lock it in a permanent state of high affinity (33). CHO/JAM-A cells were allowed to bind to JAM-A and, after a wash with binding buffer, 22.8 ± 2% of input cells remained bound (Fig. 3). In the presence of the open I-domain, adhesion of CHO/JAM-A cells was reduced by > 60% to 8.7 ± 1.2%. This large reduction in adhesion was similar to that observed after the addition of the JAM-A antibody (9.3 ± 2.2%) (Fig. 3). These results alone suggest that the binding of LFA-1 weakens the JAM-A homophilic interaction.
Figure 3.
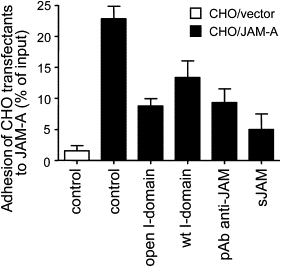
Adhesion of CHO transfectants to immobilized JAM-A. CHO cells transfected with JAM-A or vector control were allowed to adhere to JAM-A. Cell culture plate wells were incubated with open I-domain, wild-type (wt) I-domain, pAb anti-JAM-A, or soluble JAM-A. Cell adhesion is represented as the percentage of total added cells. Data are presented as mean ± SE of three separate experiments.
Our single-molecule AFM adhesion studies further confirmed the results observed in the CHO cell adhesion assay. The AFM studies also enabled us to determine whether the I-domain blocked or modulated the JAM-A homophilic interaction. JAM-A was cross-linked to the cantilever tip and allowed to interact with immobilized JAM-A in the presence and absence of the open I-domain. We used the open I-domain because it had the greatest impact on the JAM-A homophilic interaction in the adhesion assays. Fig. 4 A shows the dynamic force spectrum of the JAM-A homophilic interaction measured in the absence and presence of the open I-domain. A significant decrease of ∼20 pN was observed in the average unbinding force in the presence of the open I-domain throughout the dynamic force spectrum. In addition, the adhesion frequency decreased 20% after the addition of the open I-domain. These experiments were repeated using JAM-A·D1, which lacks the second domain where LFA-1 binding takes place. The dynamic force spectra for the JAM-A/JAM-A·D1 interaction in the absence and presence of the open I-domain are shown in Fig. 4 B. As expected, the two force spectra overlapped. No change was observed in the average unbinding force in the presence of the open I-domain for the mutant JAM-A. Interestingly, binding of the LFA-1 I-domain to JAM-A suppressed the dynamic strength of the JAM-A homophilic interaction to the level that was measured with JAM-A·D1 (Fig. 4 C). This finding suggests that binding of LFA-1 to the second Ig domain of JAM-A canceled its stabilizing effects and led to the lower adhesion forces measured for the JAM-A homophilic interaction.
Figure 4.
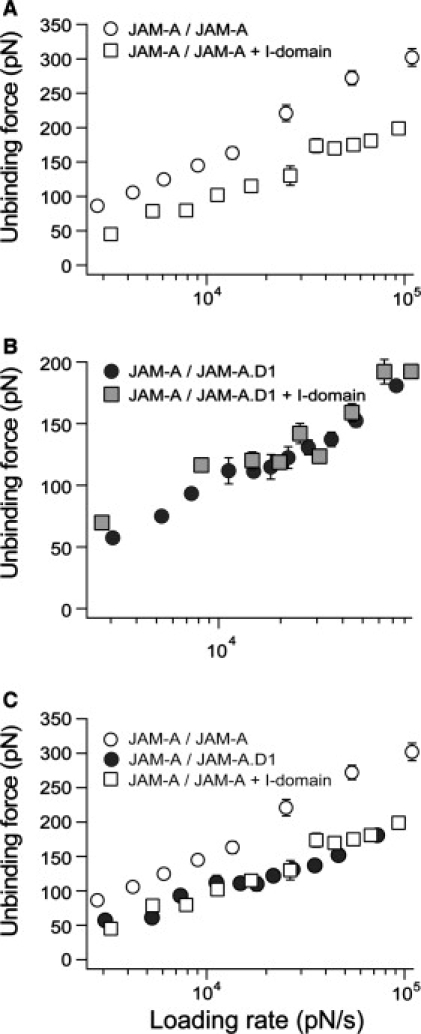
The effect of the open I-domain on the JAM-A homophilic interaction. (A) Dynamic force spectra of JAM-A/JAM-A (open circle) and JAM-A/JAM-A in the presence of the open I-domain (open square). (B) Dynamic force spectra of the JAM-A·D1/JAM-A (black circle) interaction and JAM-A/JAM-A·D1 (gray square) in the presence of the open I-domain. (C) Overlay of data from A and B: JAM-A/JAM-A (open circle) and JAM-A/JAM-A in the presence of the open I-domain (open square), and JAM-A·D1/JAM-A (black circle). In each dynamic force spectrum, the error for both loading rate and unbinding force is the standard error of at least three separate experiments.
The heterophilic interaction of LFA-1 with JAM-A is stronger than the homophilic JAM-A interaction
For TEM to proceed, the heterophilic LFA-1/JAM-A interaction between the leukocyte and endothelium must prevail, while the junctional JAM-A homophilic contacts are disrupted. Therefore, we postulated that the heterophilic LFA-1/JAM-A complex may be stronger enabling it to persevere, while the homophilic JAM-A linkages are disrupted. To determine if this were the case, we compared the dynamic strength of the JAM-A homophilic complex to its heterophilic complex with LFA-1.
To measure the adhesion strength of the LFA-1/JAM-A complex, Jurkat cells expressing LFA-1 on the cantilever tip were allowed to adhere to JAM-A on the sample dish (Fig. 5 A). Since human JAM-A is expressed on lymphocyte sets, we conducted these experiments with the JAM-A·D2 deletion mutant, which is missing the first JAM-A-binding domain. We conducted whole-cell AFM adhesion studies before proceeding with the single-molecule studies to confirm the specificity of this interaction. Representative whole-cell adhesion force scans from these experiments are shown in Fig. 5 B. Each measurement consisted of an approach trace (Fig. 5 B, top trace of the first force scan) during which the cantilever and cell were lowered onto the substrate. Contact was made, thus allowing the LFA-1/JAM-A bonds to form. Finally, the cell was withdrawn from the substrate, which allowed all the interactions to break, which is represented by the force jumps in the retract trace (Fig. 5 B, bottom trace of the first force scan). The measurements were conducted with untreated Jurkat cells and in the presence of antibodies to confirm specificity. Adhesion strength was compared by calculating the work of deadhesion, which is the work required to stretch the cell and stretch and break all the adhesive contacts. The work of deadhesion is calculated by integrating the adhesive force over the distance traveled by the cantilever. The shaded area under the top force scan in Fig. 5 B represents the work of deadhesion. The average work of deadhesion for these experiments was 1.84 ± 0.28 × 10−16 J for untreated cells (Fig. 5 C). It decreased significantly by 73% (to 4.92 ± 0.64 × 10−17 J) after treatment with an antibody directed against the second domain of JAM-A, and it decreased by 79% (to 3.91 ± 0.54 × 10−17 J) after treatment with an antibody directed against the I-domain of LFA-1 (TS1/22). These results confirmed that the LFA-1/JAM-A interaction supported the observed adhesion.
We then compared the strength of the LFA-1/JAM-A interaction with the JAM-A/JAM-A interaction by conducting single-molecule force measurements. The JAM-A/JAM-A measurements were conducted as described previously. The LFA-1/JAM-A measurements were conducted in the same manner as the whole-cell adhesion studies were conducted. LFA-1-expressing Jurkat cells were attached to the cantilever tip, and JAM-A·D2 was coated on the sample dish. Adhesion frequency was reduced to ∼30% by a short contact time (0.25 s) and low indentation force (50 pN) to ensure that we were measuring single bonds (see Methods). In control experiments, adhesion frequency was reduced further by 66% in the presence of an antibody directed against domain 2 of JAMA and by 70% with the TS1/22 antibody against LFA-1. Adhesion was not supported for Jurkat cells on BSA. Unbinding forces for LFA-1/JAM-A were compiled into force histograms, and, as described previously, the peak forces were plotted for a loading rate range of ∼500–60,000 pN/s to generate the dynamic force spectrum shown in Fig. 5 D. The average unbinding force increased with the loading rate. The dynamic force spectrum of the LFA-1/JAM-A interaction was overlaid with that of JAM-A/JAM-A demonstrating that the unbinding forces were significantly higher for the LFA-1/JAM-A interaction (Fig. 5 D). Our results indicate that the LFA-1/JAM-A interaction is better able to resist pulling forces than the JAM-A/JAM-A interaction, thus ensuring the likelihood that leukocyte TEM will proceed.
Discussion
The process of leukocyte homing involves leukocyte rolling and crawling on the endothelium, firm adhesion and, finally, TEM. During TEM, a possibility exists for the formation of a trimeric complex between LFA-1 on the migrating leukocyte, JAM-A on the endothelial cell, and a second JAM-A receptor on the opposing endothelial cell (18). One of these interactions has to be disrupted for the leukocyte to continue to migrate into the endothelial junction. Our data support a TEM model in which LFA-1 expressed on the surface of a migrating leukocyte weakens the junctional JAM-A homophilic interaction by binding to the membrane-proximal second Ig domain of JAM-A. LFA-1 is carrying out a dual purpose: it allows the migrating leukocyte to be recruited into the endothelial cell junction, and it blocks the second domain of JAM-A, which is important for stabilizing the JAM-A homophilic interaction. The weakened JAM-A interaction is likely to be disrupted, whereas the stronger LFA-1/JAM-A interaction remains and so allows the leukocyte to proceed farther down the endothelial junction.
Our results indicate that the second Ig domain of JAM-A is important for stabilizing its homophilic interaction. This was first shown in solid-phase binding assays conducted to compare the effectiveness of JAM-A and JAM-A·D1 as specific binding inhibitors for the JAM-A homophilic interaction. These assays revealed that the JAM-A mutant lacking the second domain of JAM-A could not compete for JAM-A binding as effectively as full-length JAM-A (Fig. 1). Further evidence indicating the importance of the second domain of JAM-A was obtained in the single-molecule AFM adhesion measurements of the JAM-A/JAM-A·D1 interaction in which significantly lower adhesion forces were measured than for the JAM-A/JAM-A interaction (Fig. 2 C). The greater adhesion forces measured for the JAM-A/JAM-A interaction were not the result of the second domain binding to either domain of JAM-A. Single-molecule adhesion measurements conducted with JAM-A/JAM-A·D2, JAM-A·D1/JAM-A·D2, and JAM-A·D2/JAM-A·D2 did not reveal any specific interaction of the second domain with either itself or the first domain. The second domain must therefore be stabilizing the JAM-A homophilic interaction, and its absence results in a reduction of measured adhesion forces. This finding has been demonstrated for another Ig superfamily member, namely, ICAM-1, in which the first Ig domain is involved in ligand binding and the second Ig domain stabilizes this binding (34). Therefore, by occupying the second domain of JAM-A, LFA-1 may be destabilizing the homophilic JAMA interactions, thus acting to loosen the junctional contacts and allowing leukocyte TEM to proceed.
This model is also supported by subsequent experimental data, which revealed that the binding of LFA-1 weakened the JAM-A homophilic interaction. Binding of the LFA-1 I-domain to JAM-A reduced the dynamic strength of the JAM-A homophilic interaction to the level measured with JAM-A·D1, which suggests that the binding of LFA-1 canceled the stabilizing effects of the second Ig domain of JAM-A (Fig. 4). Furthermore, our single-molecule adhesion measurements indicate that the interaction of LFA-1 with JAM-A is stronger or more resistant to pulling forces than the JAM-A homophilic interaction. This observation was evidenced by the overall lower unbinding forces measured for the JAM-A homophilic interaction compared to the LFA-1/JAM-A interaction. In addition, the dynamic force spectrum of the LFA-1/JAM-A interaction shown in Fig. 5 D exhibits two loading rate regimes. Complexes with two loading rate regimes are better able to resist pulling forces because their disruption requires overcoming two activation barriers. This finding was demonstrated for a number of interactions, including LFA-1/ICAM-1 and LFA-1/ICAM-2 (29,30). In contrast, the dynamic force spectrum of the JAM-A homophilic interaction exhibits a single regime, suggesting that only a single activation barrier would need to be overcome for the JAM-A/JAM-A interaction to be disrupted. Therefore, the heterophilic interaction is overwhelmingly the stronger and more stable one.
It is important to note that the homophilic interaction of JAM-A can take place in trans, which allows for JAM-A receptors to interact with those on opposing cells (i.e., “across” the endothelial junction), or in cis, which leads to dimer formation. Our data strongly suggest that we are measuring the JAM-A interaction in trans. Histograms compiling our measured JAM-A/JAM-A breakage forces did not reveal two peaks of adhesion forces, which would be an indication of two different types of JAM-A homophilic interactions. The absence of two peaks is an indication that either only one type of interaction was measured or that both the cis and trans interactions exhibit very similar forces and therefore the two interaction types are not resolved. Based on the most likely orientation of JAM-A receptors on the tip, it is highly probable that only the trans interaction was measured. Future studies conducted with JAM-A mutants that are unable to form dimers could further substantiate this claim. For this study, we chose not to fit the JAM-A single-molecule adhesion data to a thermodynamic model until the interaction type is confirmed.
The events of leukocyte trafficking have been the subject of many previous studies, discussed below, that have also relied on single-molecule force spectroscopy. A number of studies (31,32,35,36) have focused on the important initial step of this process, namely, leukocyte rolling, which is primarily mediated by selectin/ligand interactions as well as the integrin α4β1/VCAM-1 interaction. The subsequent firm adhesion step, primarily mediated by the LFA-1/ICAM-1 interaction and the α4β1/VCAM-1 interaction, has also been extensively studied (29,30,37). The reported dissociation rates for the unbinding of the selectin interactions were faster and the interactions were more sensitive to pulling forces than those formed between ICAM-1 and LFA-1. This finding is consistent with their physiological functions, as selectin interactions must be disrupted and reformed continuously to maintain cell rolling, while the ICAM-1/LFA-1 interaction is responsible for maintaining the firm adhesion of the leukocyte to the endothelium. Although we cannot report the dissociation rates and bond lifetimes for the JAM-A homophilic interaction due to the limitation described previously, we can note that these values for the LFA-1/JAM-A interactions very closely match those reported for the LFA-1/ICAM-1 interaction. This finding makes sense physiologically because the LFA-1/JAM-A interaction may also maintain firm adhesion on the endothelial surface before leukocyte transmigration. In terms of adhesion forces, the forces measured for the JAM-A homophilic interaction are lower than those measured for both LFA-1/JAM-A and LFA-1/ICAM-1 interactions. They are comparable to forces measured for another interaction occurring between endothelial cells, namely, that of E-cadherin, but they are higher than those for both N- and VE-cadherins (38,39). These results are all consistent with the physiological process of leukocyte TEM. For the cell to continue its migration across an endothelial junction, it makes sense for the interactions that it forms with the junctional receptors (i.e., LFA-1/JAM-A) to be stronger than those formed between the receptors found within the junction.
Future studies should focus on both cis and trans JAM-A interactions, because the weakening of both interactions may play an important role in the process of loosening junctional JAM-A contacts. Disrupting the homophilic JAM-A interactions in trans would directly open the endothelial cell junction for the migrating cell. Disrupting the cis interaction would disrupt the JAM-A dimers. Both the dimerization and clustering of receptors have been shown to strengthen cell adhesion (40–46). Therefore, it is possible that the homophilic interaction of JAM-A dimers may be stronger than that of JAM-A monomers, although this warrants further investigation. In addition, JAM-A dimers have been postulated to precede the formation of homophilic JAM-A contacts in trans in vivo (13). If this hypothesis were true, disruption of the dimers by LFA-1 would be a critical step because it would prevent the formation of junctional JAM-A homophilic contacts and keep the junction open for the migrating cell. The actual TEM process could involve a combination of these two mechanisms.
In summary, we present evidence that the second domain of JAM-A is important for stabilizing the JAM-A homophilic interaction. Our results support a model for the early events of TEM during which LFA-1 expressed on a leukocyte interacts with a JAM-A homophilic complex formed across the endothelial junction (trans). By binding to the second domain of JAM-A, LFA-1 destabilizes the JAM-A homophilic interaction. The already stronger LFA-1/JAM-A interaction persists, whereas the JAM-A homophilic interaction is disrupted, opening the junction and allowing the leukocyte to migrate into the junction.
Acknowledgments
We thank C. Freites and S. Winkler for technical assistance.
E. P. W., J. M., H. A., and V. T. M. were supported by the James and Esther King Biomedical Program (grant 06-NIR12), which is administered by the Florida Department of Health, Office of Statewide Research, and the National Institutes of Health (grant GM55611). R. R. K., L. F., and C. W. were supported by the Deutsche Forschungsgemeinschaft (DFG; grant FOR809-TP6).
References
- 1.Springer T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Hillis G.S., Flapan A.D. Cell adhesion molecules in cardiovascular disease: a clinical perspective. Heart. 1998;79:429–431. doi: 10.1136/hrt.79.5.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Springer T.A. Adhesion receptors of the immune system. Nature. 1990;346:425–434. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 4.Carman C.V., Springer T.A. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J. Cell Biol. 2004;167:377–388. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carman C.V., Sage P.T., Sciuto T.E., de la Fuente M.A., Geha R.S. Transcellular diapedesis is initiated by invasive podosomes. Immunity. 2007;26:784–797. doi: 10.1016/j.immuni.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luscinskas F.W., Lawler J. Integrins as dynamic regulators of vascular function. FASEB J. 1994;8:929–938. doi: 10.1096/fasebj.8.12.7522194. [DOI] [PubMed] [Google Scholar]
- 7.Weber C., Fraemohs L., Dejana E. The role of junctional adhesion molecules in vascular inflammation. Nat. Rev. Immunol. 2007;7:467–477. doi: 10.1038/nri2096. [DOI] [PubMed] [Google Scholar]
- 8.Martin-Padura I., Lostaglio S., Schneemann M., Williams L., Romano M. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Del Maschio A., De Luigi A., Martin-Padura I., Brockhaus M., Bartfai T. Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM) J. Exp. Med. 1999;190:1351–1356. doi: 10.1084/jem.190.9.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams L.A., Martin-Padura I., Dejana E., Hogg N., Simmons D.L. Identification and characterisation of human Junctional Adhesion Molecule (JAM) Mol. Immunol. 1999;36:1175–1188. doi: 10.1016/s0161-5890(99)00122-4. [DOI] [PubMed] [Google Scholar]
- 11.Ebnet K., Suzuki A., Ohno S., Vestweber D. Junctional adhesion molecules (JAMs): more molecules with dual functions? J. Cell Sci. 2004;117:19–29. doi: 10.1242/jcs.00930. [DOI] [PubMed] [Google Scholar]
- 12.Nourshargh S., Krombach F., Dejana E. The role of JAM-A and PECAM-1 in modulating leukocyte infiltration in inflamed and ischemic tissues. J. Leukoc. Biol. 2006;80:714–718. doi: 10.1189/jlb.1105645. [DOI] [PubMed] [Google Scholar]
- 13.Bazzoni G., Martinez-Estrada O.M., Mueller F., Nelboeck P., Schmid G. Homophilic interaction of junctional adhesion molecule. J. Biol. Chem. 2000;275:30970–30976. doi: 10.1074/jbc.M003946200. [DOI] [PubMed] [Google Scholar]
- 14.Bazzoni G. Endothelial tight junctions: permeable barriers of the vessel wall. Thromb. Haemost. 2006;95:36–42. [PubMed] [Google Scholar]
- 15.Ostermann G., Weber K.S., Zernecke A., Schroder A., Weber C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat. Immunol. 2002;3:151–158. doi: 10.1038/ni755. [DOI] [PubMed] [Google Scholar]
- 16.Fraemohs L., Koenen R.R., Ostermann G., Heinemann B., Weber C. The functional interaction of the β2 integrin lymphocyte function-associated antigen-1 with junctional adhesion molecule-A is mediated by the I domain. J. Immunol. 2004;173:6259–6264. doi: 10.4049/jimmunol.173.10.6259. [DOI] [PubMed] [Google Scholar]
- 17.Hynes R.O. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 18.Weber C. Novel mechanistic concepts for the control of leukocyte transmigration: specialization of integrins, chemokines, and junctional molecules. J. Mol. Med. 2003;81:4–19. doi: 10.1007/s00109-002-0391-x. [DOI] [PubMed] [Google Scholar]
- 19.Ostermann G., Fraemohs L., Baltus T., Schober A., Lietz M. Involvement of JAM-A in mononuclear cell recruitment on inflamed or atherosclerotic endothelium: inhibition by soluble JAM-A. Arterioscler. Thromb. Vasc. Biol. 2005;25:729–735. doi: 10.1161/01.ATV.0000157154.14474.3b. [DOI] [PubMed] [Google Scholar]
- 20.Binnig G., Quate C.F., Gerber C. Atomic force microscope. Phys. Rev. Lett. 1986;56:930–933. doi: 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- 21.Benoit M., Gabriel D., Gerisch G., Gaub H.E. Discrete interactions in cell adhesion measured by single-molecule force spectroscopy. Nat. Cell Biol. 2000;2:313–317. doi: 10.1038/35014000. [DOI] [PubMed] [Google Scholar]
- 22.Willemsen O.H., Snel M.M., Cambi A., Greve J., De Grooth B.G. Biomolecular interactions measured by atomic force microscopy. Biophys. J. 2000;79:3267–3281. doi: 10.1016/S0006-3495(00)76559-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heinz W.F., Hoh J.H. Spatially resolved force spectroscopy of biological surfaces using the atomic force microscope. Trends Biotechnol. 1999;17:143–150. doi: 10.1016/s0167-7799(99)01304-9. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X., Chen A., Wojcikiewicz E.P., Moy V.T. Probing ligand-receptor interactions with atomic force microscopy. In: Golemis E.A., editor. Protein-Protein Interactions: A Molecular Cloning Manual. Cold Spring Harbor Laboratory Press; Woodbury, NY: 2002. pp. 241–254. [Google Scholar]
- 25.Wojcikiewicz E.P., Zhang X., Moy V.T. Force and compliance measurements on living cells using atomic force microscopy (AFM) Biol. Proced. Online. 2004;6:1–9. doi: 10.1251/bpo67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wojcikiewicz E.P., Zhang X., Chen A., Moy V.T. Contributions of molecular binding events and cellular compliance to the modulation of leukocyte adhesion. J. Cell Sci. 2003;116:2531–2539. doi: 10.1242/jcs.00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tees D.F., Waugh R.E., Hammer D.A. A microcantilever device to assess the effect of force on the lifetime of selectin-carbohydrate bonds. Biophys. J. 2001;80:668–682. doi: 10.1016/S0006-3495(01)76047-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans E. Probing the relation between force–lifetime and chemistry in single molecular bonds. Annu. Rev. Biophys. Biomol. Struct. 2001;30:105–128. doi: 10.1146/annurev.biophys.30.1.105. [DOI] [PubMed] [Google Scholar]
- 29.Zhang X., Wojcikiewicz E., Moy V.T. Force spectroscopy of the leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction. Biophys. J. 2002;83:2270–2279. doi: 10.1016/S0006-3495(02)73987-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wojcikiewicz E.P., Abdulreda M.H., Zhang X., Moy V.T. Force spectroscopy of LFA-1 and its ligands, ICAM-1 and ICAM-2. Biomacromolecules. 2006;7:3188–3195. doi: 10.1021/bm060559c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X., Bogorin D.F., Moy V.T. Molecular basis of the dynamic strength of the sialyl Lewis X selectin interaction. ChemPhysChem. 2004;5:175–182. doi: 10.1002/cphc.200300813. [DOI] [PubMed] [Google Scholar]
- 32.Zhang X., Craig S.E., Kirby H., Humphries M.J., Moy V.T. Molecular basis for the dynamic strength of the integrin alpha4beta1/VCAM-1 interaction. Biophys. J. 2004;87:3470–3478. doi: 10.1529/biophysj.104.045690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu C., Shimaoka M., Ferzly M., Oxvig C., Takagi J. An isolated, surface-expressed I domain of the integrin alphaLbeta2 is sufficient for strong adhesive function when locked in the open conformation with a disulfide bond. Proc. Natl. Acad. Sci. USA. 2001;98:2387–2392. doi: 10.1073/pnas.041606398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stanley P., McDowall A., Bates P.A., Brashaw J., Hogg N. The second domain of intercellular adhesion molecule-1 (ICAM-1) maintains the structural integrity of the leucocyte function-associated antigen-1 (LFA-1) ligand-binding site in the first domain. Biochem. J. 2000;351:79–86. doi: 10.1042/0264-6021:3510079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans E., Leung A., Hammer D., Simon S. Chemically distinct transition states govern rapid dissociation of single L-selectin bonds under force. Proc. Natl. Acad. Sci. USA. 2001;98:3784–3789. doi: 10.1073/pnas.061324998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanley W.D., Wirtz D., Konstantopoulos K. Distinct kinetic and mechanical properties govern selectin-leukocyte interactions. J. Cell Sci. 2004;117:2503–2511. doi: 10.1242/jcs.01088. [DOI] [PubMed] [Google Scholar]
- 37.Shimaoka M., Lu C., Palframan R.T., von Andrian U.H., McCormack A. Reversibly locking a protein fold in an active conformation with a disulfide bond: integrin alphaL I domains with high affinity and antagonist activity in vivo. Proc. Natl. Acad. Sci. USA. 2001;98:6009–6014. doi: 10.1073/pnas.101130498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Panorchan P., George J.P., Wirtz D. Probing intercellular interactions between vascular endothelial cadherin pairs at single-molecule resolution and in living cells. J. Mol. Biol. 2006;358:665–674. doi: 10.1016/j.jmb.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 39.Panorchan P., Thompson M.S., Davis K.J., Tseng Y., Konstantopoulos K. Single-molecule analysis of cadherin-mediated cell-cell adhesion. J. Cell Sci. 2006;119:66–74. doi: 10.1242/jcs.02719. [DOI] [PubMed] [Google Scholar]
- 40.Woska J.R., Morelock M.M., Jeanfavre D.D., Bormann B.J. Characterization of molecular interactions between intercellular adhesion molecule-1 and leukocyte function- associated antigen-1. J. Immunol. 1996;156:4680–4685. [PubMed] [Google Scholar]
- 41.Dustin M.L., Springer T.A. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature. 1989;341:619–624. doi: 10.1038/341619a0. [DOI] [PubMed] [Google Scholar]
- 42.Weber C., Lu C.F., Casasnovas J.M., Springer T.A. Role of alpha L beta 2 integrin avidity in transendothelial chemotaxis of mononuclear cells. J. Immunol. 1997;159:3968–3975. [PubMed] [Google Scholar]
- 43.Pyszniak A.M., Welder C.A., Takei F. Cell surface distribution of high avidity LFA-1 detected by soluble ICAM-1-coated microspheres. J. Immunol. 1994;152:5241–5249. [PubMed] [Google Scholar]
- 44.Stewart M., Hogg N. Regulation of leukocyte integrin function: affinity vs. avidity. J. Cell. Biochem. 1996;61:554–561. doi: 10.1002/(sici)1097-4644(19960616)61:4<554::aid-jcb8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 45.van Kooyk Y., Figdor C.G. Avidity regulation of integrins: the driving force in leukocyte adhesion. Curr. Opin. Cell Biol. 2000;12:542–547. doi: 10.1016/s0955-0674(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 46.van Kooyk Y., Weder P., Heije K., Figdor C.G. Extracellular Ca2+ modulates leukocyte function-associated antigen-1 cell surface distribution on T lymphocytes and consequently affects cell adhesion. J. Cell. Biol. 1994;124:1061–1070. doi: 10.1083/jcb.124.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]