

Abstract
The composite and versatile structure of the cytoskeleton confers complex mechanical properties on cells. Actin filaments sustain the cell membrane and their dynamics insure cell shape changes. For example, the lamellipodium moves by actin polymerization, a mechanism that has been studied using simplified experimental systems. Much less is known about the actin cortex, a shell-like structure underneath the membrane that contracts for cell movement. We have designed an experimental system that mimicks the cell cortex by allowing actin polymerization to nucleate and assemble at the inner membrane of a liposome. Actin shell growth can be triggered inside the liposome, which offers a useful system for a controlled study. The observed actin shell thickness and estimated mesh size of the actin structure are in good agreement with cellular data. Such a system paves the way for a thorough characterization of cortical dynamics and mechanics.
Introduction
Characterizing the mechanical properties of cells has been of growing interest in the last 10 years with the motivation of understanding how cytoskeletal polymers dynamically rearrange to produce cell shape changes. However, the mechanical characterization of whole cells is difficult to interpret due to their various complex cytoskeletal structures. In vitro reconstitution of “functional modules” of the cytoskeleton has been developed over the last few years with the goal of building up, step by step, the complexity of cells (1). Once integrated into the cellular context, these modules can give highly versatile properties to the cell and organize the various constituents of the cytoskeleton for highly specialized functions. During cell motility, for example, two actin structures are involved, a lamellipodium that elongates and constitutes the advancing front and an actomyosin cortex that contracts, detaches the cell rear from the cell cortex, and moves the cell mass forward. In both structures, actin polymerization is nucleated in the presence of actin nucleation activating factors. Among these are the Wiskott Aldrich syndrome protein (WASP) family of proteins and formins that act locally next to the membrane (see (2) for a review). Proteins from the WASP family activate a protein complex, Arp2/3, which was found to localize at both the leading edge of Acanthamoeba and at its cortex (3), a feature that was also found in animal cells (4).
Mimicking actin polymerization that occurs in the extending lamellipodium has been achieved in the last 10 years with the biomimetic study of Listeria motility (5), which consists of designing and characterizing stripped-down systems that reproduce the same behavior in vitro (6). Such biomimetic systems have well-controlled physical properties and are powerful tools to decipher cellular dynamics and the importance of the gel-like structure of actin assemblies. Actin gels have mechanical properties characterized by in vitro experiments in three dimensions (1).
The actin cortex of cells has received far less attention than lamellipodia and filopodia, although its role is physiologically important for cell division or cell movements through narrow gaps of three-dimensional networks (7). A cell cortex is a thin shell roughly 1 μm thick located just underneath the plasma membrane and made up of short branched filaments that can lie from parallel to orthogonal to the plasma membrane, as observed by electron microscopy (8). It is an actin meshwork put under tension by molecular motors such as myosin filaments (in particular myosin II). The resulting contraction of the actin cortex is one of the key steps for cell motility. How the actin cortex is formed at the plasma membrane is not yet clear, although it appears that actin dynamics takes place preferentially close to the plasma membrane, suggesting that there must be an actin polymerization promoting factor there (9). Some of the proteins that participate in the linkage of the cytoskeleton to the membrane in cells have been identified as being part of the ERM (ezrin-radixin-moesin) family of proteins that are activated at the membrane and linked indirectly to microfilaments. Moreover, ezrin was found to be present in cell blebs, membrane extensions that retract under the action of cortical contraction (9).
The interactions between actin filaments and an artificial membrane have been addressed by the encapsulation in giant liposomes of actin networks made of long actin filaments cross-linked by proteins like filamin and α-actinin (10). In that case, actin filaments assemble into rings confined inside the liposome and sometimes appear next to the membrane when filaments are longer than the liposome size, which is explained by bending-energy arguments. However, these filaments are long (about ten micrometers) compared to the ones of a cell cortex (≤1μm). We designed a new experimental setup that allows to reproduce an actin structure mimicking the actin cortex by activating actin polymerization specifically at the inner liposome membrane. Actin polymerization occurs in situ as it is triggered by ATP and salts delivered inside the liposome through pores inserted in the membrane. The actin structure is cortical, as shown by fluorescence labeling of actin, and relies on local actin polymerization at the membrane. The actin shell thickness underneath the membrane can be explained by a mechanical model that takes into account actin gel growth inward in spherical geometry. Note that the goal of our work is not to study spontaneous actin assembly at an interface, but rather to reconstitute, as closely as possible to that of a cell, an actin meshwork that grows specifically from the inner leaflet of a liposome membrane, and thus build a controlled system for the study of cell mechanics.
Materials and Methods
Proteins and reagents
Actin, the Arp2/3 complex, gelsolin, actin depolarizing factor (ADF)-cofilin, and profilin were purchased from Cytoskeleton (Denver, CO) and used without further purification. Protein concentrations did not always correspond to product data, and were determined by SDS-PAGE using a bovine serum albumin standard. Alexa Fluor 568-labeled actin (red actin) and Alexa Fluor 488-labeled actin (green actin), obtained by labeling amine residues on F-actin before depolymerization, were purchased from Molecular Probes (Eugene, OR). All commercial proteins were delivered in buffers indicated by the manufacturers. N-WASP fragment WWA (aa400–501, also called VVCA) was expressed as a recombinant protein with an N-terminal his6 tag out of a plasmid given by D. Mullins' laboratory (11). We call this fragment VVCA-His in the following. The fragment was expressed in Escherichia coli BL21 (DE3) Codon Plus cells (Invitrogen, Carlsbad, CA). Proteins were expressed in the presence of 1 mM IPTG for 2 h at 37°C and purified by chromatography on Ni-NTA Agarose (Qiagen, Venlo, The Netherlands) according to the manufacturer's instructions. The proteins were then dialyzed with phosphate-buffered saline (PBS), pH 7.4 (PBS, 130 mM NaCl, 2.7 mM KCl, 5.4 mM Na2HPO4, 1.8 mM KH2PO4) and stored at −80°C. Rhodamine-labeled phalloidin, latrunculin A, casein (β-casein from bovine milk), dextran (from Leuconostoc mesenteroides average mol wt 35,000–45,000), and the pore-forming protein α-hemolysin (powder) from Staphylococcus aureus were obtained from Sigma Aldrich (St. Louis, MO). The protein α-hemolysin was dissolved in 150 mM KCl, 10 mM HEPES and kept at 4°C for up to 6 months.
Lipids
L-α-phosphatidylcholine from egg yolk (EPC), 1,2-dioleoyl-sn-glycero-3- {[N(5-amino-1-carboxypentyl)iminodiacetic acid]succinyl} nickel salt (DOGS-NTA-Ni), cholesterol (ovine wool) were purchased from Avanti Polar Lipids (Alabaster, AL).
Buffers, inside and outside solutions
The solution inside the liposomes must correspond to nonpolymerizing conditions with the constraint that the histidine-nickel interaction used for binding the protein to the membrane had to be preserved. The inside buffer (I-buffer) was thus made of 0.1 mM CaCl2, 10 mM HEPES (pH 7.6), 100 mg/mL dextran, 0.2 mM ATP, 6 mM dithiothreitol (DTT), 0.13 mM diazabicyclo[2,2,2]octane (Dabco), 280 mM sucrose. The inside solution (I-solution) consisted of proteins diluted in I-buffer at the final concentrations of 0.12 μM for Arp2/3, 50 nM for gelsolin, 2 μM for ADF-cofilin, 1 μM for profilin, 6.5 μM for G-actin (including 20% fluorescently labeled actin) and 0.64 μM VVCA-His. Experiments were also carried out with twice those protein concentrations. Note that the final solute concentrations of the I-solution were slightly changed after protein incorporation from the ones of the I-buffer (since proteins are kept in different buffers) to 0.1 mM CaCl2, 9.4 mM HEPES, 94 mg/mL dextran, 0.2 mM ATP, 5.6 mM DTT, 0.12 mM diazabicyclo[2,2,2]octane (Dabco), 271 mM sucrose CaCl2, as calculated from the manufacturer's buffer description. The outside buffer (O-buffer) for liposome production contained 10 mM HEPES (pH 7.5), 2 mM MgCl2, 0.2 mM CaCl2, 2 mM ATP, 6 mM DTT, 0.13 mM Dabco, 275 mM glucose, and 0.5 mg/mL casein. The polymerization buffer (P-buffer) consisted of 150 mM KCl, 2 mM CaCl2 and 5 mM HEPES (pH 7.5), 2 mM ATP, 6 mM DTT, 0.13 mM Dabco. For liposome observation in actin polymerization conditions, one-third of the O-buffer was replaced by P-buffer. The osmolarity of all three buffers (I-buffer, O-buffer, and P-buffer) was kept constant at 320 mOsm. Unless otherwise stated, all products were obtained from Sigma Aldrich.
Liposome preparation
Lipids (EPC, DOGS-NTA-Ni phospholipids, and cholesterol) were first dissolved in mineral oil in a molar ratio EPC/DOGS-NTA-Ni/cholesterol of 95:5:0 or 58:5:37 at a total concentration of 0.5 mg/mL. A volume of 5–10 mL of oil-lipid mixture was then sonicated in a bath at room temperature for 30 min at a power of 30 W. Then, the oil-lipid mixture was heated to 50°C for 3 h and finally cooled to room temperature and stored at 4°C for up to a week.
A few microliters of I-solution was added to the phospholipid-containing oil at a ratio of 1/200 and suspended by gentle back-and-forth pumping with a syringe. At this step, vortexing or sonicating must be avoided to preserve protein integrity. The obtained emulsion looks cloudy and is stabilized by lipid adsorption within a few minutes. In the meantime, 30 μl of the oil-lipid mixture was placed in a tube on top of the same volume of O-buffer to allow for the assembly of a monolayer of lipids at the interface. A volume of 50 μL of the emulsion was then slowly poured on top of the oil-lipid mixture, thus resulting in a three-level sample with O-buffer at the bottom, the oil-lipid mixture in the middle, and the emulsion on top. The whole tube was then centrifuged allowing the emulsion drops to pass through the lipid monolayer (100 × g for 12 min then 350 × g for 8 min, to progressively centrifuge droplets of all sizes). Finally, I-solution-filled liposomes were obtained in a bath of O-buffer (see Fig. 1).
Figure 1.
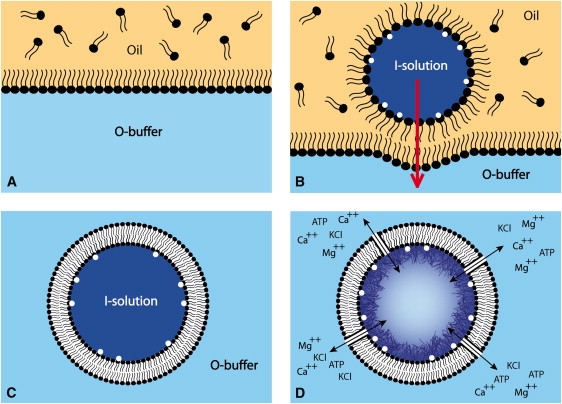
Schematic illustration of liposome preparation. (A) Assembly of the outer layer. (B) The inner layer, created by emulsion that was sedimented through the monolayer. (C) The liposome obtained by the assembly described in A and B, with different solutions inside and outside. (D) Polymerization was triggered when the pores were added, thus allowing salt and ATP to flow into the liposome. This figure is adapted from Pautot et al. (16).
To induce actin polymerization, one third of the O-buffer was replaced by P-buffer and 0.6 μL of 1 mg/mL α-hemolysin solution was added directly to a 6-μL sample on the microscope slide. Note that the addition of the α-hemolysin solution decreased the final concentrations of ATP, DTT, and Dabco by 10%. All the same, the final solute concentrations of the O- and P-buffer mix were reduced by 10% except for KCl and HEPES, which were already in the α-hemolysine buffer. The mixture was observed by either phase contrast or epifluorescence microscopy.
Observation and quantification of the actin cortex inside liposomes
Liposomes were observed with an IX70 Olympus inverted microscope and an Olympus 100/N.A. 1.35 phase-contrast objective (Olympus, Tokyo, Japan). Fluorescently labeled molecules were excited by a 200-W mercury lamp (OSRAM, Munich, Germany). Images were recorded with a charge-coupled device camera (Roper Scientific, Trenton, NJ) driven by Meta-Morph software (Universal Imaging, Downingtown, PA).
Fluorescent images were deconvoluted with the corresponding point-spread function and analyzed using ImageJ software (http://rsb.info.nih.gov/ij). The maximum intensity value along a radius starting from the center of the liposome was measured 360 times by rotating the radius, and the mean maximal intensity () was calculated (see Supplementary Material). The background intensity (m) inside the liposome was measured as the average fluorescence intensity of a centered disk of half the diameter of the liposome (see Fig. S1 in the Supplementary Material). For each experimental condition, 10–30 liposomes were analyzed, and results were gathered in a histogram. The thickness of the actin shell inside the liposome was measured as the thickness of the shell on an image where the intensity threshold was set at half maximum (see Supplementary Material). The actin contrast in a liposome was defined as , where and m were measured on rough images (before deconvolution).
Results and Discussion
Strategy for the design of an artificial actin cortex
To reconstitute actin dynamics underneath the membrane, we chose to use the (N-WASP) subdomain WWA (or VVCA), an activation-promoting factor of actin polymerization that is well characterized in other in vitro systems, such as motile beads (12). Moreover, WASP-family proteins recruit the ARP2/3 complex that has been detected in cell cortices (3). An alternative possibility would have been to use formin, another well studied activator of actin polymerization (2), but in vitro, formins form long and bundled actin filaments that do not resemble those of the cell cortex observed by electron microscopy (8). Instead, the use of the WASP-family proteins provides a branched actin network. The addition of gelsolin limited the filament length by capping the filament barbed ends. We used the histidine tag to bind VVCA to the membrane through a complexation with nickel lipid heads in the liposome membrane (see Materials and Methods). Besides having VVCA localized at the membrane, the liposome internal solution must contain the actin machinery needed for actin dynamics. A minimal protein medium that is able to reconstitute actin-based artificial movements was incorporated into the liposomes and consisted of G-actin, the Arp2/3 complex, gelsolin, ADF-cofilin, and profilin (13), in concentrations given in Materials and Methods. The actin machinery dynamics inside the liposome must be triggered by the presence of salt and ATP that must be delivered inside the liposome. We chose to allow salt and ATP to exchange through pores inserted in the bilayer. The pores must be big enough to allow salt and ATP to flow through, but small enough to keep the proteins trapped inside. We chose α-hemolysin, an extracellular protein secreted by Staphylococcus aureus, which assembles into a ring structure on a membrane and forms transmembrane pores (14), previously used by Noireaux et al. (15) to render liposomes selectively permeable for nutrients. Those pores are permeable to ions and small metabolites and have a molecular mass cutoff of 3 kDa, which is smaller than for any protein of the actin machinery.
Encapsulation of the actin machinery inside a liposome using an inverted emulsion technique
We used an inverted emulsion technique first proposed by Pautot et al. (16). This technique is well adapted to incorporate precious quantities of proteins inside liposomes. Moreover, this technique allows the use of drastically different buffers inside and outside the liposome, unlike other techniques like electroformation or spontaneous swelling.
In addition, the inverted emulsion technique preserves protein integrity, whereas proteins might be altered during the electroformation process.
The experimental principle of liposome production is schematized in Fig. 1. Our goal was to obtain liposomes filled with the I-solution and place them in an O-buffer containing salt and ATP, corresponding to actin polymerization conditions. The compositions of I-solution and O-buffer are given in Materials and Methods. A volume of phospholipid-containing oil was placed on top of an equal volume of O-buffer to allow the formation of a single phospholipid layer at the interface (Fig. 1 A). At the same time, an inverted emulsion was made consisting of droplets of I-solution dispersed in phospholipid-containing oil by gentle syringe pumping. This emulsion was centrifuged through the single phospholipid layer (Fig. 1 B) to finally obtain liposomes containing I-solution and surrounded with O-buffer (Fig. 1 C). Liposomes were monolamellar (see below), with a size distribution ranging from 1 to 8 μm in diameter, and were easily observed by phase contrast microscopy (Fig. 2 A) due to the refractive index difference between sucrose (inside) and glucose (outside). Such liposomes (Fig. 1 C and Fig. 2 A) filled with I-solution containing 1/5 fluorescently labeled actin displayed a bulk actin fluorescence (Fig. 2 B), because the I-solution maintains actin in its globular form.
Figure 2.
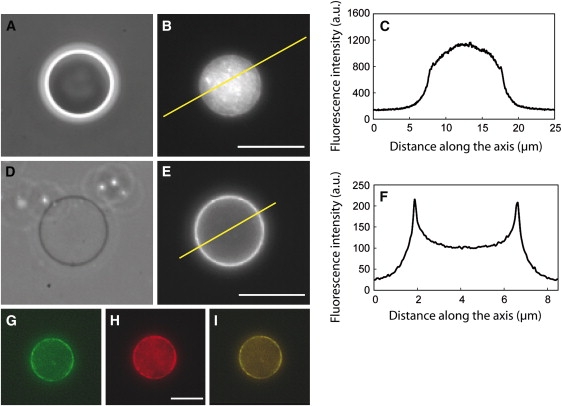
Triggering polymerization at the membrane. (A–D) Phase contrast microscopy (A and D) and epifluorescence microscopy (B and E) of actin. Without pores (A), the inside of the liposomes appeared denser and correlated to mass fluorescence of the actin (B). (C) Fluorescence profile along the yellow line drawn in B. In the presence of pores (D), sucrose left the liposome, and the contrast observed in phase contrast vanished, whereas actin fluorescence was localized at the membrane (E), which is shown by peaks (F) on the profile along the line shown in E. A fraction of 10% of actin was marked with Alexa Fluor 488 (G) and actin filaments were revealed by rhodamine-phalloidin staining (H) that colocalized with the actin shell (I). Scale bars: (A and B) 10 μm; (D and E) 5 μm, (G–I) 10 μm.
Triggering actin polymerization inside the liposomes
To trigger actin polymerization, the inside buffer must be changed into a higher salt concentration solution that favors actin polymerization. We simultaneously loaded the outside buffer with a polymerization buffer containing KCl, magnesium, calcium, and ATP (see Materials and Methods) and incorporated pores (α-hemolysin) to allow salt and ATP to flow into the liposome (Fig. 1 D). The pores were incorporated into the membrane by adding 0.6 μL of 1 mg/mL α-hemolysin directly into the sample before sealing the slide and coverslip chamber for observation. α-Hemolysin was in large excess, which allowed instantaneous exchange. In these conditions, we observed a loss of phase contrast due to sucrose leakage (compare Fig. 2, A and D). Note that this observation confirms that the liposomes are monolamellar. Indeed, if they were multilamellar, α-hemolysin could insert in the outer membrane, but would not trigger liposome leakage, since the content of the liposome would be protected by the other membrane layers. The loss of contrast correlated with the appearance of a fluorescent shell at the membrane (Fig. 2, D and E). Cortices were determined by actin shells displaying a contrast of C > 0.01. The time at which pores were added to the sample was the zero time reference for experiment characterization, since it corresponds to the addition of salt and ATP allowing actin polymerization. Experiments were performed in two lipid conditions, either in the absence of cholesterol (Figs. 2 and 3 A) or in the presence of cholesterol (Fig. 3 B), which was found to reduce the nonspecific interactions (see below).
Figure 3.
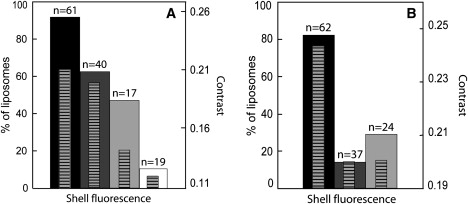
Effect of the presence of cholesterol, the Arp2/3 complex, and VVCA-His or drugs on cortex formation in the absence (A) and presence (B) of cholesterol (x = 0.37), in the lipid composition. Plain bars represent the percentage of liposomes displaying a fluorescent shell (actin contrast, C > 0.01), and hatched bars refer to the contrast. Solid bars, the complete system of proteins (see Materials and Methods); dark shaded bars, VVCA-His is omitted; light shaded bars, both VVCA and Arp2/3 complex are omitted; open bar, in the presence of latrunculin A. For each condition, we give the total number, n, of observed liposomes. The shell fluorescence percentages in the different conditions either in the absence or in the presence of cholesterol were proven to differ significantly using the χ2 statistic.
Characterization of the actin cortex inside the liposomes
The specificity of actin recruitment at the membrane through VVCA-His was checked by carrying out experiments in the same conditions as described, but without VVCA-His and then without either VVCA-His or the Arp2/3 complex (Fig. 3). In the presence of all proteins, under epifluorescence microscopy, 95% or 80% of the liposomes displayed a clear actin shell in the absence or presence, respectively, of cholesterol (Fig. 3, solid bars). In both cases, the removal of VVCA from the membrane, and of Arp2/3 complex from the solution, caused a decrease in the number of liposomes displaying an actin shell (Fig. 3, solid and shaded bars), accompanied with a decrease of the fluorescence contrast that was more pronounced in the presence of cholesterol (Fig. 3, hatched bars). These data show that actin shell assembly significantly depends on the presence of actin polymerization activation at the liposome membrane. In the absence of cholesterol, the observation that ∼60% of the liposomes still display an actin cortex in the absence of VVCA (Fig. 3 A) indicates that there are nonspecific interactions between the membrane and the actin filaments grown in the bulk of the liposome. When cholesterol was added, we found that this nonspecific interaction was drastically reduced, an effect that was observed with long actin filaments encapsulated in giant vesicles (17). Finally, in the presence of cholesterol, no cortex was observed when the Ni-lipid was absent in the membrane (counted on 15 liposomes), thus preventing VVCA from attaching to the membrane.
To confirm that the actin cortex was the result of a dynamic polymerization of actin, the assay was carried out in the presence of 4.7 μM latrunculin A, which inhibits actin polymerization by sequestering G-actin. The drug was introduced into the outside solution after 20 min and is small enough to travel through the pores. The number of liposomes displaying an actin shell after the incorporation of latrunculin was significantly reduced in the presence of the drug (Fig. 3 A, open bar). This inhibition shows that the actin shell is dynamic and that filaments are constantly polymerizing. Moreover, in the presence of 2 μM rhodamine-phalloidin, which specifically marks F-actin, the fluorescence signal was colocalized with the actin signal underneath the membrane (Fig. 2, G–I).
The actin shell thicknesses were constant after 5 min (see Fig. S2), at which time they were measured on a population of various sized liposomes. As displayed in Fig. 4, the thickness globally increases with liposome size. Note that increasing the protein concentration by a factor of 2 slightly increased the shell thickness (Fig. 4, shaded crosses versus solid “×” symbols).
Figure 4.
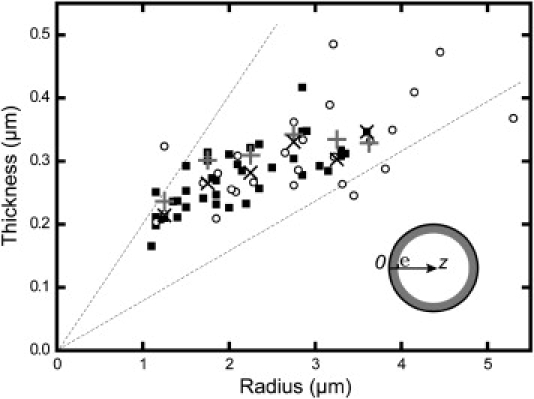
Evolution of the gel thickness as a function of liposome size. Solid squares or open circles correspond to one single liposome measurement. Solid-square points were obtained under concentration conditions of 0.12 μM for the Arp2/3 complex, 50 nM for gelsolin, 2 μM for ADF-cofilin, 1 μM for profilin, 6.5 μM for G-actin (including 20% fluorescently labeled actin), and 0.64 μM VVCA-His. Solid “×”symbols are thickness values averaged over each 0.5-μm radius increase. Shaded crosses are thickness values averaged over each 0.5-μm radius increase in the case of twice the protein concentration. Open circles are shell thicknesses for liposomes in the presence of cholesterol in the same protein conditions as solid points. Most points lie between the slopes 0.08 and 0.2 (dashed lines). (Inset) Scheme of notations used in the text.
Actin shell thickness modeling
To estimate the structural characteristics of the actin shell, we derive from a study using a slightly different geometry (18) a simple geometrical model in which actin growth might be limited not only by monomer diffusion or stress-induced depolymerization but also by the restricted amount of actin due to confinement in the liposome.
Polymerization occurs at the liposome membrane with a velocity Vp:
where the z axis is perpendicular to the membrane (origin), a is the size of a monomer, ϕ represents an angle of polymerization relative to the z axis, dn/dt is the number of monomers polymerized per unit time, kp the rate constant of polymerization at the barbed end, and C(z) the concentration. For the sake of simplicity, we take in the following cosϕ=1, which corresponds to perpendicular polymerization at the membrane surface.
The conservation of the total number of monomers in the vesicle reads:
where e is the actin gel thickness inside the liposome, R the liposome radius, ρgel the mass density of actin inside the gel, M the mass of one actin monomer, the initial bulk unpolymerized monomer concentration, and Ci the bulk monomer concentration (e<z<R). All values are expressed in SI units. We have assumed here that e<<R, which is indeed the case experimentally (see Fig. 4). We then calculate C(0) at steady state considering that the monomer consumption at the membrane is equal to the diffusive flux of monomers J through the actin layer:
Here, ξ is the average distance between activators, which we consider the same as the gel mesh size.
Then the polymerization velocity reads:
The depolymerization velocity is given by
where kd is the rate constant of depolymerization at the pointed end, which we consider as constant, thus neglecting its dependence on stress. Note that kp depends on the stress of a second order in , and is also considered here as constant.
At steady state, the polymerization and depolymerization velocities are equal, which leads to
For , the thickness is proportional to R, e=pR, with a slope
For an estimate of , we take , (19), and (or twice ) in our experiments, and find that , and thus p simplifies to
for geometrical reasons.
In experiments (Fig. 4), p lies between 0.08 and 0.2, leading (with a = 5 nm) to a mesh size on the order of 150 nm, which is a reasonable value compared to the measured mesh size in a cell cortex (8). Note that the effect of increasing by a factor of 2 does not double the value of p, thus indicating that ξ slightly decreases, which could be explained by the presence of more branches due to increasing amounts of Arp2/3 complex. Note that a lower estimate of the mesh size can be obtained simply by considering that all the actin inside a liposome is consumed in the actin shell of thickness e grown next to the membrane. In these conditions, the surface S of the liposome reads
with Nfil the total number of filaments on the liposome, which is given by the total number of actin monomers in the liposome divided by the number of monomers in a filament of length e. The number of monomers per filament is given by the length of a filament (e) divided by the monomer size (a = 2.5 nm). With e on the order of 250 nm and a liposome of radius R = 5 μm, one finds
and thus, a lower limit of ξ equal to 100 nm, which is in good agreement with the value found above.
For e>>e∗, the thickness is diffusion-limited and saturates to a plateau
With the same values as above, a diffusion coefficient of 10−8 cm2 s−1 (20) and the estimated value of ξ, we find ep to be on the order of 400 μm, which is outside the range of our experiments.
Conclusion
We used here a protein mixture that provides an actin structure mimicking that of the cell cortex. Such an actin shell can be reconstituted inside a liposome by triggering actin dynamics at the liposome membrane. This system constitutes an important step toward the reconstitution of the different actin structures in well-controlled systems that would then be compared with cells. Such an approach will allow for unraveling cytoskeletal functions in important cellular events like cell motility and division.
Supplementary Material
Two figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(08)00038-6.
Supporting Material
Acknowledgments
We thank Sophie Pautot for fruitful discussions, Arkun Akin and Dyche Mullins for the gift of the VVCA-his plasmid, Julie Plastino for many fruitful discussions, and John Manzi for the purification of the VVCA-His protein. We thank the lab of Laurent Blanchoin for their advice on biochemistry, and the lab of Loïc Auvray for advice on the use of α-hemolysin. We thank Timo Betz and Ewa Paluch for critical reading.
This work was funded by a grant from the Human Frontiers Science Program and a grant from the Agence Nationale pour la Recherche, France.
Footnotes
Jasper van der Gucht's present address is Laboratory of Physical Chemistry and Colloid Science, Wageningen University, Wageningen, The Netherlands
Julien Heuvingh's present address is Laboratoire de Physique et Mécanique des Milieux Hétérogènes, CNRS UMR 7636, ESPCI, Paris, France, and Université Paris Diderot, Paris, France.
References
- 1.Bausch A.R., Kroy K. A bottom-up approach to cell mechanics. Nat. Phys. 2006;2:231–238. [Google Scholar]
- 2.Chhabra E.S., Higgs H.N. The many faces of actin: matching assembly factors with cellular structures. Nat. Cell Biol. 2007;9:1110–1121. doi: 10.1038/ncb1007-1110. [DOI] [PubMed] [Google Scholar]
- 3.Machesky L.M., Atkinson S.J., Ampe C., Vandekerckhove J., Pollard T.D. Purification of a cortical complex containing two unconventional actins from Acanthamoeba by affinity chromatography on profilin agarose. J. Cell Biol. 1994;127:107–115. doi: 10.1083/jcb.127.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Machesky L.M., Reeves E., Wientjes F., Mattheyse F., Grogan A. Mammalian Arp2/3 complex localizes to regions of lamellipodial protrusion and is composed of conserved subunits. Biochem. J. 1997;328:105–112. doi: 10.1042/bj3280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loisel T.P., Boujemaa R., Pantaloni D., Carlier M.F. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- 6.Plastino J., Sykes C. The actin slingshot. Curr. Opin. Cell Biol. 2005;17:62–66. doi: 10.1016/j.ceb.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Lammermann T., Bader B.L., Monkley S.J., Worbs T., Wedlich-Soldner R. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 2008;453:51–55. doi: 10.1038/nature06887. [DOI] [PubMed] [Google Scholar]
- 8.Morone N., Fujiwara T., Murase K., Kasai R.S., Ike H., Yuasa S. Three-dimensional reconstruction of the membrane skeleton at the plasma membrane interface by electron tomography. J. Cell Biol. 2006;174:851–862. doi: 10.1083/jcb.200606007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charras G.T., Hu C.K., Coughlin M., Mitchison T.J. Reassembly of contractile actin cortex in cell blebs. J. Cell Biol. 2006;175:477–490. doi: 10.1083/jcb.200602085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Limozin L., Sackmann E. Polymorphism of cross-linked actin networks in giant vesicles. Phys. Rev. Lett. 2002;89:168103.1–168103.4. doi: 10.1103/PhysRevLett.89.168103. [DOI] [PubMed] [Google Scholar]
- 11.Zalevsky J., Lempert L., Kranitz H., Mullins R.D. Different WASP family proteins stimulate different Arp2/3 complex-dependent actin-nucleating activities. Curr. Biol. 2001;11:1903–1913. doi: 10.1016/s0960-9822(01)00603-0. [DOI] [PubMed] [Google Scholar]
- 12.Bernheim-Groswasser A., Wiesner S., Golsteyn R.M., Carlier M.-F., Sykes C. The dynamics of actin-based motility depend on surface parameters. Nature. 2002;417:308–311. doi: 10.1038/417308a. [DOI] [PubMed] [Google Scholar]
- 13.van der Gucht J., Paluch E., Plastino J., Sykes C. Stress release drives symmetry breaking for actin-based movement. Proc. Natl. Acad. Sci. USA. 2005;102:7847–7852. doi: 10.1073/pnas.0502121102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song L., Hobaugh M.R., Shustak C., Cheley S., Bayley H. Structure of Staphylococcal α-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1865. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 15.Noireaux V., Libchaber A. A vesicle bioreactor as a step toward an artificial cell assembly. Proc. Natl. Acad. Sci. USA. 2004;101:17669–17674. doi: 10.1073/pnas.0408236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pautot S., Frisken B.J., Weitz D.A. Production of unilamellar vesicles using an inverted emulsion technique. Langmuir. 2003;19:2870–2879. [Google Scholar]
- 17.Limozin L., Barmann M., Sackmann E. On the organization of self-assembled actin networks in giant vesicles. Eur. Phys. J.E. 2003;10:319–330. doi: 10.1140/epje/i2002-10118-9. [DOI] [PubMed] [Google Scholar]
- 18.Noireaux V., Golsteyn R.M., Friederich E., Prost J., Antony C. Growing an actin gel on spherical surfaces. Biophys. J. 2000;278:1643–1654. doi: 10.1016/S0006-3495(00)76716-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pollard T.D. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. J. Cell Biol. 1986;103:2747–2754. doi: 10.1083/jcb.103.6.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plastino J., Lelidis I., Prost J., Sykes C. The effect of diffusion, depolymerization and nucleation promoting factors on actin gel growth. Eur. Biophys. J. 2004;33:310–320. doi: 10.1007/s00249-003-0370-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.