

Abstract
The 18.5 kDa isoform of myelin basic protein (MBP) is a peripheral membrane protein that maintains the structural integrity of the myelin sheath of the central nervous system by conjoining the cytoplasmic leaflets of oligodendrocytes and by linking the myelin membrane to the underlying cytoskeleton whose assembly it strongly promotes. It is a multifunctional, intrinsically disordered protein that behaves primarily as a structural stabilizer, but with elements of a transient or induced secondary structure that represent binding sites for calmodulin or SH3-domain-containing proteins, inter alia. In this study we used solid-state NMR (SSNMR) and Fourier transform infrared (FTIR) spectroscopy to study the conformation of 18.5 kDa MBP in association with actin microfilaments and bundles. FTIR spectroscopy of fully 13C,15N-labeled MBP complexed with unlabeled F-actin showed induced folding of both protein partners, viz., some increase in β-sheet content in actin, and increases in both α-helix and β-sheet content in MBP, albeit with considerable extended structure remaining. Solid-state NMR spectroscopy revealed that MBP in MBP-actin assemblies is structurally heterogeneous but gains ordered secondary structure elements (both α-helical and β-sheet), particularly in the terminal fragments and in a central immunodominant epitope. The overall conformational polymorphism of MBP is consistent with its in vivo roles as both a linker (membranes and cytoskeleton) and a putative signaling hub.
Introduction
The 18.5 kDa isoform of myelin basic protein (MBP) is one of the major protein components in human adult central nervous system (CNS) myelin, where it maintains the tight multilamellar packing of the sheath by adhesion of the cytoplasmic faces of the oligodendrocyte membrane in close apposition. It is an intrinsically disordered protein (IDP) (1,2) that is believed to be multifunctional because it also interacts with a diversity of other proteins and ligands, principally calmodulin (CaM), actin, tubulin, and SH3-domain containing proteins (3–6). The associations of MBP with lipids and other proteins are modified by a “dynamic molecular barcode” of combinatorial posttranslational modifications, primarily single-site methylation, and multisite deimination and phosphorylation (5,7).
IDPs have extended conformations that offer a large effective surface area for target interaction, and their intermolecular associations may involve preformed structural elements, such as α-helices, and/or an induced fit, i.e., coupled folding and binding (8–10). The 18.5 kDa MBP isoform first displays a disorder-order transition by association with the myelin membrane, and several segments form α-helices as ascertained by numerous spectroscopic studies (circular dichroism (CD) (11), electron paramagnetic resonance (EPR) (12,13), and solution NMR (14,15)). Solid-state NMR studies of rmMBP reconstituted with lipid vesicles have revealed that at least a third of the protein remains disordered in association with lipids (16). These regions of the protein may be available for association with CaM or SH3-domains, where a localized target may undergo an induced fit according to the generalized molecular recognition fragments (MoRF) paradigm postulated by Mohan et al. (17). The α-helix forming segments of MBP were initially postulated to be canonical CaM-binding targets (18,19), but both EPR and solution NMR spectroscopy indicate that the entire protein interacts with CaM and that the carboxy-terminus of MBP appears to be the primary binding site, but not necessarily in a canonical way (15,19). The association of MBP with SH3-domains appears to be mediated by the transient or induced formation of a polyproline type II helix (6).
The interaction of the 18.5 kDa MBP isoform with actin is the subject of this study. In vitro, MBP interacts with G-actin to cause it to polymerize into filaments (F-actin), even under otherwise nonpolymerizing conditions, and to assemble these F-actin fibers into bundles (20,21). The polymerization and bundling activities of MBP can be reversed by the calcium-dependent binding of MBP to CaM (20–22), and are modulated by deimination, methylation, and phosphorylation (4,23,24). Additionally, it has been shown that MBP is capable of binding actin and anionic phospholipid bilayers simultaneously (4,22,23), indicating that MBP could potentially serve as a link between microfilaments and the cytoplasmic leaflet of the oligodendrocyte membrane. Similarly, MBP polymerizes and bundles microtubules (25,26), and may also tether microtubules to the membrane or to actin microfilaments (3).
The conformation of MBP in association with actin (MBP-F-actin bundles) can be studied by means of solid-state NMR (SSNMR) spectroscopy. Magic angle spinning (MAS) SSNMR spectroscopy of nonoriented samples can be used to determine complete three-dimensional (3D) arrangements of insoluble and noncrystalline peptides and proteins (27–33). We recently applied SSNMR spectroscopy to 18.5 kDa rmMBP reconstituted with lipid vesicles, thereby emulating the physiological environment and mimicking the lipid-protein electrostatic and hydrophobic interactions (16). Here, we use MAS SSNMR and Fourier transform infrared (FTIR) spectroscopy to demonstrate that the association of rmMBP with actin induces the formation of ordered secondary structure elements, albeit with some degree of polymorphism and disorder. This conformational “fuzziness” probably allows MBP to interact simultaneously with lipids and actin, as well as with CaM, SH3-domains, tubulin, and various modifying enzymes.
Materials and Methods
Materials
Electrophoresis-grade acrylamide, ultrapure Tris base, and ultrapure Na2EDTA were purchased from ICN Biomedicals (Costa Mesa, CA). Most other chemicals were reagent grade and acquired from either Fisher Scientific (Unionville, Ontario, Canada) or Sigma-Aldrich (Oakville, Ontario, Canada). Electrophoresis-grade sodium dodecyl sulfate (SDS) was obtained from Bio-Rad Laboratories (Mississauga, Ontario, Canada). The Ni2+-NTA (nitrilotriacetic acid) agarose beads were purchased from Qiagen (Mississauga, Ontario, Canada). For uniform labeling of protein for NMR spectroscopy, the stable isotopic compounds 2H2O (D2O), 15NH4Cl, and 13C6-glucose were obtained from Cambridge Isotope Laboratories (Cambridge, MA).
Expression and purification of rmMBP
The unmodified 18.5 kDa recombinant murine MBP isoform (rmMBP, or component rmC1, with a C-terminal LEH6 tag) was expressed in Escherichia coli and purified by nickel-affinity chromatography as described previously (34,35). An additional step of ion exchange chromatography served to remove minor contaminating material (36). Protein eluate from the column was dialyzed (using tubing with an Mr cutoff of 6000–8000) twice against 2 L of buffer (50 mM Tris-HCl, pH 7.4, 250 mM NaCl), twice against 2 L of 100 mM NaCl, and finally four times against 2 L of ddH2O. Uniformly 13C,15N-labeled protein was cultured in cells grown in M9 minimal media (37). The protein concentration was determined by measuring the absorbance at 280 nm, using the extinction coefficient ɛ = 0.627 Lg−1cm−1 for rmMBP (as calculated by SwissProt for protein in 6.0 M guanidium hydrochloride, 0.02 M phosphate buffer, pH 6.5). The purity of the protein preparation was assayed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and the results (a major band representing undegraded, unmodified rmMBP) were consistent with our previous investigations (34,35).
Purification of actin from chicken muscle
Chicken muscle acetone powder was prepared, and actin was extracted from 8 g of it at a time as described elsewhere (38). The purity of the actin was checked using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry, supported by SDS-PAGE, and it was clear that no further purification steps were required. Protein concentration was determined by measuring the absorbance at 280 nm, using the extinction coefficient ɛ = 0.62 Lg−1cm−1. The polymerization activity of the globular actin was assessed using salt (50 mM KCl, 1 mM EGTA, and 2 mM MgCl2). The solution was kept for 1 h at room temperature, and then centrifuged for 20 min at 340,000 × g to collect polymerized actin. The actin content in both the supernatant and pellet was assessed using SDS-PAGE. In addition, an equivalent volume of the G-actin stock was centrifuged at the same speed and for the same time, and SDS-PAGE of the supernatant and pellet was used to check for actin content. The active protein suspended in G-buffer (2 mM Tris, pH 8.0, 0.2 mM ATP, 0.2 mM CaCl2, and 0.2 mM 2-mercaptoethanol) was then aliquoted into 1.5 mL microfuge tubes, flash frozen in liquid nitrogen, and stored at −80°C.
NMR sample preparation
Protein samples were prepared for SSNMR spectroscopy in two different ways. In sample 1, the labeled rmMBP was allowed to interact with G-actin in the presence of polymerizing salts (50 mM KCl, 1 mM EGTA, and 2 mM MgCl2). In sample 2, G-actin was first polymerized into filaments by salt, and then labeled rmMBP was added to bundle it into larger assemblies.
In sample 1, freeze-dried rmMBP was dissolved in G-buffer (2 mM Tris, pH 8.0, 0.2 mM ATP, 0.2 mM CaCl2, and 0.2 mM 2-mercaptoethanol) and mixed with G-actin in G-buffer to a final G-actin concentration of 10 μM and a [G-actin]/[rmMBP] molar ratio of 1:1.6. To obtain more compact bundles, KCl (from a 3 M stock solution) was added dropwise to the initial rmMBP preparation before mixing it with G-actin, to give a final KCl concentration of 50 mM. The solution was incubated at room temperature overnight to ensure complete polymerization to F-actin and bundling of the microfilaments by the rmMBP, and was then centrifuged for 1 h at 205,000 × g to collect the bundles. The pellet was then centrifuged for 20 min at 340,000 × g before it was packed into a regular wall 3.2 mm MAS rotor for NMR measurements. The total amount of rmMBP packed into the rotor was ∼6 mg. This amount was obtained from a pellet with a total protein content of 15 mg (actin + MBP), and it filled the whole volume of the rotor (20 μL).
Sample 1 showed some branching at the boundaries of formed bundles under the electron microscope. To further quantify probable inhomogeneity introduced by this branching, we used salt-polymerized actin at a higher KCl concentration to produce more compact bundles in sample 2. For this sample, G-actin was first polymerized into F-actin by addition of KCl to a final concentration of 50 mM, along with 1 mM EGTA and 2 mM MgCl2. To ensure maximal polymerization, the final solution was incubated overnight at room temperature. Freeze-dried rmMBP was dissolved in G-buffer containing 50 mM KCl, 1 mM EGTA, and 2 mM MgCl2. Both solutions were mixed together, and the KCl concentration was raised to 75 mM to enhance bundle compactness. The sample was incubated for 4 h at room temperature, initially centrifuged for 30 min at 600 × g to collect the bundles, and finally centrifuged for 20 min at 340,000 × g. The resulting compact pellet (with a final rmMBP content of ∼9 mg) was then center-packed between two cylindrical Vespel inserts into a Bruker thin-walled MAS rotor. The rotor has a total volume of 60 μL and the sample was packed into 44% of this volume to minimize the radiofrequency field inhomogeneity at both edges of the rotor.
Transmission electron microscopy
Transmission electron microscopy (TEM) was used to examine the morphology of the rmMBP-actin assemblies before NMR analysis. The samples used for TEM measurements were prepared in exactly the same way as both NMR samples, without the last two centrifugation steps, and were also diluted four times with G-buffer. The samples were negatively stained with uranyl acetate, dried, and examined on a Philips CM10 transmission electron microscope.
FTIR spectroscopic measurement of hydrogen-deuterium exchange
FTIR spectroscopy was performed on a Bruker IFS66vs spectrometer equipped with a diamond attenuated total reflection (ATR) cell operating at 4 cm−1 resolution. The rmMBP-actin assembly was prepared exactly the same way as for sample 2, except without the last two centrifugation steps. To distinguish the vibrational bands of rmMBP from those of actin, uniformly 13C,15N-labeled rmMBP was used in the preparation of the FTIR sample. The sample was suspended in 10 μL H2O-based G-buffer, spread evenly onto the diamond prism, and dehydrated completely under vacuum for 10 min. An absorption spectrum was taken on the dry film. The dry film was then incubated with 10 μL of D2O and sealed to prevent D2O evaporation exchange with the atmosphere during the measurement time. A series of absorption spectra were recorded at different times after exposure to D2O under the “rapid scan” mode. The time base extended over a course of 6 h. The measurements were done as follows: First, 100 measurements with zero delay were performed during the first 20 s. After that, another 100 measurements with 10 s delays between consecutive measurements were done during the next 17 min. The last stage of 160 measurements had 120 s delays between consecutive measurements for the rest of the 6 h. Each measurement was done as an average of four scans (total scan time of 200 ms). The first 100 measurements were considered to have an effective delay of 200 ms, and the rest of the 360 measurements were corrected to reflect the scan time. The timescale of the FTIR measurements was referenced to the moment when D2O was added to the sample. All spectra were normalized by the integrated peak intensity of the amide I band, to correct for the reduction in intensity due to swelling of the sample after the addition of D2O. The normalization was done by multiplying each spectrum by the ratio of the integrated intensity of the amide I band of the spectrum before adding D2O to that of each respective spectrum. An in-house-written FORTRAN script was used to carry out this normalization. The normalization of FTIR spectra was only necessary to display the data better in the early stages of exchange, by eliminating the fast-decaying component as a result of swelling. To extract all time constants of swelling, as well as exchange processes in rmMBP and actin, a global exponential fitting to all spectra in the range of 1350–1700 cm−1 was done.
NMR spectroscopy
All experiments were performed on Bruker Avance III spectrometers (Bruker BioSpin, Karlsruhe, Germany)—one operating at 600.130 MHz proton frequency and equipped with a standard Bruker triple-resonance 1H-13C-15N 3.2 mm BioSolids MAS probe (Bruker USA, Billerica, MA), and the other operating at 800.23 MHz proton frequency, equipped with a Bruker triple-resonance 1H-13C-15N 3.2 mm E-Free MAS probe (39–41). The one-dimensional (1D) 13C spectra were collected at 800 MHz 1H frequency, using either through-space cross-polarization (CP) (42,43) or through-bond insensitive nuclei enhanced by polarization transfer (INEPT) (44) excitations. In both cases, a proton field of 77 kHz was applied for SPINAL-64 decoupling (45). All 1D spectra presented in this work were measured on sample 1.
The two-dimensional (2D) 13C-13C correlation spectra were measured with the 13C-13C mixing accomplished using either through-space dipolar assisted rotational resonance (DARR) (46) or SPC53 (47,48) mechanisms, or through-bond correlation spectroscopy (TOBSY) (49,50).
The through-space DARR and SPC53 spectra were acquired on both samples 1 and 2 at a MAS rate of 12 kHz on the 600 MHz spectrometer, with a recycle delay of 2.9 s. The DARR mixing was of 50 ms duration. The band-selective through-space SPC53 irradiation was 1.26 ms, which is sufficient to generate one-bond transfers. The 100 kHz CW proton decoupling was applied during mixing. The through-bond TOBSY (49,50) experiment was modified to start with 1H/13C CP and a π/2 13C pulse to ensure that both mobile and immobile parts of the spectra were excited with maximum efficiency.
The TOBSY spectrum was collected from sample 2 on the 800 MHz Bruker Avance III spectrometer at a MAS rate of 14.3 kHz and with a pulse delay of 1.7 s. The TOBSY mixing was performed using the sequence (50) with a total irradiation time of 8.4 ms. Continuous-wave (CW) proton decoupling of 100 kHz was applied during mixing. The 13C carrier frequency was set to 40 ppm. Additional experimental parameters are given in the figure captions. Carbon chemical shifts were indirectly referenced to 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS) through the 13C adamantane downfield peak resonating at 40.48 ppm (51). The nitrogen chemical shifts were referenced indirectly by using the ratio of γN/γC = 0.402979946, taken without temperature factor correction (52). All temperature values reported here represent the temperature of the cooling gas in the close vicinity of the sample. All 2D spectra were processed with NMRPipe (53).
Results and Discussion
Sample preparation: mass spectrometry, SDS-PAGE, and TEM
Actin was prepared from chicken skeletal muscle using standard protocols (38). The purity of actin was characterized using mass spectrometry, which indicated one band at 42.027 kDa corresponding to singly ionized monomers, another band corresponding to doubly ionized monomers, and two extra bands corresponding to singly and doubly ionized dimers (results not shown). The measured molecular mass was quite close to that of the full-length unmodified chicken actin (377 residues, 42.051 kDa), and was slightly higher than for the mature α-skeletal actin, which is modified by N-terminal deletion of two residues and has a reduced molecular mass of 41.817 kDa (Swiss-Prot database entry P02568). The absence of a 38 kDa band showed that the final actin product was highly pure and not affected by proteolysis (38).
The polymerization and bundling of actin by salts or by rmMBP was assessed by SDS-PAGE. Lanes 1 and 2 in Fig. 1 A show single protein bands corresponding to actin and rmMBP, respectively, whereas lanes 3 and 4 demonstrate that essentially all actin remains in the supernatant upon ultracentrifugation and is in the globular form in the G-buffer. It is polymerized by salts (lanes 5 and 6) and is therefore biologically active. The evaluation of rmMBP/G-actin interactions and stoichiometry of rmMBP/actin binding is shown in Fig. 1 B (lanes 7–12). We found that rmMBP induces polymerization and bundling of G-actin in a molar ratio of 1:1.6 of G-actin/rmMBP, in agreement with previous measurements (20,21). This stoichiometry is different if rmMBP interacts with filamentous actin polymerized by salts (Fig. 1 C). In this case, the interaction occurs in a 1:1 molar ratio (lanes 15 and 16).
Figure 1.

(A) Assessment of actin polymerization by salt using SDS-PAGE. Lanes 1 and 2 are the actin and rmMBP controls, respectively. Lanes 3 and 4 are the supernatant and pellet, respectively, obtained by ultracentrifugation of the G-actin sample. The absence of any actin in the pellet indicates minimal polymerization in the absence of rmMBP. Lanes 5 and 6 represent the supernatant and pellet fractions, respectively, of salt-induced F-actin, confirming the integrity of the original G-actin preparation; 20 μL of each protein sample (actin at 5 μM concentration) were applied to lanes 1–6. (B) Interaction of rmMBP with G-actin. In lanes 7–12, each adjacent pair of lanes marked S and P represents supernatant and pellet, respectively, obtained by ultracentrifugation of the complex formed by the interaction of rmMBP and G-actin in G-buffer under nonpolymerizing conditions. The molar ratio of [G-actin]/[MBP] was 1:1.6 (lanes 7 and 8), 1:1.7 (lanes 9 and 10), and 1:1.8 (lanes 11 and 12). (C) Interaction of rmMBP with F-actin. Lanes 13–20 represent the result of bundling of salt-produced F-actin by rmMBP. The molar ratios of [actin]/[MBP] were 1:0.9 (lanes 13 and 14), 1:1.0 (lanes 15 and 16), 1:1.1 (lanes 17 and 18), and 1:1.2 (lanes 19 and 20). The final actin concentration in all samples was 10 μM. The samples were mixed with loading dye at 1:1 volume ratio, reducing the actin concentration in all samples to 5 μM, and the rmMBP concentration was as indicated above. Lanes 7–20: 10 μL of sample were applied to each lane.
The morphology of the rmMBP/F-actin assemblies was examined by means of TEM on fourfold-diluted and negatively stained (with uranyl acetate) samples. The TEM experiments were performed on two samples (described in detail in Materials and Methods): G-actin polymerized and bundled with rmMBP (sample 1), and G-actin polymerized with salts to form F-actin, which was then bundled with rmMBP (sample 2). The electron micrographs of sample 1 (Fig. 2) showed large bundles that appear to be ordered, along with pronounced branching of many filaments at the boundaries of these bundles. The appearance of these bundles in sample 1 suggests that there could be some microscopic structural inhomogeneity associated with the branching.
Figure 2.
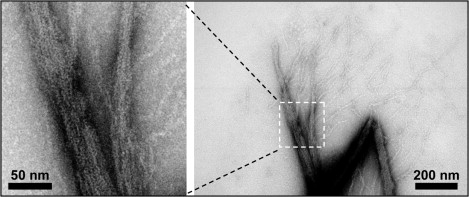
TEM of rmMBP-actin bundles obtained at a 1:1.5 molar ratio of actin to rmMBP and negatively stained with uranyl acetate. The image was captured on a Philips CM10 transmission electron microscope, using a fourfold-diluted fraction of a preparation typical of that used for NMR spectroscopy.
On the other hand, micrographs from sample 2 (not shown) did not display the branching of filaments at the boundaries of the bundles, indicating that the bundles were more compact than the ones in sample 1, in agreement with previously reported observations (23,24). As discussed below, SSNMR measurements, which are sensitive to the degree of microscopic order, yielded similar spectra for both samples, indicating the minor effect of the appearance of these filaments on the atomic level structural order.
Amide-proton exchange by FTIR spectroscopy
The amide-proton exchange of the rmMBP-F-actin samples was examined by FTIR spectroscopy to probe different timescale exchange processes, as well as the existence of different secondary structure elements in rmMBP. Previous FTIR (54) and solution NMR (55) studies of MBP in aqueous solution showed that MBP adopts an extended conformation without a significant proportion of regular secondary structure elements. Fig. 3 A shows two ATR FTIR spectra of rmMBP-F-actin films with the band assignments. To study amide-proton exchange separately in actin and in MBP, we used fully isotopically labeled rmMBP, which results in shifts of the C- and N-containing vibrational bands due to rmMBP to lower frequencies (up to 40 cm−1). In the dry film before the addition of D2O, the amide I and amide II bands for protonated actin and rmMBP dominate the spectrum. Amide I bands for actin indicate the presence of more than one type of secondary structure (α-helices, β-sheets, and β-turns, and extended structure), as can be inferred from the multiple peaks in the range of 1680–1615 cm−1 (56). This conclusion is also confirmed by the appearance of multiple amide II peaks in protonated actin in the range of 1558–1533 cm−1, especially shortly after the addition of D2O, when many overlapping bands due to the fast-exchanging extended structures are quickly removed in both actin and rmMBP. The two major amide I peaks at 1653 and 1631 cm−1 were previously assigned to α-helices and β-sheets of F-actin, respectively (57). Of interest, in our spectra, the 1631 cm−1 peak corresponding to β-sheets dominates the spectra of rmMBP-associated actin, similar to membrane-associated actin but in contrast to free F-actin (57). Thus, the FTIR results suggest that the interaction with MBP induces a higher content of β-sheets in F-actin, similar to the interaction with lipids in the presence of divalent cations.
Figure 3.
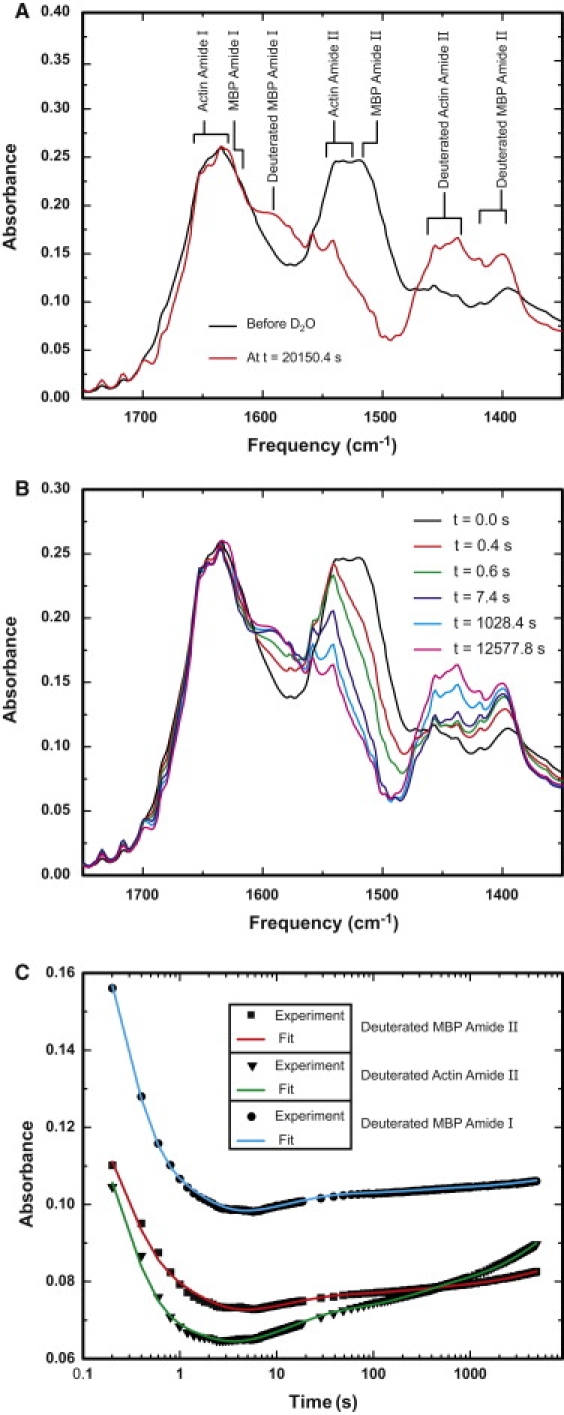
ATR-FTIR spectroscopic measurements of amide-proton exchange in the rmMBP-F-actin assemblies. (A) Spectra for the dry film before soaking with D2O (black curve) and at 6 h after adding D2O (red curve). Amide I and II bands for both fully isotopically labeled rmMBP and unlabeled actin (two major bands for β-and α-structures are observed) in the protonated state, as well as amide I′ and II′ bands for the deuterated fractions of both rmMBP and actin, are indicated. (B) A series of FTIR spectra at different time points after adding D2O as indicated. The spectra in both panels A and B are normalized to the integrated peak intensity of the amide I band. The amide II′ band for deuterated rmMBP increases very rapidly within the first second of the time course, as compared to the increase in the corresponding band in actin. Later on, the intensity of the rmMBP amide II′ band very quickly reaches near-saturation, whereas that of the actin amide II′ band continues to increase almost linearly. (C) The absorbance as a function of time for deuterated rmMBP amide II′ (red curve), deuterated actin amide II′ (green curve), and deuterated rmMBP amide I′ (blue curve). The solid lines are the global exponential fitting results, whereas the scattered points are the experimental values of the absolute absorbance. The points are not normalized. The initial drop in intensity is due to the swelling effect on the addition of D2O to the dry film.
The bands due to both deuterated rmMBP and actin in the amide II′ region in the range of 1471–1400 cm−1 in Fig. 3 B give rise to at least six peaks. This large number of peaks indicates the presence of different types of secondary structure in deuterated actin (1471–1437 cm−1). In addition, the observation of more than one peak between 1437 and 1400 cm−1 implies the presence of different types of secondary structure elements in rmMBP in this assembly. Moreover, the bands due to extended structures of isotopically labeled rmMBP centered at 1400 cm−1 have a very fast buildup in the first second after the addition of D2O, much faster than the corresponding buildup rate for all actin bands in the range of 1471–1437 cm−1. At longer times, the amplitude at 1400 cm−1 comes to saturation, whereas the amplitudes of actin bands in the range of 1471–1437 cm−1 continue to increase.
Fig. 3 C compares the experimental absorbances measured as a function of time after the addition of D2O, and the results of global exponential fitting. The absorbances were measured for the deuterated rmMBP amide II′ band at 1400 cm−1, corresponding to the random coil conformation, to the deuterated actin amide II′ band at 1437 cm−1, corresponding to β-sheet or extended structure, and to the deuterated rmMBP amide I′ band at 1595 cm−1 for random coil. It should be noted that a substantial (>10 cm−1) deuterium-induced downshift of the amide I band of rmMBP indicates the predominance of an extended (disordered) structure, as both α-helices and β-sheets display only minor shifts (58). The values of absorbance reported in the plots in Fig. 3 C are the absolute values given without normalization. Although the initial intensity reduction, due to sample swelling after the addition of D2O, distorts the spectra in the initial time stage in all of the spectral ranges and hinders the accurate determination of time constants of exchange within the fast (subsecond) timescale, the rest of the time course can be used to differentiate proton-deuteron exchange processes in both rmMBP and actin. The buildup of absorbance at the three given frequencies shows that most of the rmMBP exchanges very quickly during the first 12 s after the addition of D2O and reaches a near-saturation state, as can be inferred from the small increase in amplitudes for both curves corresponding to rmMBP. Actin displays similar behavior in the initial 12 s, when it displays a pronounced increase in absorbance. However, during the rest of the time course, an even larger increase in absorbance is pronounced, indicating the continuity of exchange processes in actin in a longer timescale regime (of the order of 500 s).
The vibrational bands observed by FTIR spectroscopy are very sensitive to hydrogen bonding and couplings between transition dipoles, which makes it a very powerful technique for probing secondary structure in proteins, and for monitoring hydrogen-deuterium exchange by analysis of the amide II band (56,59). Protein-protein interactions (involving IDPs) are usually accompanied by conformational changes that range from local environmental changes to the induction of new secondary structure elements and induced folding (8–10). Here, the FTIR spectra suggest the formation of secondary structure elements in rmMBP in complex with F-actin bundles. The large number of peaks in the amide II′ region due to both actin and rmMBP indicate that actin, as is well known, contains all the different types of secondary structure elements, including α-helices, β-sheets, β-turns, and extended structures such as random coil. The remaining peaks (especially the one at 1417 cm−1) cannot be due to random coil or other extended structures in rmMBP. Moreover, the buildup of absorbance at 1417 cm−1 indicates that the polypeptide chain due to this band is more protected than the other part that gives rise to the band at 1400 cm−1. This result indicates that a significant fraction of rmMBP in complex with F-actin gains some ordered secondary structure, and that more solvent-protected parts of the protein give rise to the band centered at 1417 cm−1.
SSNMR spectroscopy—1D 13C MAS experiments
SSNMR spectroscopy provides information on dynamics and sequence-specific formation of ordered secondary structure in actin-bound rmMBP. The temperature dependence of the intensities and resolution in the CP-MAS spectra demonstrate differential mobility in the protein. Two representative CP-MAS spectra measured at 280K and 250K (Fig. 4, A and B, respectively) differ in both intensity and resolution for all resonances. The low-temperature spectrum has a substantially higher intensity (by approximately a factor of 2), yet significantly lower resolution, especially in the aliphatic region from 15 to 35 ppm. Both observations are attributed to the effects of molecular motions, which result in conformational averaging and better resolution at higher temperatures, but also have averaging effects on the proton-carbon dipolar couplings. Consequently, the temperature-dependent resolution and intensity changes are not uniform across the spectrum: the averaging of dipolar interactions results in stronger intensity attenuation for carbonyl atoms (not shown), which are relatively weakly coupled to protons, compared to the proton-bonded aliphatic spins. Furthermore, the 1D 13C CP-MAS spectra at temperatures lower than 250K (not shown) showed little improvement in CP enhancement, which thus had little effect on the improvement of the signal/noise ratio of multidimensional spectra collected at the same temperature (not shown).
Figure 4.
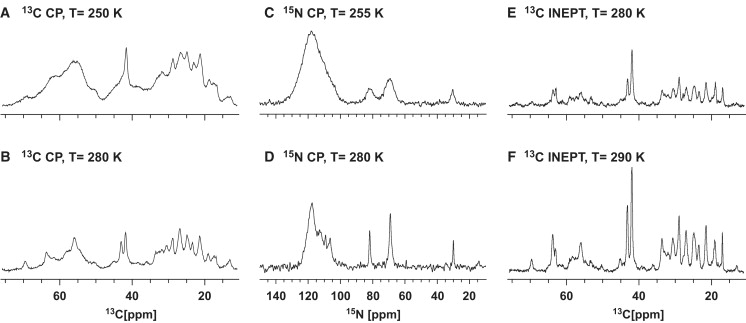
13C CP-MAS spectra (A and B), 15N CP-MAS spectra (C and D), and INEPT-MAS spectra (E and F) of uniformly 13C,15N-labeled 18.5 kDa rmMBP in rmMBP-actin complex as a function of temperature, as indicated in the figure. The 13C CP-MAS spectra were collected with an acquisition time of 30 ms, with 256 scans and a CP proton-carbon contact time of 2 ms. The 15N CP-MAS spectra were collected with an acquisition time of 25 ms, with 64 and 256 scans for C and D, respectively, and with a CP contact time of 2 ms. The INEPT-MAS spectra were collected with an acquisition time of 30 ms and 512 scans. All 13C spectra were acquired on a Bruker Avance III 800 MHz spectrometer equipped with a triple-resonance 3.2 mm E-free probe at a 14 kHz MAS rate, whereas 15N spectra were collected on a Bruker Avance III 600 MHz equipped with a triple-resonance 3.2 mm probe at 12 kHz MAS. Data were processed with 10 Hz exponential line-broadening before Fourier transformation.
The 1D CP-MAS 15N spectra in Fig. 4, C and D, exhibit a similar type of temperature dependence: the spectral intensity increases nonuniformly with decreasing temperature, whereas the resolution becomes worse. The region in the 15N spectrum affected the most by the temperature increase is in the range of 100–115 ppm, which corresponds to the backbone resonances of Gly, Ser, and Thr. In agreement with this result is the resolution enhancement in the region of Ser/Thr Cβ resonances around 60–70 ppm, and the region of Gly Cα resonances around 45 ppm in Fig. 4, A and B. Moreover, the side-chain nitrogen atoms of amino acids such as Lys and Arg display extensive line-broadening, accompanied by a pronounced intensity reduction as the temperature is reduced from 280K to 255K. This phenomenon can be attributed only to the quenching of global motions that result in freezing of different conformations at these sites.
The INEPT experiment provides an alternative means to preferentially excite spectral regions that correspond to protein fragments of increased mobility. The observed temperature dependence of INEPT spectra is quite different from that of CP-MAS spectra: both intensity and resolution enhancements are observed at higher temperature (Fig. 4, E and F), consistent with a higher degree of mobility. The lines' positions and shapes in the INEPT-MAS spectra are very similar to those in the CP-MAS spectra, most likely indicating that the motional averaging of proton-carbon dipolar couplings is incomplete, so one can observe mobile species in both through-space and through-bond excited spectra, as previously demonstrated for membrane-associated MBP (16).
SSNMR spectroscopy—2D 13C-13C correlations
The line-broadening observed in 1D spectra at low temperatures is most likely due to the conformational inhomogeneity introduced by quenching of motions upon freezing. To further assess mobility, conformational heterogeneity, and the formation of ordered secondary structure in rmMBP upon binding actin, we acquired a series of 2D 13C-13C correlation spectra at different temperatures, using both through-space and through-bond interactions as mechanisms for establishing correlations.
In Fig. 5, A and B, we show 13C-13C 2D spectra acquired with through-space DARR (46) mixing of 50 ms at 274K and 255K, respectively. Both spectra were acquired on sample 1 (G-actin polymerized and bundled with 13C,15N-rmMBP). The spectrum that was obtained at 274K (Fig. 5 A) has a lower signal/noise ratio, with fewer groups of crosspeaks. This result is in fair agreement with the preliminary picture derived from 1D experiments: the increased mobility at higher temperatures causes averaging of carbon-carbon dipolar interactions and decreases the polarization transfer efficiency.
Figure 5.
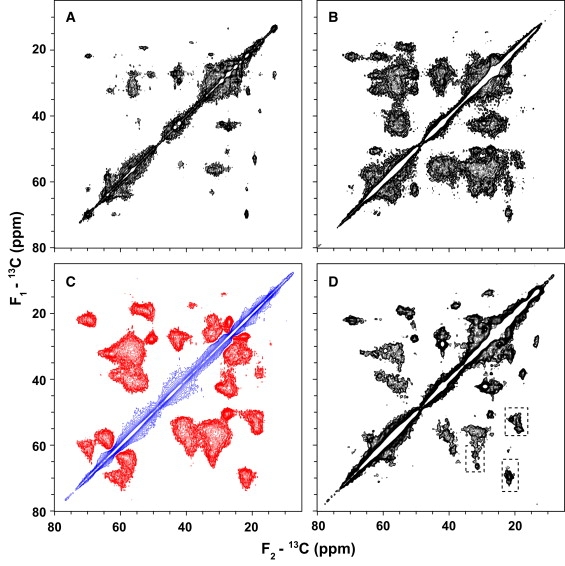
Series of 2D 13C-13C correlation spectra of uniformly 13C,15N-labeled rmMBP in the rmMBP-actin complex. In all experiments, the indirect and direct acquisition times were 7.6 and 12 ms, and the spectra were apodized with a π/2-shifted sine-squared window function before Fourier transformation. The first contour is cut at 4.5 times the noise floor and each subsequent contour level is multiplied by 1.15. The regions indicated by dashed rectangles are expanded in Fig. 6 for secondary structure analysis. (A and B) Aliphatic regions of DARR experiments collected at 274K and 255K, respectively, at 600 MHz. Both DARR spectra were acquired with 64 scans per t1 point. (C) Aliphatic region of the SPC53 double-quantum correlation spectrum collected at 600 MHz, 248K, and with 56 scans per t1 point. All crosspeaks in this spectrum are negative, indicating that they are all due to one-bond correlations. (D) Aliphatic region of TOBSY spectrum collected at 278K, at 800 MHz, with 128 scans per t1 point.
In contrast, the spectrum at 255K in Fig. 5 B shows all the expected backbone-backbone, backbone-side chain, and side chain-side chain correlations, although it also displays dramatically limited spectral resolution except for some isolated peaks. Because of the nature and length of DARR mixing, the low-temperature spectrum in Fig. 5 B likely shows multibond and possible interresidue correlations. An additional low-temperature carbon-carbon correlation that uses short SPC53 (47,48) mixing of 1.26 ms (Fig. 5 C) shows only one-bond correlations. All crosspeaks are very broad in this spectrum, reflecting the heterogeneity of the actin/MBP complex.
From the resolution of the DARR and SPC53 spectra, it is already apparent that actin-bound MBP is likely to be structurally heterogeneous. We further used through-bond TOBSY (49,50) spectroscopy to establish amino acid specific correlations at 5°C, where spectra have higher spectral resolution. The TOBSY mixing was chosen to be 8.4 ms to result in mainly one-bond correlations (Fig. 5 D). The spectrum has higher resolution, but the transfer efficiency varies between different peaks. Most notably, Cα/Cβ correlations due to threonines around 60/70 ppm in F1/F2 dimensions are quite weak, although the Cβ/Cγ correlations around 70/20 ppm are very pronounced.
MBP is an IDP and yields poorly dispersed NMR spectra when in unbound form, with most backbone chemical shifts having random coil values (55). The information content and the resolution of the TOBSY spectrum are sufficient to enable conclusions to be drawn about the formation of secondary structure in MBP in actin-associated form. Fig. 6 displays well-isolated Ala Cα-Cβ, Thr Cβ-Cγ, and Pro/Val Cα-Cβ regions of the TOBSY spectra, which we use to analyze the presence of secondary structure in MBP. The binding to actin induces substantial structural rearrangements in rmMBP, which in turn result in better-dispersed chemical shifts with both α-helical and β-strand elements present. In particular, the region corresponding to Ala Cα-Cβ correlations provides an excellent example of the presence of different secondary structure types in the protein. The detected chemical shift ranges for alanines are 50.6–56.0 ppm for Cα, and 17.8–22.0 ppm for Cβ, considerably more dispersed than the random coil values of 52.67 ± 0.7 ppm and 19.03 ± 0.7 ppm for Cα and Cβ, respectively (60). Furthermore, both types of secondary structure, α-helical and β-strand, are detected. Similarly, the presence of induced secondary structure is observed in the Cα/Cβ regions of threonines, and of valines and prolines (Fig. 6).
Figure 6.
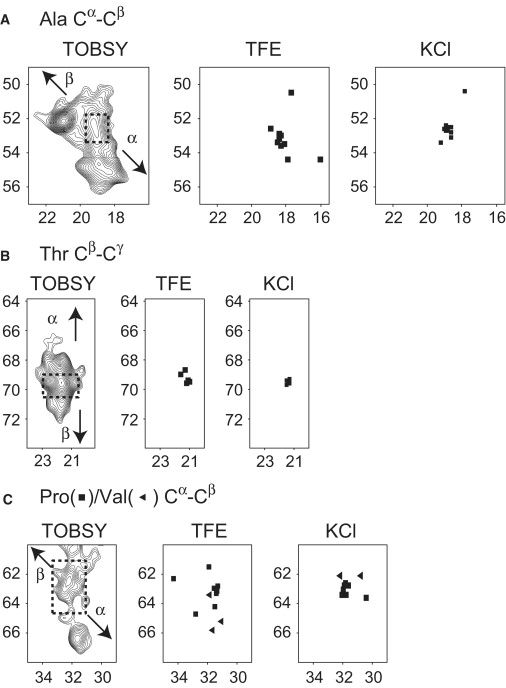
Formation of the secondary structure in rmMBP bound to actin and comparisons with the 2D plots constructed using chemical shift assignments of rmMBP dissolved in 30% TFE (37) (BMRB accession number 6100) and in aqueous 100 mM KCl solution (55) (BMRB accession number 15131). (A) The Ala Cα-Cβ region extracted from the TOBSY spectrum shown in Fig. 5D. (B) The Thr Cβ-Cγ region. (C) The Pro/Val Cα-Cβ region. The dashed squares indicate random coil regions, and the arrows indicate expected trends for crosspeaks due to α-helices and β-strands (60).
All of the examples above prove the formation of different regular secondary structure elements, both α-helices and β-strands. This is the first unequivocal experimental demonstration of the formation of β-strands in 18.5 kDa MBP in association with proteins, as opposed to lipids (11). Although site-specific information is not available at this stage, further insights into secondary structure formation can be derived from comparisons of the carbon chemical shifts of rmMBP in its actin-bound form with the recent chemical shift assignment obtained for rmMBP in aqueous solution containing 100 mM KCl (55), where the protein exists in extended unstructured form, and with the assignment in 30% trifluoroethanol (TFE) (37), where MBP contains several restrained regions, particularly three α-helical segments (15).
There are 11 alanines in the recombinant murine MBP. Almost all of the alanyl peaks are poorly dispersed and clustered around the random coil values in both KCl and 30% TFE solutions (37,55). In contrast, the alanyl Cα shifts become dispersed over a 5–6 ppm range, unambiguously showing the presence of both α-helical and β-strand structures. Another example is provided by well-dispersed Cβ shifts in the range of 66.8–71.9 ppm of the Thr Cβ-Cγ region, for actin-bound MBP, as depicted in Fig. 6 B. In contrast, in KCl and 30% TFE solutions, no threonines are found in any of the locally ordered elements, and the coil chemical shifts of these residues suggest they are in a random coil formation (60).
Of 10 Thr residues in full-length rmMBP, seven are located in the N-terminus (Thr15–Thr77), one (Thr147) is in the C-terminus, and the remaining two (Thr92 and Thr95) are within or in the vicinity of the immunodominant epitope (Pro82–Pro93) (12–16). Similarly, of 11 alanines, five are located in the N-terminus (Ala1–Ala49) and the remaining six are in the C-terminal half of the protein (Ala115–His176). The high population of these residues in the N- and C-termini implies that the terminal regions are particularly affected by the interaction with actin.
A more site-specific analysis of induced order can be obtained with the aid of Val resonances. There are only three valines in the 18.5 kDa murine MBP isoform (Val84, Val85, and Val90), and all are located within the primary immunodominant epitope (12–16). In Fig. 6 C, the unambiguous identification of valines cannot be performed based on the Cα/Cβ region alone, because of the overlap with the Cα/Cβ resonances of the 11 prolines. However, a direct comparison with the corresponding region constructed based on the assignments in TFE indicates strong correlations between positions of two of the valines, Val83 and Val84, in TFE with some of the resonances in the TOBSY spectrum. The residues Val83 and Val84 are known to be part of the α-helix in the immunodominant epitope in various membrane-mimetic conditions (TFE, lipid vesicles, or dodecylphosphocholine micelles) (12–16). This correlation suggests, albeit circumstantially, the formation of an α-helix in the proximity of the immunodominant epitope, but this time in association with actin, not lipids. Fig. 7 shows a schematic comparison of induced secondary structure elements for rmMBP associated with lipid vesicles, and the potential regions containing ordered secondary structure elements as predicted here from the chemical shift information of Val, Ala, and Thr.
Figure 7.
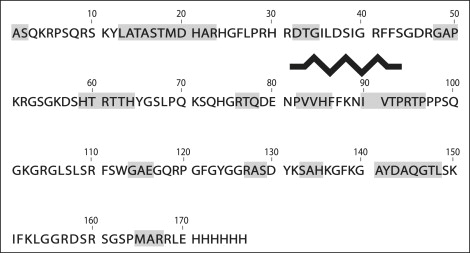
Comparison between the secondary structure elements formed in rmMBP upon association with lipids or with actin. The black zigzag represents the amphipathic α-helix that forms in the immunodominant epitope (Asn81–Thr92) upon binding lipid vesicles (12,13). The proline-rich segment immediately following (and slightly overlapping) this epitope forms a transient polyproline type II helix (6). The potential regions that might undergo changes in secondary structure upon binding actin are shaded.
Biological significance
Because of its genetic diversity (different transcription start sites and numerous translocatable splice isoforms) and extensive posttranslational modifications, it is believed that MBP must play further roles in myelin development and maintenance beyond mere membrane adhesion (3). Shiverer transgenic mice that lack the MBP gene exhibit both substantial hypomyelination of CNS neurons and myelination of inappropriate targets (61), whereas oligodendrocytes cultured from these mice display extensive cytoskeletal disorganization and an abundance of aberrant processes (62). Polymerization of actin in the CNS causes movement of the leading edge of membranous cellular processes. The extension of these cellular processes, in turn, forms myelin (63,64). Such in vivo evidence, collectively with in vitro studies of the 18.5 kDa protein's interactions with cytoskeletal proteins (see Introduction), suggests that it is involved in cytoskeletal assembly during myelin formation, and in its maintenance (assembly and tethering to the membrane) in mature myelin.
This multifunctionality is directly related to the intrinsically disordered nature of MBP. Some IDPs are thought to function merely as springs or linkers (i.e., entropic chains). Often IDPs associate rapidly (but still specifically) with one or multiple binding partners (65), and thus function as hubs in protein interaction and signaling networks, or as molecular switches in transcriptional and translational control. Their binding affinities can be regulated by modifications such as phosphorylation. The intermolecular associations of IDPs may involve preformed structural elements such as α-helices, potentially transient, and/or an induced fit, i.e., coupled folding and binding (8–10,17).
Conclusion
Here, we have used FTIR and MAS SSNMR spectroscopy to show that 18.5 kDa rmMBP in association with F-actin exhibits induced secondary structure formation, albeit with a high degree of polymorphism and disorder remaining. These results are reminiscent of the protein's conformational behavior upon association with lipid vesicles, which was shown in our previous study (also by MAS SSNMR) in which at least one-third of the protein remained exposed to solvent and was mainly disordered (16). In our solution NMR investigations of MBP binding to Ca2+-CaM (15), we discussed the interaction in the framework of the MoRF hypothesis (17). In the framework of the more extensive discussion of “fuzzy complexes” by Tompa and Fuxreiter (66), the MBP-Ca2+-CaM (and potentially MBP-SH3-domain) interaction can best be described by a “dynamic fuzziness” (flanking) model, whereas the MBP-actin association studied here may represent an example of “static fuzziness” (polymorphism).
In conclusion, if MBP regulates or maintains cytoskeletal assembly in developing and mature myelin, it must have both a static nature (to maintain membrane adhesion) and a significant dynamic component to allow it to interact with actin, tubulin, CaM, SH3-domains, and various modifying enzymes. In terms of the prevailing paradigms of IDPs, MBP behaves like both an entropic spring or linker, and an induced fit protein target that may have additional roles in signaling.
Acknowledgments
We are grateful to Mr. Robert Harris (manager, Guelph Regional Microscopy Facility) for his assistance with sample preparation and TEM. We thank Dr. Joan Boggs, Hospital for Sick Children, University of Toronto, for carefully reading the manuscript.
This work was supported by the Canadian Institutes of Health Research (CIHR, operating grant MOP 74468) and the Natural Sciences and Engineering Research Council of Canada (NSERCC). V.L. holds a Tier II Canada Research Chair and is a recipient of an Early Researcher Award from the Ontario Ministry of Research and Innovation. M.A. is a recipient of a doctoral studentship from the Ministry of Higher Education and Scientific Research of Egypt.
Contributor Information
George Harauz, Email: gharauz@uoguelph.ca.
Vladimir Ladizhansky, Email: vladimir@physics.uoguelph.ca.
References
- 1.Radivojac P., Iakoucheva L.M., Oldfield C.J., Obradovic Z., Uversky V.N. Intrinsic disorder and functional proteomics. Biophys. J. 2007;92:1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Receveur-Brechot V., Bourhis J.M., Uversky V.N., Canard B., Longhi S. Assessing protein disorder and induced folding. Proteins. 2006;62:24–45. doi: 10.1002/prot.20750. [DOI] [PubMed] [Google Scholar]
- 3.Boggs J.M. Myelin basic protein: a multifunctional protein. Cell. Mol. Life Sci. 2006;63:1945–1961. doi: 10.1007/s00018-006-6094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boggs J.M., Rangaraj G., Gao W., Heng Y.M. Effect of phosphorylation of myelin basic protein by MAPK on its interactions with actin and actin binding to a lipid membrane in vitro. Biochemistry. 2006;45:391–401. doi: 10.1021/bi0519194. [DOI] [PubMed] [Google Scholar]
- 5.Harauz G., Ishiyama N., Hill C.M.D., Bates I.R., Libich D.S. Myelin basic protein—diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron. 2004;35:503–542. doi: 10.1016/j.micron.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Polverini E., Rangaraj G., Libich D.S., Boggs J.M., Harauz G. Binding of the proline-rich segment of myelin basic protein to SH3 domains: spectroscopic, microarray, and modeling studies of ligand conformation and effects of posttranslational modifications. Biochemistry. 2008;47:267–282. doi: 10.1021/bi701336n. [DOI] [PubMed] [Google Scholar]
- 7.Kim J.K., Mastronardi F.G., Wood D.D., Lubman D.M., Zand R. Multiple sclerosis—an important role for post-translational modifications of myelin basic protein in pathogenesis. Mol. Cell. Proteomics. 2003;2:453–462. doi: 10.1074/mcp.M200050-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Oldfield C.J., Cheng Y., Cortese M.S., Romero P., Uversky V.N. Coupled folding and binding with α-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 9.Sugase K., Dyson H.J., Wright P.E. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447:1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 10.Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–3354. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 11.Polverini E., Fasano A., Zito F., Riccio P., Cavatorta P. Conformation of bovine myelin basic protein purified with bound lipids. Eur. Biophys. J. 1999;28:351–355. doi: 10.1007/s002490050218. [DOI] [PubMed] [Google Scholar]
- 12.Bates I.R., Feix J.B., Boggs J.M., Harauz G. An immunodominant epitope of myelin basic protein is an amphipathic α-helix. J. Biol. Chem. 2004;279:5757–5764. doi: 10.1074/jbc.M311504200. [DOI] [PubMed] [Google Scholar]
- 13.Musse A.A., Boggs J.M., Harauz G. Deimination of membrane-bound myelin basic protein in multiple sclerosis exposes an immunodominant epitope. Proc. Natl. Acad. Sci. USA. 2006;103:4422–4427. doi: 10.1073/pnas.0509158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fares C., Libich D.S., Harauz G. Solution NMR structure of an immunodominant epitope of myelin basic protein—conformational dependence on environment of an intrinsically unstructured protein. FEBS J. 2006;273:601–614. doi: 10.1111/j.1742-4658.2005.05093.x. [DOI] [PubMed] [Google Scholar]
- 15.Libich D.S., Harauz G. Backbone dynamics of the 18.5 kDa isoform of myelin basic protein reveals transient α-helices and a calmodulin-binding site. Biophys. J. 2008;94:4847–4866. doi: 10.1529/biophysj.107.125823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong L., Bamm V.V., Ahmed M.A., Harauz G., Ladizhansky V. Solid-state NMR spectroscopy of 18.5 kDa myelin basic protein reconstituted with lipid vesicles: spectroscopic characterisation and spectral assignments of solvent-exposed protein fragments. Biochim. Biophys. Acta. 2007;1768:3193–3205. doi: 10.1016/j.bbamem.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohan A., Oldfield C.J., Radivojac P., Vacic V., Cortese M.S. Analysis of molecular recognition features (MoRFs) J. Mol. Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 18.Libich D.S., Hill C.M.D., Bates I.R., Hallett F.R., Armstrong S. Interaction of the 18.5-kDa isoform of myelin basic protein with Ca2+-calmodulin: effects of deimination assessed by Trp fluorescence spectroscopy, dynamic light scattering, and circular dichroism. Protein Sci. 2003;12:1507–1521. doi: 10.1110/ps.0303603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polverini E., Boggs J.M., Bates I.R., Harauz G., Cavatorta P. Electron paramagnetic resonance spectroscopy and molecular modelling of the interaction of myelin basic protein (MBP) with calmodulin (CaM)—diversity and conformational adaptability of MBP CaM-targets. J. Struct. Biol. 2004;148:353–369. doi: 10.1016/j.jsb.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 20.Barylko B., Dobrowolski Z. Ca2+-calmodulin-dependent regulation of F-actin-myelin basic protein interaction. Eur. J. Cell Biol. 1984;35:327–335. [PubMed] [Google Scholar]
- 21.Dobrowolski Z., Osinska H., Mossakowska M., Barylko B. Ca2+-calmodulin-dependent polymerization of actin by myelin basic protein. Eur. J. Cell Biol. 1986;42:17–26. [PubMed] [Google Scholar]
- 22.Boggs J.M., Rangaraj G. Interaction of lipid-bound myelin basic protein with actin filaments and calmodulin. Biochemistry. 2000;39:7799–7806. doi: 10.1021/bi0002129. [DOI] [PubMed] [Google Scholar]
- 23.Boggs J.M., Rangaraj G., Hill C.M.D., Bates I.R., Heng Y.M. Effect of arginine loss in myelin basic protein, as occurs in its deiminated charge isoform, on mediation of actin polymerization and actin binding to a lipid membrane in vitro. Biochemistry. 2005;44:3524–3534. doi: 10.1021/bi0473760. [DOI] [PubMed] [Google Scholar]
- 24.Hill C.M.D., Harauz G. Charge effects modulate actin assembly by classic myelin basic protein isoforms. Biochem. Biophys. Res. Commun. 2005;329:362–369. doi: 10.1016/j.bbrc.2005.01.151. [DOI] [PubMed] [Google Scholar]
- 25.Galiano M.R., Andrieux A., Deloulme J.C., Bosc C., Schweitzer A. Myelin basic protein functions as a microtubule stabilizing protein in differentiated oligodendrocytes. J. Neurosci. Res. 2006;84:534–541. doi: 10.1002/jnr.20960. [DOI] [PubMed] [Google Scholar]
- 26.Hill C.M.D., Libich D.S., Harauz G. Assembly of tubulin by classic myelin basic protein isoforms and regulation by post-translational modification. Biochemistry. 2005;44:16672–16683. doi: 10.1021/bi050646+. [DOI] [PubMed] [Google Scholar]
- 27.Baldus M. Molecular interactions investigated by multi-dimensional solid-state NMR. Curr. Opin. Struct. Biol. 2006;16:618–623. doi: 10.1016/j.sbi.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Castellani F., van Rossum B., Diehl A., Schubert M., Rehbein K. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 29.Jaroniec C.P., MacPhee C.E., Bajaj V.S., McMahon M.T., Dobson C.M. High-resolution molecular structure of a peptide in an amyloid fibril determined by magic angle spinning NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 2004;101:711–716. doi: 10.1073/pnas.0304849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDermott A.E. Structural and dynamic studies of proteins by solid-state NMR spectroscopy: rapid movement forward. Curr. Opin. Struct. Biol. 2004;14:554–561. doi: 10.1016/j.sbi.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 31.Thompson L.K. Solid-state NMR studies of the structure and mechanisms of proteins. Curr. Opin. Struct. Biol. 2002;12:661–669. doi: 10.1016/s0959-440x(02)00374-3. [DOI] [PubMed] [Google Scholar]
- 32.Tycko R. Biomolecular solid state NMR: advances in structural methodology and applications to peptide and protein fibrils. Annu. Rev. Phys. Chem. 2001;52:575–606. doi: 10.1146/annurev.physchem.52.1.575. [DOI] [PubMed] [Google Scholar]
- 33.Wasmer C., Lange A., Van Melckebeke H., Siemer A.B., Riek R. Amyloid fibrils of the HET-s(218–289) prion form a β solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 34.Bates I.R., Libich D.S., Wood D.D., Moscarello M.A., Harauz G. An Arg/Lys -> Gln mutant of recombinant murine myelin basic protein as a mimic of the deiminated form implicated in multiple sclerosis. Protein Expr. Purif. 2002;25:330–341. doi: 10.1016/s1046-5928(02)00017-7. [DOI] [PubMed] [Google Scholar]
- 35.Bates I.R., Matharu P., Ishiyama N., Rochon D., Wood D.D. Characterization of a recombinant murine 18.5-kDa myelin basic protein. Protein Expr. Purif. 2000;20:285–299. doi: 10.1006/prep.2000.1307. [DOI] [PubMed] [Google Scholar]
- 36.Kaur J., Libich D.S., Campagnoni C.W., Wood D.D., Moscarello M.A. Expression and properties of the recombinant murine Golli-myelin basic protein isoform J37. J. Neurosci. Res. 2003;71:777–784. doi: 10.1002/jnr.10547. [DOI] [PubMed] [Google Scholar]
- 37.Libich D.S., Robertson V.J., Monette M.M., Harauz G. Backbone resonance assignments of the 18.5 kDa isoform of murine myelin basic protein (MBP) J. Biomol. NMR. 2004;29:545–546. doi: 10.1023/B:JNMR.0000034348.99658.d7. [DOI] [PubMed] [Google Scholar]
- 38.Pardee J.D., Spudich J.A. Purification of muscle actin. Methods Enzymol. 1982;85:164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 39.Gor'kov P.L., Chekmenev E.Y., Li C., Cotten M., Buffy J.J. Using low-E resonators to reduce RF heating in biological samples for static solid-state NMR up to 900 MHz. J. Magn. Reson. 2007;185:77–93. doi: 10.1016/j.jmr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 40.Gor'kov P.L., Witter R., Chekmenev E.Y., Nozirov F., Fu R. Low-E probe for 19F-1H NMR of dilute biological solids. J. Magn. Reson. 2007;189:182–189. doi: 10.1016/j.jmr.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 41.Stringer J.A., Bronnimann C.E., Mullen C.G., Zhou D.H., Stellfox S.A. Reduction of RF-induced sample heating with a scroll coil resonator structure for solid-state NMR probes. J. Magn. Reson. 2005;173:40–48. doi: 10.1016/j.jmr.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 42.Pines A., Gibby M.G., Waugh J.S. Proton-enhanced nuclear induction spectroscopy. A method for high resolution NMR of dilute spins in solids. J. Chem. Phys. 1972;56:1776–1777. [Google Scholar]
- 43.Pines A., Gibby M.G., Waugh J.S. Proton-enhanced NMR of dilute spins in solids. J. Chem. Phys. 1973;59:569–590. [Google Scholar]
- 44.Morris G.A., Freeman R. Enhancement of nuclear magnetic resonance signals by polarization transfer. J. Am. Chem. Soc. 1979;101:760–762. [Google Scholar]
- 45.Fung B.M., Khitrin A.K., Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 46.Takegoshi K., Nakamura S., Terao T. C13-H1 dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 2001;344:631–637. [Google Scholar]
- 47.Hohwy M., Rienstra C.M., Jaroniec C.P., Griffin R.G. Fivefold symmetric homonuclear dipolar recoupling in rotating solids: application to double quantum spectroscopy. J. Chem. Phys. 1999;110:7983–7992. [Google Scholar]
- 48.Hohwy M., Rienstra C.M., Griffin R.G. Band-selective homonuclear dipolar recoupling in rotating solids. J. Chem. Phys. 2002;117:4973–4987. [Google Scholar]
- 49.Baldus M., Meier B.H. Total correlation spectroscopy in the solid state. The use of scalar couplings to determine the through-bond connectivity. J. Magn. Reson. A. 1996;121:65–69. [Google Scholar]
- 50.Hardy E.H., Verel R., Meier B.H. Fast MAS total through-bond correlation spectroscopy. J. Magn. Reson. 2001;148:459–464. doi: 10.1006/jmre.2000.2258. [DOI] [PubMed] [Google Scholar]
- 51.Morcombe C.R., Zilm K.W. Chemical shift referencing in MAS solid state NMR. J. Magn. Reson. 2003;162:479–486. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 52.Markley J.L., Bax A., Arata Y., Hilbers C.W., Kaptein R. Recommendations for the presentation of NMR structures of proteins and nucleic acids–IUPAC-IUBMB-IUPAB Inter-Union Task Group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. Eur. J. Biochem. 1998;256:1–15. doi: 10.1046/j.1432-1327.1998.2560001.x. [DOI] [PubMed] [Google Scholar]
- 53.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 54.Surewicz W.K., Moscarello M.A., Mantsch H.H. Fourier transform infrared spectroscopic investigation of the interaction between myelin basic protein and dimyristoylphosphatidylglycerol bilayers. Biochemistry. 1987;26:3881–3886. doi: 10.1021/bi00387a021. [DOI] [PubMed] [Google Scholar]
- 55.Libich D.S., Monette M.M., Robertson V.J., Harauz G. NMR assignment of an intrinsically disordered protein under physiological conditions: the 18.5 kDa isoform of murine myelin basic protein. Biomol. NMR Assign. 2007;1:61–63. doi: 10.1007/s12104-007-9016-1. [DOI] [PubMed] [Google Scholar]
- 56.Haris P.I., Chapman D. The conformational-analysis of peptides using Fourier-transform IR spectroscopy. Biopolymers. 1995;37:251–263. doi: 10.1002/bip.360370404. [DOI] [PubMed] [Google Scholar]
- 57.Gicquaud C., Wong P. Mechanism of interaction between actin and membrane-lipids—a pressure-tuning infrared-spectroscopy study. Biochem. J. 1994;303:769–774. doi: 10.1042/bj3030769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Susi H., Timasheff S.N., Stevens L. Infrared spectra and protein conformations in aqueous solutions. I. The amide I band in H2O and D2O solutions. J. Biol. Chem. 1967;242:5460–5466. [PubMed] [Google Scholar]
- 59.Haris P.I., Chapman D., Benga G. A Fourier-transform infrared spectroscopic investigation of the hydrogen-deuterium exchange and secondary structure of the 28-kDa channel-forming integral membrane-protein (Chip28) Eur. J. Biochem. 1995;233:659–664. doi: 10.1111/j.1432-1033.1995.659_2.x. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y.J., Jardetzky O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 2002;11:852–861. doi: 10.1110/ps.3180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Inoue Y., Nakamura R., Mikoshiba K., Tsukada Y. Fine structure of the central myelin sheath in the myelin deficient mutant Shiverer mouse, with special reference to the pattern of myelin formation by oligodendroglia. Brain Res. 1981;219:85–94. doi: 10.1016/0006-8993(81)90269-9. [DOI] [PubMed] [Google Scholar]
- 62.Dyer C.A., Philibotte T.M., Billings-Gagliardi S., Wolf M.K. Cytoskeleton in myelin-basic-protein-deficient shiverer oligodendrocytes. Dev. Neurosci. 1995;17:53–62. doi: 10.1159/000111273. [DOI] [PubMed] [Google Scholar]
- 63.Wilson R., Brophy P.J. Role for the oligodendrocyte cytoskeleton in myelination. J. Neurosci. Res. 1989;22:439–448. doi: 10.1002/jnr.490220409. [DOI] [PubMed] [Google Scholar]
- 64.Baumann N., Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- 65.Oldfield C.J., Cheng Y., Cortese M.S., Brown C.J., Uversky V.N. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44:1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- 66.Tompa P., Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]