

Abstract
We demonstrate beam scanning-stimulated emission depletion microscopy with in vivo labeled cells. A red emitting fluorescent dye is introduced into membrane protein fused to a multifunctional reporter protein (HaloTag, Promega, Madison, WI) in live cells. This approach allows superresolution stimulated emission depletion imaging without the limitations of immunofluorescence-based staining.
Noninvasive optical imaging is still an indispensable and elementary tool in modern life sciences. Unfortunately, the predominant obstacle of classical microscopes is limited spatial resolution due to the diffractive nature of light. Distinct technologies like electron and x-ray microscopy as well as near-field approaches evolved during the past decades to circumvent this major limitation. Furthermore, various techniques emerged in recent years breaking the diffraction resolution barrier of far-field optical microscopy (1–3). In particular, stimulated emission depletion (STED) microscopy became a breakthrough technology (4,5) for fluorescence imaging and is now available commercially (see Supplemetary Material). Commercial systems are fully integrated in optimized multispectral, high sensitivity confocal scanning microscopes for all needs of modern life science research. Since STED microscopy has evolved from high-end technology in specialist's labs to a reliable and easy-to-use instrument for life science, the expected widespread use of STED certainly will push the technology as well as applications to a next level.
Thus far, biological relevant imaging applications of STED microscopy were based on fixed immunolabeling techniques (e.g., (4–8)). Additionally, certain fluorescent membrane markers as well as genetically engineered fluorescent proteins that exhibited good performance when highly overexpressed (9) were used as in vivo labels in proof-of-principle applications (10,11). However, optimal results can only be obtained with selected fluorescent markers that fulfill spectroscopic needs for stimulated emission depletion. To date, best performing (amino-reactive) dyes are only available for standard immunostaining procedures. Hence, application of STED microscopy in live cell imaging has been hindered by a lack of protein-labeling technology. Here we present a flexible technology that, for the first time, allows fluorescent dyes optimized for STED microscopy to be linked onto reporter protein in living cells. The reporter protein (HaloTag, Promega, Madison, WI) is an engineered, catalytically inactive derivative of a bacterial hydrolase that can be fused to a protein of interest and is designed to covalently bind synthetic HaloTag ligands (12,13). The HaloTag ligands comprised two parts: a common reactive linker that forms the covalent bond with the reporter protein; and a functional group such as a fluorescent dye, affinity tag, or bead. Covalent bond formation is highly specific, occurs rapidly under physiological conditions, and is essentially irreversible. The synthetic chemistry associated with the ligands enables their multifunctional nature, allowing addition of various functional groups including amino-reactive dyes optimized to support STED microscopy. The interchangeable design of the ligands allows adaptation of the reporter protein to different experimental requirements without altering the underlying genetic construct (12,13).
To demonstrate that STED microscopy can be efficiently applied to study proteins labeled in live cells we used previously developed a β1-integrin-HaloTag model (14). Integrins are trans-membrane proteins that play a central role in cellular adhesion and migration; they are involved in development, inflammation, and disease, and remain a focus of both life science search and drug development (15,16). Previously, fusing the HaloTag reporter protein to an extracellular domain of a truncated human β1-integrin (β1Int-HaloTag), we were able to convert the HaloTag protein into transmembrane protein and expose it on the cell surface. Using sequential labeling of live cells expressing the β1Int-HaloTag with cell impermeant and cell permeant ligands of different colors, we demonstrated spatial separation of plasma membrane and internal pools of the β1Int-HaloTag fusion protein (14). We were also able to monitor bidirectional trafficking of these proteins over time, i.e., endocytosis of the surface-exposed pool and translocation of the internal pool to the cell surface (14).
In this study, we designed a cell impermeant ligand carrying a fluorescent dye optimized to support STED microscopy, labeled live cells expressing a β1-integrin-HaloTag fusion protein, fixed cells to prevent cell movement during imaging, and imaged cells using STED microscopy. Generated images revealed localization of the β1-integrin-HaloTag fusion protein in unprecedented details (Fig. 1), that proteins labeled in the complex environment of living cells can be studied using STED microscopy. The β1-integrins populate a variety of plasma membrane formations including filopodia, and are internalized as a part of endocytic vesicles (17).
Figure 1.
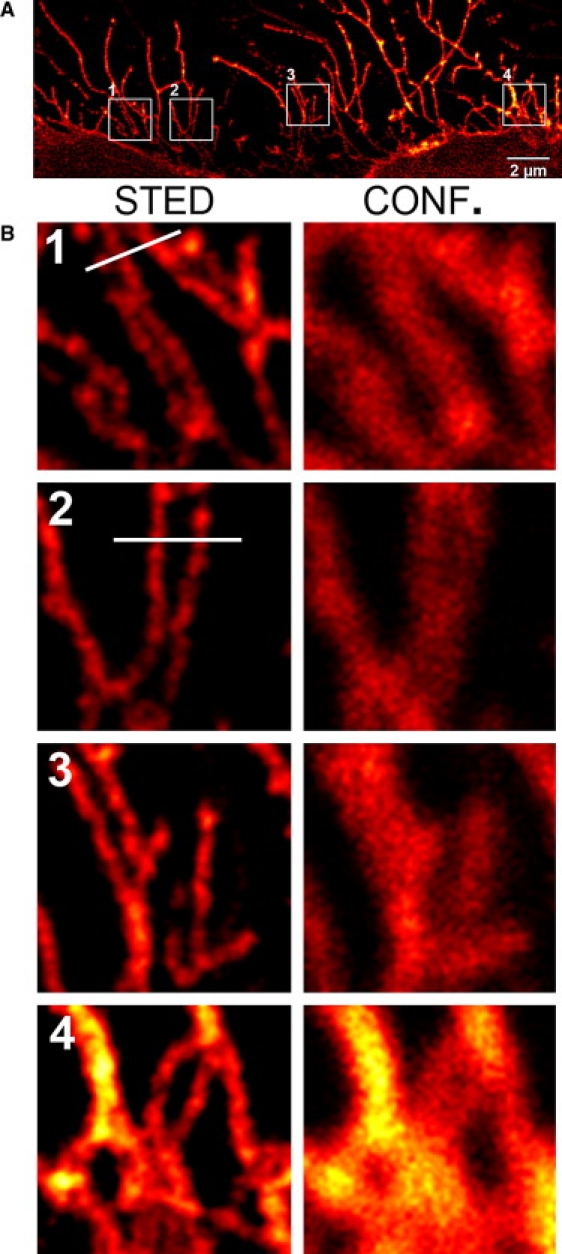
STED image showing an in vivo labeled HeLa cell (A). Detailed areas (B) showing filopodia imaged in STED mode (left) or confocal mode (right). Each region is 2-μm-square size. Profiles in 1 and 2 are referring to Fig. 2.
To further elucidate precise subcellular distribution of the β1-integrin-HaloTag fusion protein, we transiently expressed this protein in HeLa cells, known to form filopodia (18) and internalizing the β1-integrin-HaloTag fusion protein (14). We labeled cells with the HaloTag 655 ligand, and then imaged them using confocal and then STED microscopy. Confocal microscopy shows that the β1-integrin-HaloTag fusion protein populates the cell surface and filopodia (Fig. 1).
Using STED microscopy, diameters of fluorescently-labeled filopodia between 90 and 130 nm and distances of ∼130 nm can be resolved whereas confocal images revealed diameters of ∼300 nm and could often not resolve adjacent filopodia (Figs. 1 and 2). Although we believe our membrane-bound reporter to label all of the plasma membrane, including the minute filopodia (Fig. 1) known to cover cells such as HeLa, and we have suspected the observed endocytotic-like structures to be vesicular in nature, such detail has not been shown in cells labeled live, to date. In fact, conventional confocal microscopy, which has very successfully been used in the study of cellular trafficking, is limited in resolution such that finer subcellular detail has had to come from complimentary use of electron microscopy in the past. Here the use of HaloTag technology with STED-capable fluorophores combined with STED microscopy provides images of clear filopodia on the surface (Fig. 1) of the cells and opens new perspectives to elucidate the vesicular nature of the endocytotic-like structures labeled inside of the cells.
Figure 2.
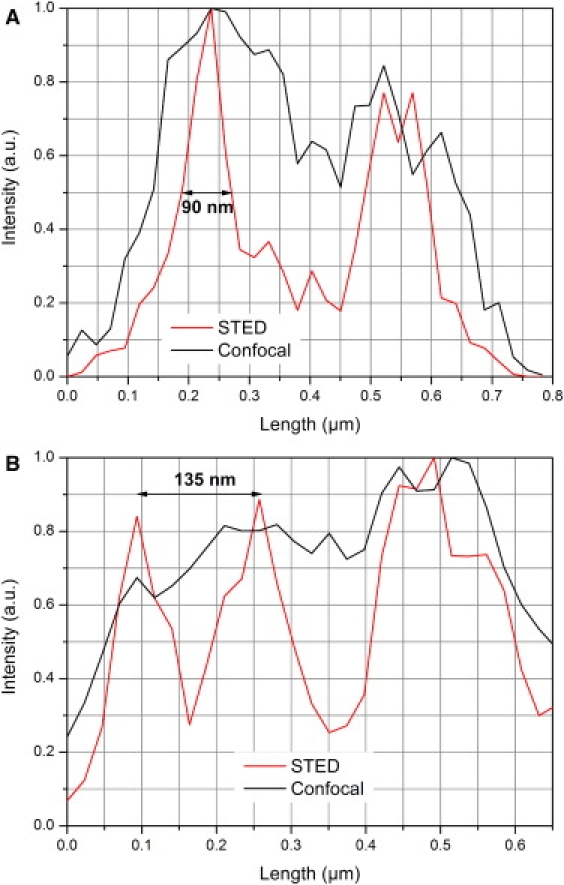
Showing the intensity profiles of Fig. 1B. (A) Thickness of one filopodium in detailed area 2. (B) Distance between to adjacent filopodia in detailed area 1.
Fluorescent reporters and confocal microscopy, allowing for live intracellular imaging, have greatly advanced the study of protein trafficking. As an extension of fluorescent-labeling technology, HaloTag represents a step forward both by limiting the necessary cloning to just one construct while providing the choice of using multiple reporters, i.e., spectrally different fluorescent labels as well as dedicated labels optimized for advanced imaging or sensing methods. Such a choice allows more precise study of protein dynamics by adding the flexibility of labeling a single protein in more than one color, highlighting both its precise localization(s) and its directed translocation, or trafficking, over time. Further, as the increased knowledge of cell trafficking also stems from our ability to resolve the resulting fluorescent signal within living cells, confocality has become a standard tool in cell biology labs. STED microscopy provides best superresolution results in combination with spectroscopic-optimized fluorophores and diverse fluorescent or affinity tags. STED microscopy, as shown here, exceeds traditional confocality by offering a strongly enhanced spatial resolution. Thus, combining the above technological advances in this study, we are able to further elucidate the localization of a membrane-bound protein directly and for the first time by live cell labeling. Specifically we clearly show that, when at the cell surface, this integrin actually does inhabit the entirety of the plasma membrane, including all filopodia. Also, when internalized, β1-integrin is clearly seen in the membrane of endocytotic vesicles.
Supplementary Material
Three figures and Materials and Methods are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(08)00041-6.
Supplementary Material
Acknowledgments
We gratefully acknowledge Chad Zimprich and Shana Svendsen for the cloning, characterization of β1-integrin-HaloTag and providing cells for this work. We are also very grateful to Mark McDougall for synthesizing the HaloTag655 ligand.
References and Footnotes
- 1.Hell S.W., Stelzer E.H.K. Properties of a 4Pi-confocal fluorescence microscope. J. Opt. Soc. Am. A. 1992;9:2159–2166. [Google Scholar]
- 2.Hell S.W., Wichmann J. Breaking the diffraction resolution limit by stimulated emission. Opt. Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- 3.Betzig E., Patterson G.H., Sougrat R., Lindwasser O.W., Olenych S. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 4.Willig K.I., Rizzoli S.O., Westphal V., Jahn R., Hell S.W. STED-microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature. 2006;440:935–939. doi: 10.1038/nature04592. [DOI] [PubMed] [Google Scholar]
- 5.Kittel R.J., Wichmann C., Rasse T.M., Fouquet W., Schmidt M. Bruchpilot promotes active zone assembly, Ca2+-channel clustering, and vesicle release. Science. 2006;312:1051–1054. doi: 10.1126/science.1126308. [DOI] [PubMed] [Google Scholar]
- 6.Sieber J.J., Willig K.I., Heintzmann R., Hell S.W., Lang T. The SNARE-motif is essential for the formation of syntaxin clusters in the plasma membrane. Biophys. J. 2006;90:2843–2851. doi: 10.1529/biophysj.105.079574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sieber J.J., Willig K.I., Kutzner C., Gerding-Reimers C., Harke B. Anatomy and dynamics of a supramolecular membrane protein cluster. Science. 2007;317:1072–1076. doi: 10.1126/science.1141727. [DOI] [PubMed] [Google Scholar]
- 8.Kellner R., Baier J., Willig K.I., Hell S.W., Barrantes F.J. Nanoscale organization of nicotinic acetylcholine receptors revealed by STED microscopy. Neuroscience. 2007;144:135–143. doi: 10.1016/j.neuroscience.2006.08.071. [DOI] [PubMed] [Google Scholar]
- 9.Willig K.I., Kellner R.R., Medda R., Hein B., Jakobs S. Nanoscale resolution in GFP-based microscopy. Nat. Methods. 2006;3:721–723. doi: 10.1038/nmeth922. [DOI] [PubMed] [Google Scholar]
- 10.Klar T.A., Jakobs S., Dyba M., Egner A., Hell S.W. Fluorescence microscopy with diffraction resolution limit broken by stimulated emission. Proc. Natl. Acad. Sci. USA. 2000;97:8206–8210. doi: 10.1073/pnas.97.15.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dyba M., Hell S.W. Photostability of a fluorescent marker under pulsed excited-state depletion through stimulated emission. Appl. Opt. 2003;42:5123–5129. doi: 10.1364/ao.42.005123. [DOI] [PubMed] [Google Scholar]
- 12.Los G.V., Wood K. The HaloTag: a novel technology for cell imaging and protein analysis. In: Guliano K., Taylor D.L., Haskin J., editors. Methods in Molecular Biology—High Content Screening: A Powerful Approach to System Cell Biology and Drug Discovery. Humana Press; Totowa, NJ: 2007. [DOI] [PubMed] [Google Scholar]
- 13.Los G.V., Encell L.P., McDougall M.G., Hartzell D.D., Karassina N. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ASC. Chem. Biol. 2008;3:351–361. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 14.Svendsen S., Zimprich C., McDougall M.G., Klaubert D.H., Los G.V. Spatial separation and bidirectional trafficking of proteins using a multi-functional reporter. BMC Cell Biol. 2008;9:1–14. doi: 10.1186/1471-2121-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo B., Carman C., Springer T. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hilden T., Nurmi S., Fagerholm S., Gahmberg C. Interfering with leukocyte integrin activation—a novel concept in the development of anti-inflammatory drugs. Ann. Med. 2006;38:503–511. doi: 10.1080/07853890600969130. [DOI] [PubMed] [Google Scholar]
- 17.Roberts M., Barry S., Woods A., van der Sluijs P., Norman J. PDGF-regulated Rab4-dependent recycling of αvβ3 integrin from early endosomes is necessary for cell adhesion and spreading. Curr. Biol. 2001;11:1392–1402. doi: 10.1016/s0960-9822(01)00442-0. [DOI] [PubMed] [Google Scholar]
- 18.Laakkonen P., Auvinen P., Kujala P., Kaariainen L. Alpha-Virus Replicase protein NSP1 induces filopodia and rearrangement of actin filaments. J. Virol. 1998;72:10265–10269. doi: 10.1128/jvi.72.12.10265-10269.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.