

Abstract
Objectives:
Neuroendocrine tumors (NETs) are uncommon tumors that exhibit a wide range of neuroendocrine differentiation and biological behavior. Primary NETs of the kidney, including carcinoid tumor, small cell carcinoma (SCC), and large cell neuroendocrine carcinoma (LCNEC) are exceedingly rare.
Materials and Methods:
The clinicopathologic features of renal NETs diagnosed at a single institution were reviewed along with all reported cases in the worldwide literature.
Results:
Eighty renal NETs have been described, including nine from our institution. Differentiation between renal NETs and the more common renal neoplasms (renal cell carcinoma, transitional cell carcinoma) can be difficult since clinical, radiographic, and histopathologic features overlap. Immunohistochemical staining for neuroendocrine markers, such as synaptophysin and chromogranin, can be particularly helpful in this regard. Renal carcinoids are typically slow-growing, may secrete hormones, and pursue a variable clinical course. In contrast, SCC and LCNEC often present with locally advanced or metastatic disease and carry a poor prognosis. Nephrectomy can be curative for clinically localized NETs, but multimodality treatment is indicated for advanced disease.
Conclusions:
A spectrum of NETs can rarely occur in the kidney. Renal carcinoids have a variable clinical course; SCC and LCNEC are associated with poor clinical outcomes. Diagnosis of NETs, especially LCNEC, requires awareness of their rare occurrence and prudent use of immunohistochemical neuroendocrine markers.
Keywords: Carcinoid tumor, kidney neoplasm, large-cell neuroendocrine carcinoma, metastasis, neuroendocrine tumor, small cell carcinoma
INTRODUCTION
Neuroendocrine tumors (NETs) are rare neoplasms that share a phenotype notable for neuroendocrine and neural differentiation.[1] Although previous studies have suggested a neural crest origin for all NETs, more recent studies have demonstrated that a few have embryologic origin from the neuroectoderm.[1] NETs can arise from any tissue or organ, including organs that do not normally contain neuroendocrine cells.[1] NETs can be subdivided into epithelial and neural types, both of which express a common set of neuroendocrine markers, including synaptophysin. Neural- type NETs include neuroblastomas, paragangliomas and pheochromocytomas. The classification of epithelial- type NETs is controversial, however, and the nomenclature depends upon the anatomic site of origin. Nevertheless, it has been recognized that NETs represent a spectrum of diseases with a varying degree of biological potential, including well-differentiated NET (carcinoid), well-differentiated neuroendocrine carcinoma, poorly differentiated neuroendocrine carcinoma (large cell neuroendocrine carcinoma (LCNEC)), and small cell carcinoma (SCC).[2,3] NETs may occur in pure forms or may be admixed with non-endocrine components, such as adenocarcinoma or squamous cell carcinoma. The majority of the references to NETs in the literature describe epithelial-type NETs.
The clinical behavior of NETs is heterogeneous and can be difficult to predict based on histology alone. In general, patients with well-differentiated NETs fare better than those with poorly-differentiated NETs. Many well-differentiated NETs are cured by surgical resection alone, although some well-differentiated NETs can pursue an aggressive clinical course. In contrast, poorly differentiated NETs are uniformly aggressive and often fatal.
Renal NETs are exceedingly rare and have been reported as case reports in the literature. Carcinoid tumors of the kidney are unusual because neuroendocrine cells are not found within normal renal parenchyma. The first reported case of a renal carcinoid tumor was over 40 years ago. Ever since, only 62 cases have been reported in the English language literature including six cases from our institution. [4–7] Primary renal SCC not associated with renal pelvic urothelial carcinoma is even rarer, with 18 cases reported in 13 studies. [7–13] Only one case of LCNEC has been reported in the literature so far.[7] The clinical behavior of renal NETs remains unclear due to the rarity of these lesions. In this article, we review the histogenesis, the clinicopathological characteristics, prognostic factors and outcomes of these tumors based on previously published studies.
MATERIALS AND METHODS
A literature search for all articles listed in MEDLINE since 1966 was performed in 2007 using the key words: “Neuroendocrine tumor”, “carcinoid”, “small cell carcinoma”, or “large cell neuroendocrine carcinoma” and “renal” or “kidney”. Epidemiological, clinical, diagnostic, histopathological, therapeutic and prognostic data were evaluated. The surgical pathology database at the authors' institute was queried for any renal neoplasm that was diagnosed as carcinoid tumor, neuroendocrine carcinoma, SCC, or had immunohistochemical stains positive for neuroendocrine immunohistochemical stains (synaptophysin, chromogranin, CD56).
DISCUSSION
Pathogenesis
The pathogenesis of primary NETs of the kidney is still controversial. Neuroendocrine cells have been identified in the renal pelvis but not in the normal renal parenchyma. [14] Different theories support the fact that NETs arise from primitive totipotential stem cells that subsequently differentiate in a neuroendocrine direction. Several mechanisms have been used to explain the origin of such tumors, including metastasis from an occult primary tumor site to the kidney, activation of aberrant gene sequences in a totipotential stem cell line that differentiates into aberrant neuroendocrine tumor cells, and concurrent renal congenital abnormalities. [14,15] El-Naggar et al., reported a loss of heterozygocity at one locus on Chromosome 3p21 in one case of carcinoid tumor and suggested that this anomaly (which is frequent in renal cell carcinoma (RCC)) is a preliminary event that is common to all renal neoplasms including carcinoid tumors.[14] NETs, essentially carcinoid tumors, are frequently associated with horseshoe kidney and teratomas. Romero et al., reported horseshoe kidney in 10 patients (17.8%) and renal teratomas in eight patients (14.3%) concomitantly with carcinoid tumors.[15] In our series, three patients with carcinoid tumors had horseshoe kidney.[7] NETs in a horseshoe kidney were located in midportion of one moiety or the vicinity of the isthmus. This association is likely due to teratogenic events involving the migration of the posterior nephrogenic cells which coalesce to form the isthmus.
Classification
NETs can have a variable degree of neuroendocrine differentiation based on morphological, immunohistochemical, and ultrastructural studies, and are best studied in the gastrointestinal tract and lung. The nomenclature used in the classification of NETs located in different anatomic sites remains controversial.[1] Much of this debate focuses on the use of “carcinoid”, as the term may imply a benign nature, even though some carcinoid tumors exhibit aggressive clinical behavior. In some organ systems, “low-grade neuroendocrine tumor” has been used instead of “carcinoid” to avoid potential confusion. Nevertheless, pathologists and clinicians have become increasingly aware that NETs constitute a spectrum of disease with a wide range of biological behavior, and pathological terminology may not fully reflect clinical outcomes, especially for low-grade NETs.
Prevalence
Renal NETs are extremely rare. There have been 62 renal carcinoid tumors and 18 renal SCC reported so far in the English literature.[7,8,15] Only one LCNEC has been reported to our knowledge.[7] We reported nine cases of renal neuroendocrine neoplasms, including six carcinoid tumors, two SCC and one LCNEC, which accounted for 0.3% of all the primary renal tumors encountered at our institution during a seven-year period.[7]
Epidemiology
The peak age of incidence for NET is between the fifth and sixth decades. Older age, notably older than 40 years, was found to be significantly related to advanced disease at diagnosis and subsequent worse prognosis. The distribution of NETs reflects no predilection to either gender. Nevertheless, carcinoid tumors in horseshoe kidneys are more frequently reported in men, likely reflecting the higher prevalence of horseshoe kidneys in male patients.[15]
Clinical features
Carcinoid tumors tend to grow slowly and may remain silent for many years before manifesting any symptoms. A diagnosis of carcinoid tumor is rarely suspected in patients with renal cortical neoplasms prior to surgery as the clinical features of these patients are similar to those with other renal neoplasms, with the exception of a disproportionate number occurring in horseshoe kidneys (17.8% of previously reported cases[15] and three of six carcinoids in our series[7]). These tumors may present with flank or abdominal pain, hematuria, weight loss, or abdominal mass, but about 27% of patients are detected incidentally.
Carcinoids have the potential to secrete hormonal substances and to induce neuroendocrine syndromes. Carcinoid syndrome characterized by flushing, dyspnea, diarrhea, is the most common form.[15] Other syndromes are rare and include one case of oncocytic carcinoid tumor presenting with episodic Cushing syndrome due to ectopic adrenocorticotrophic hormone secretion (ACTH) secretion,[16] one case of watery diarrhea – hypokalemia – achlorhydia due to vasoactive intestinal peptide secretion,[17] and a case of symptoms related to glucagon secretion by tumor.[18] Clinically documented neuroendocrine syndromes have been reported in 12.8% of patients with carcinoid tumors,[15] but none of our patients presented with such syndromes.
Our review of the currently published literature confirms that renal SCCs are highly aggressive tumors, as is renal LCNEC, like their counterparts in other anatomic sites. These patients often present with locally advanced disease and may have distant metastases with associated symptoms. Despite multi-modality treatment, the prognosis is usually dismal and patients often succumb to the disease shortly after diagnosis. SCC and LCNEC may respond to chemotherapy that improves patient's survival, however, making it important for pathologists to recognize and diagnose these tumors.
Macroscopic features
NETs are usually unilateral and solitary. Carcinoid tumors typically have a well-circumscribed yellow or tan brown cut surface with some foci of hemorrhage, calcification or necrosis. Rarely, they may present with a grossly diffuse necrotic, multilobulated and hemorrhagic friable cut surface [Figure 1]. Necrosis and multilobulation are more frequently seen in SCC and LCNEC.
Figure 1.
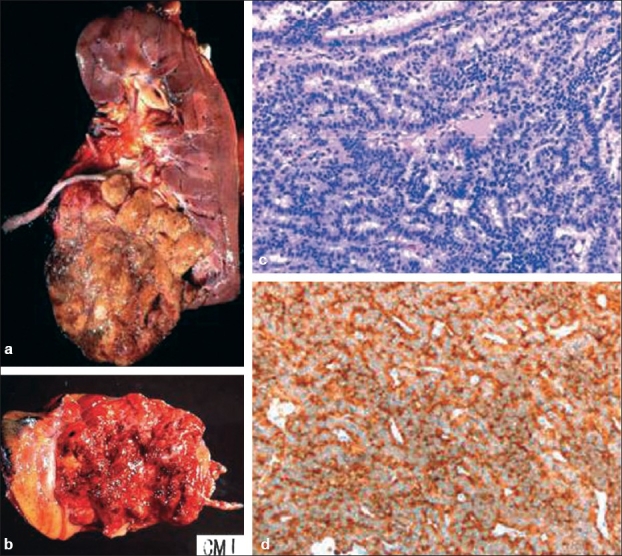
(a) Renal carcinoid tumor. Grossly it forms a circumscribed mass with a yellow cut surface with foci of hemorrhage. (b) Occasionally, it can have a hemorrhagic and friable cut surface. (c) Microscopically it comprises ribbon and trabeculae of uniform tumor cells, (d) which are positive for synaptophysin immunostain
Microscopic features
Renal carcinoid tumors typically present with a trabecular or ribbon-like growth pattern along with peripheral palisading. This leads to the typical rosette-like arrangement which is characteristic of these tumors [Figure 2]. Cells are polygonal with homogenous chromatin repartition and round to oval nuclei. Rare mitotic events are usually seen (<2 per high- power fields HPF). Necrosis is usually lacking.[19] However, the occasional pseudo-papillary appearance of carcinoid tumors may lead to misdiagnosis.[15,19] In our study, one renal carcinoid was initially misdiagnosed as papillary tubular carcinoma and the diagnosis was changed to carcinoid tumor only after a metastatic carcinoid tumor was found in the liver.[7] Of the reported cases in the medical literature 14.5% were initially misdiagnosed histopathologically because of the rarity of such tumors and the lack of a fully blown neuroendocrine syndrome. Only after retrospective analysis, were these tumors characterized as carcinoid tumors. Necrosis, calcification and hemorrhage are often identified in carcinoid tumors. While necrosis and hemorrhage are associated with a more aggressive course and a poorer prognosis, calcification is considered a stigmata of long-term tumor growth and is associated with a more indolent course.[15] None of these differences were proven to be statistically significant.[15]
Figure 2.
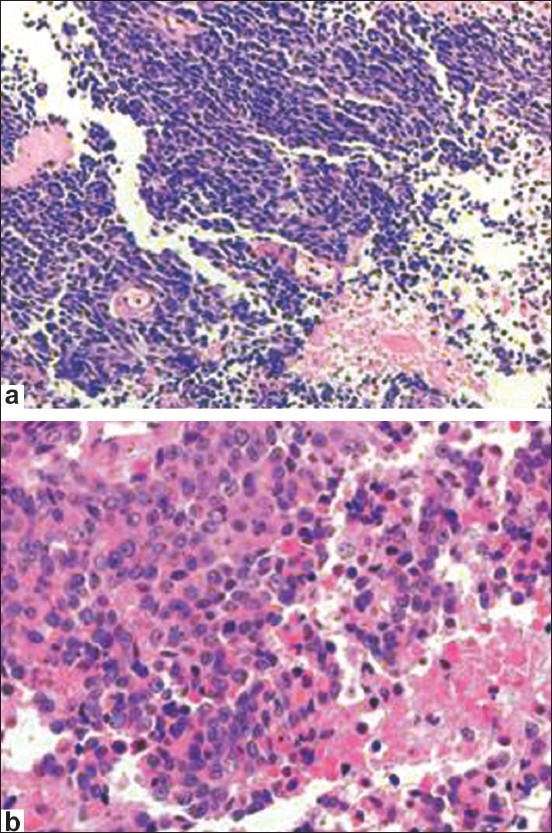
Small cell carcinoma (a) has tumor cells with scant cytoplasm, finely granular chromatin with inconspicuous nucleoli, high mitotic rate and tumor necrosis. Large cell neuroendocrine carcinoma (b) has a solid growth pattern, large zone necrosis, large cell size, low nuclear to cytoplasmic ratio, vesicular chromatin, and frequent mitosis
LCNEC has a typical neuroendocrine growth pattern (organoid nesting, palisading, rosettes, trabeculae), a mitotic rate >2 per 10 HPF, large zone necrosis, cytological features of non-small cell carcinoma (large cell size, low nuclear to cytoplasmic ratio, vesicular or fine chromatin, and/or frequent nucleoli), and positive immunohistochemical staining for one or more neuroendocrine markers.[7] SCC has tumor cells with scant cytoplasm, finely granular chromatin with inconspicuous nucleoli, high mitotic rate and frequent large zone necrosis. LCNEC can be difficult to diagnose and can be mistaken for a high-grade renal cell carcinoma, or urothelial carcinoma, as the morphology overlaps between these entities. A correct diagnosis relies on the awareness of the occasional occurrence of LCNEC in the kidney and careful sampling of atypical tumors. Extensive sampling often will reveal areas with histological features characteristic for RCC or urothelial carcinoma in an otherwise poorly differentiated RCC. In these cases, immunostains for neuroendocrine and renal markers are required to establish the diagnosis. The expression of these markers has not been studied in renal LCNEC, but has been studied in LCNEC and neuroendocrine carcinomas of other organs. LCNEC is positive for at least one of the neuroendocrine markers (synaptophysin, chromogranin and CD56), but rarely positive for CD10.[20–22] In contrast, RCC and urothelial carcinoma is often positive for CD10, but negative for neuroendocrine markers.[20–22] Therefore, a combination of neuroendocrine markers and CD10 will help establish a diagnosis of renal LCNEC. In the one reported case of renal LCNEC, tumor cells were diffusely positive for synaptophysin, but negative for CD10. Synaptophysin seems to be the most sensitive marker for diagnostic confirmation of a renal NET. Prostatic Acid Phosphatase positivity has been reported in some cases of carcinoid tumors but Prostatic Specific Antigen is negative.[14] Vimentin and S100 are also negative. In our previously published series, synaptophysin was positive in all renal NETs, including carcinoid tumors, SCC and LCNEC. Other markers were less sensitive in our study, including chromogranin, which was positive in one of three SCC/LCNEC, and negative in four of four carcinoid tumors. A recent meta-analysis of the neuroendocrine markers in renal carcinoid tumors in published case reports demonstrated 100% (15/15) sensitivity for synaptophysin, and 97.2% (34/35) sensitivity for chromogranin.[15] The difference in chromogranin sensitivity in our study may reflect a difference in the laboratory protocols.[7]
Radiological findings
Several radiographic findings are commonly present in carcinoid tumors, including no or minimal enhancement, heterogeneity, and calcifications, which were present in 75%, 60%, and 26% of reported lesions, respectively. [15,23–26] These findings are neither sensitive nor specific for renal carcinoid tumor and cannot distinguish these tumors from RCC. Typically, carcinoid tumors present with a well- circumscribed, non-enhancing or slightly enhancing mass on computed tomography (CT) with a solid component. [26] Cystic components or calcifications may be present in some cases. The typical finding on renal angiography is a hypovascular or avascular lesion. Somatostatin scintigraphy is useful for diagnosis, staging, and monitoring for the development of recurrence or metastases of NETs after treatment. It has been reported to be a sensitive diagnostic tool, as 85% of primary carcinoid tumors and metastases have high affinity for somatostatin. One limiting aspect of octreotide scintigraphy is that the tracer material is taken up by the normal renal parenchyma, which may lead to an underestimation of potentially suspicious lesions.
Staging
Renal carcinoid tumors, although considered to be low- grade NETs, can pursue an aggressive clinical course. Patients with primary renal carcinoid tumors frequently present with metastatic disease (45.6% of reported cases) and the risk of metastasis is directly related to the size of the primary tumor.[15] This can be explained by the fact that carcinoid tumors develop in a potentially expandable space; the retroperitoneum. In our series, two of five patients with renal carcinoids developed lymph node or distant metastases, including one patient with liver metastases after complete resection of a 4.5-cm carcinoid tumor confined to the kidney. The other patient had synchronous bilateral disease with a 6-cm tumor in the left kidney, two pathologically-proven locoregional lymph node metastases, and three tumors (0.6-3.5 cm) in the right kidney. It is not clear whether this patient had bilateral primary tumors or the tumors in the right kidney were metastatic from the larger tumor in the left kidney, although the tumors from both kidneys were histologically identical and the patient lacked clinical or radiographic evidence of other metastases, including an octreotide scan. The pathologic findings that have been associated with metastasis are large tumor size, high mitotic rate (>1 per 10 HPF) and lack of confinement to the kidney. [15,27] In our series, however, none of the carcinoids had any appreciable mitosis. One carcinoid tumor with hepatic metastasis was organ-confined (Stage T1b), and the other tumor with synchronous contralateral carcinoid tumors and lymph node metastases extended out of the kidney. Therefore, the histological features are unreliable in predicting the biological nature of renal carcinoid tumors and all patients should undergo rigorous oncologic surveillance.
Evaluation after diagnosis
After initial diagnosis, octreotide scintigraphy is the most important primary investigation. Serum chromogranin and 5-HIAA should be requested even in the absence of an overt carcinoid syndrome to evaluate for this condition. These tumor markers can also help monitor tumor progression or relapse after treatment. Ultrasonography is the single most sensitive diagnostic tool to diagnose liver metastases, especially when combined with biopsy. CT and magnetic resonance imaging are both needed to identify lymph node metastases and pulmonary metastases.
Treatment
Surgical excision with regional lymph node dissection has been proven to be curative for renal carcinoid tumors and low-stage renal NETs of other histology. Nephrectomy should be viewed as the primary mode of therapy for these patients, but many will require additional treatments for the best chance at cure. In the reviewed literature, 47% of patients who underwent nephrectomy for renal carcinoids were disease-free after a mean follow-up of 43 months.[15] Unfortunately, SCC and LCNEC often present with extrarenal extension and may not be amenable to complete resection. Due to the rarity of these tumors, no standard treatment has been approved for locally advanced or metastatic disease.[27] In cases with liver metastases treatment is controversial, as nephrectomy rarely cures the disease but may help to relieve symptoms. Systemic and trans-arterial chemotherapy have been used in some patients with NETs with liver metastases.[27] Residual local disease can be treated with radiotherapy, and chemotherapy with platin-based agents can be used for treatment or palliation in the setting of metastatic disease. Although multimodality treatment appears to be warranted, outcome in the three patients with SCC or LCNEC in our series has been suboptimal despite this approach.[7]
Prognosis
In our study, four of five patients with carcinoid tumors were free of disease at the most recent follow-up.[7] These results compared favorably with the combined reported 63% cancer-free survival at 34 months follow-up (median), but are limited by the small number of cases, more limited follow-up (median: Nine months), and smaller average tumor size (4.8cm) compared with the 56 previously reported cases (8.4cm).[15] In fact, three major prognostic factors have been identified for patients with renal carcinoids.[15] Age above 40 years is associated with a more rapid progression and a more severe initial presentation. Tumors measuring less than 4cm or confined to the renal parenchyma tend to metastasize less and thus are associated with a better prognosis. Purely solid tumors and those with a mitotic rate higher than 1/10HPF have also been reported to be associated with a worse prognosis.[15]
CONCLUSION
In summary, a spectrum of NETs can occur in the kidney. Renal carcinoids have a variable clinical course, and SCC and LCNEC are associated with poor clinical outcomes. Diagnosis of NETs, especially LCNEC, requires keen awareness of their rare occurrence in the kidney and prudent use of immunohistochemical neuroendocrine markers.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.DeLellis RA, Osamura RY. Neuroendocrine tumors: An overview. Path Case Rev. 2006;11:229–34. [Google Scholar]
- 2.Oberg K. Neuroendocrine tumors of the gastrointestinal tract: Recent advances in molecular genetics, diagnosis, and treatment. Curr Opin Oncol. 2005;17:386–91. doi: 10.1097/01.cco.0000167739.56948.a9. [DOI] [PubMed] [Google Scholar]
- 3.Travis WD. The concept of pulmonary neuroendocrine tumoures. In: Travis WD, Bramibilla E, Muller-Hermelink HK, Harris CC, editors. Pathology and genetics: Tumours of the lung, pleura, thymus and heart. Lyon: IARC Press; 2004. pp. 19–20. [Google Scholar]
- 4.Resnick ME, Unterberger H, McLoughlin PT. Renal carcinoid producing the carcinoid syndrome. Med Times. 1966;94:895–6. [PubMed] [Google Scholar]
- 5.Mufarrij P, Varkarakis IM, Studeman KD, Jarrett TW. Primary renal carcinoid tumor with liver metastases detected with somatostatin receptor imaging. Urology. 2005;65:1002. doi: 10.1016/j.urology.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 6.Shurtleff BT, Shvarts O, Rajfer J. Carcinoid tumor of the kidney: Case report and review of the literature. Rev Urol. 2005;7:229–33. [PMC free article] [PubMed] [Google Scholar]
- 7.Lane BR, Chery F, Jour G, Sercia L, Magi-Galluzzi C, Novick AC, et al. Renal neuroendocrine tumours: A clinicopathological study. BJU Int. 2007;100:1030–5. doi: 10.1111/j.1464-410X.2007.07116.x. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Lois C, Madero S, Redondo P, Alonso I, Salas A, Angeles Montalbαn M. Small cell carcinoma of the kidney: A case report and review of the literature. Arch Pathol Lab Med. 2001;125:796–8. doi: 10.5858/2001-125-0796-SCCOTK. [DOI] [PubMed] [Google Scholar]
- 9.Majhail NS, Elson P, Bukowski RM. Therapy and outcome of small cell carcinoma of the kidney: Report of two cases and a systematic review of the literature. Cancer. 2003;97:1436–41. doi: 10.1002/cncr.11199. [DOI] [PubMed] [Google Scholar]
- 10.Kilicarsalan Akkaya B, Mustafa U, Esin O, Turker K, Gulten K. Primary small cell carcinoma of the kidney. Urol Oncol. 2003;21:11–3. doi: 10.1016/s1078-1439(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 11.Kim JH, Lee SH, Park J, Kim HY, Lee SI, Nam EM, et al. Extrapulmonary small-cell carcinoma: A single-institution experience. Jpn J Clin Oncol. 2004;34:250–4. doi: 10.1093/jjco/hyh052. [DOI] [PubMed] [Google Scholar]
- 12.Karadeniz-Bilgili MY, Semelka RC, Hyslop WB, Pamuklar E, Rivero H, Firat Z, et al. MRI findings of primary small-cell carcinoma of kidney. Magn Reson Imaging. 2005;23:515–7. doi: 10.1016/j.mri.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 13.Chung CH, Park CY. Small cell carcinoma of the kidney. Korean J Intern Med. 2006;21:191–3. doi: 10.3904/kjim.2006.21.3.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.el-Naggar AK, Troncoso P, Ordonez NG. Primary renal carcinoid tumor with molecular abnormality characteristic of conventional renal neoplasms. Diagn Mol Pathol. 1995;4:48–53. doi: 10.1097/00019606-199503000-00009. [DOI] [PubMed] [Google Scholar]
- 15.Romero FR, Rais-Bahrami S, Permpongkosol S, Fine SW, Kohanim S, Jarrett TW. Primary carcinoid tumors of the kidney. J Urol. 2006;176:2359–66. doi: 10.1016/j.juro.2006.07.129. [DOI] [PubMed] [Google Scholar]
- 16.Hannah J, Lippe B, Lai-Goldman M, Bhuta S. Oncocytic carcinoid of the kidney associated with periodic Cushing's syndrome. Cancer. 1988;61:2136–40. doi: 10.1002/1097-0142(19880515)61:10<2136::aid-cncr2820611034>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 17.Hamilton I, Reis L, Bilimoria S, Long RG. A renal vipoma. Br Med J. 1980;281:1323–4. doi: 10.1136/bmj.281.6251.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gleeson MH, Bloom SR, Polak JM, Henry K, Dowling RH. Endocrine tumour in the kidney affecting small bowel structure, motility and absorptive function. Gut. 1971;12:773–82. doi: 10.1136/gut.12.10.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huettner PC, Bird DJ, Chang YC, Seiler MW. Carcinoid tumor of the kidney with morphologic and immunohistochemical profile of a hindgut endocrine tumor: Report of a case. Ultrastruct Pathol. 1991;15:655–61. doi: 10.3109/01913129109023195. [DOI] [PubMed] [Google Scholar]
- 20.Kaufmann O, Georgi T, Dietel M. Utility of 123C3 monoclonal antibody against CD56 (NCAM) for the diagnosis of small cell carcinomas on paraffin sections. Hum Pathol. 1997;28:1373–8. doi: 10.1016/s0046-8177(97)90226-4. [DOI] [PubMed] [Google Scholar]
- 21.Chu P, Arber DA. Paraffin-section detection of CD10 in 505 nonhematopoietic neoplasms: Frequent expression in renal cell carcinoma and endometrial stromal sarcoma. Am J Clin Pathol. 2000;113:374–82. doi: 10.1309/8VAV-J2FU-8CU9-EK18. [DOI] [PubMed] [Google Scholar]
- 22.Chu PG, Arber DA, Weiss LM. Expression of T/NK-cell and plasma cell antigens in nonhematopoietic epithelioid neoplasms: An immunohistochemical study of 447 cases. Am J Clin Pathol. 2003;120:64–70. doi: 10.1309/48KC-17WA-U69B-TBXQ. [DOI] [PubMed] [Google Scholar]
- 23.Sahin A, Demirbas M, Ozen H, Sungur A, Küηükali T, Aygün N, et al. Primary carcinoid of the kidney. Scand J Urol Nephrol. 1996;30:325–7. doi: 10.3109/00365599609182316. [DOI] [PubMed] [Google Scholar]
- 24.Joshi VV, Nord KS, Hanna M. Case 3: Renal gastrinoma with zollinger- ellison syndrome. Pediatr Pathol. 1986;6:475–6. doi: 10.3109/15513818609041563. [DOI] [PubMed] [Google Scholar]
- 25.Moulopoulos A, DuBrow R, David C, Dimopoulos MA. Primary renal carcinoid: Computed tomography, ultrasound, and angiographic findings. J Comput Assist Tomogr. 1991;15:323–5. [PubMed] [Google Scholar]
- 26.Yoo J, Park S, Jung Lee H, Jin Kang S, Kee Kim B. Primary carcinoid tumor arising in a mature teratoma of the kidney: A case report and review of the literature. Arch Pathol Lab Med. 2002;126:979–81. doi: 10.5858/2002-126-0979-PCTAIA. [DOI] [PubMed] [Google Scholar]
- 27.Kawajiri H, Onoda N, Ohira M, Nakatani T, Wakasa K, Ishikawa T, et al. Carcinoid tumor of the kidney presenting as a large abdominal mass: Report of a case. Surg Today. 2004;34:86–9. doi: 10.1007/s00595-003-2644-x. [DOI] [PubMed] [Google Scholar]